
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-isolated manganese carbonyl on bipyridine-functionalities of periodic mesoporous organosilicas: efficient CO2 photoreduction and detection of key reaction intermediates†
Xia
Wang
a,
Indre
Thiel
b,
Alexey
Fedorov
 b,
Christophe
Copéret
b,
Victor
Mougel
*a and
Marc
Fontecave
*a
b,
Christophe
Copéret
b,
Victor
Mougel
*a and
Marc
Fontecave
*a
aLaboratoire de Chimie des Processus Biologiques, UMR 8229, CNRS, Collège de France, Université P. et M. Curie, PSL Research University, 11 Place Marcelin Berthelot, 75231 Paris Cedex 05, France. E-mail: victor.mougel@college-de-france.fr; marc.fontecave@college-de-france.fr
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog-Weg 1-5, CH-8093 Zürich, Switzerland
First published on 9th October 2017
Abstract
Well-defined and fully characterized supported CO2 reduction catalysts are developed through the immobilization of an earth abundant Mn complex on bpy-PMO (bpy = bipyridine; PMO = Periodic Mesoporous Organosilica) platform materials. The resulting isolated Mn-carbonyl centers coordinated to bipyridine functionalities of bpy-PMO catalyze the photoreduction of CO2 into CO and HCOOH with up to ca. 720 TON in the presence of BIH (1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzoimidazole), used as the electron donor. A broad range of photochemical conditions (varying solvents, sacrificial electron donors, photosensitizer type and concentration, catalyst loading as well as the Mn loading within the PMO) are investigated, demonstrating high activity even for simple organic dyes and Zn-porphyrin as photosensitizers. Spectroscopic and catalytic data also indicate that site isolation of the Mn complex in the PMO framework probably inhibits bimolecular processes such as dimerisation and disproportionation and thus allows the spectroscopic observation of key reaction intermediates, namely the two meridional isomers of the carbonyl complexes and the bipyridine radical anion species.
Introduction
Sunlight-driven conversion of carbon dioxide into carbon-based energy carriers is a highly attractive approach currently envisioned as an alternative to using non-renewable fossil fuels.1,2 A large number of molecular catalysts have been investigated for CO2 photoreduction, for instance the cationic [Re(bpy)(CO)3]+ and [Ru(bpy)2(CO)2]2+ systems, yielding mainly carbon monoxide and formic acid, respectively.3,4 Although these catalysts exhibit reasonable efficiencies with up to 20 and close to 400 productive turnover numbers (TON), their further development is limited by the scarcity and price of the precious metals used. In this context, earth-abundant metal complexes such as [Mn(bpy)(CO)3]+, an analogue of [Re(bpy)(CO)3]+, have been recently studied as catalysts for both electro- and photocatalytic CO2 reduction: while electrocatalytic CO2 reduction using [Mn(bpy)(CO)3]+ yields CO as the only product with quantitative faradaic yields,5 this catalyst also affords formic acid under photocatalytic conditions.6 The difference in product selectivity between electro- and photocatalysis is explained by the formation of distinct catalytically active species. In electrolysis, the dimer [Mn(bpy)(CO)3]2, formed after the one-electron reduction of the starting [Mn(bpy)(CO)3]Br complex, is further reduced at a more negative potential to [Mn(bpy)(CO)3]−, which then reacts with CO2 to give CO and regenerate the dimeric complex.5,7 Under photocatalytic conditions, this reaction competes with the light-induced homolysis of the Mn–Mn bond of the dimeric species, affording a putative monomeric [Mn(bpy)(CO)3]˙ radical species.6 The latter, predominant at the steady state under photocatalytic reaction conditions, has been proposed to catalytically reduce CO2 to formic acid, presumably via an H atom abstraction and formation of a transient Mn–H hydride intermediate. Interestingly, formation of the Mn–Mn dimer can be prevented using diimine ligands bearing bulky substituents or by substituting the halide ligand with strongly coordinating anions such as cyanide.8–11 In these cases, the radical species formed upon one-electron reduction does not dimerize but instead readily disproportionates to generate the doubly reduced species [Mn(L)(CO)3]− (L is a diimine ligand). This mechanism lowers the overpotential required for CO2 electroreduction.8,9 In addition, the stability of the radical species can be controlled by the solvent, and selectivity of the reaction can be tuned under photocatalytic conditions; for instance, formate is the major product in N,N-DMF (DMF coordination stabilizes the singly reduced radical species) while CO is the main product in MeCN.10In that context, immobilisation of Mn carbonyl catalysts has been investigated, improving the robustness, reprocessing and recyclability of the catalysts, by using TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 and Metal–Organic Frameworks (MOFs)13 as supports. Periodic Mesoporous Organosilicas (PMOs) is an alternative class of highly stable organosilica support materials with high surface area and organic functionalities uniformly distributed within the pores and the walls of the silica matrix.14–16 Recent effort towards the immobilization of molecular complexes have shown that such materials constitute ideal platform towards robust molecularly-defined heterogeneous catalysts,17,18 including for the photoreduction of CO2 with Re- and Ru-based PMO materials.19,20
12 and Metal–Organic Frameworks (MOFs)13 as supports. Periodic Mesoporous Organosilicas (PMOs) is an alternative class of highly stable organosilica support materials with high surface area and organic functionalities uniformly distributed within the pores and the walls of the silica matrix.14–16 Recent effort towards the immobilization of molecular complexes have shown that such materials constitute ideal platform towards robust molecularly-defined heterogeneous catalysts,17,18 including for the photoreduction of CO2 with Re- and Ru-based PMO materials.19,20
We thus reasoned that PMOs having bipyridine ligands would be particularly suitable for the immobilisation of Mn carbonyl catalysts, by providing a robust matrix with uniformly-distributed21 well-defined metal sites. The formation of isolated metal sites in molecularly defined environment would combine the advantages of molecular and heterogeneous catalyst, i.e. high activity and selectivity of molecular catalysts and stability of heterogeneous catalysts by preventing bimolecular deactivation pathways.22,23
Herein, we report the synthesis, characterisation and photocatalytic CO2 reduction activity of bipyridyl PMO materials containing earth-abundant [Mn(bpyPMO)(CO)3Br] active sites. Exploring a large set of conditions (type and concentration of photosensitizer, sacrificial electron donors, solvents and metal site density) helps to demonstrate the high catalytic activity of this tailored catalyst, which can reach ca. 720 cumulated turnovers in CO and HCOOH. The site isolation provided by the PMO support also results in opening an original reaction path necessitating two successive one electron transfer steps. The potential necessary to doubly reduce Mn centers is hence lowered, and in situ generated strong reducing agents are required to enable catalytic activity. Furthermore, the high stability of this catalyst enables detecting highly reactive intermediates generated upon photo-irradiation, namely the meridional isomers and the bipyridine radical anion species, thereby providing insight about the reaction mechanism of the photoreduction of CO2.
Results and discussion
Synthesis and characterization of Mn@bpy-PMO materials
The mesoporous bipyridine-containing PMO (bpy-PMO) was synthesized by hydrolysis and condensation of 5,5′-bis(triisopropoxysilyl)-2,2′-bipyridine in the presence of a trimethylstearylammonium surfactant. The porous structure of the material was confirmed by transmission electron microscopy (TEM) and nitrogen adsorption and desorption experiments (Fig. S1†). Analysis according to the BET and Barrett–Joyner–Halenda models revealed a surface area of 596 m2 g−1 and a maximum pore-size distribution of 2.4 nm respectively. As expected, this material shows a high degree of ordering demonstrated by sharp peaks in powder X-ray diffraction with an interpore distance of 1.16(1) nm (Fig. S2†). The Fourier transform infrared (FT-IR) spectrum of the material, presented in Fig. S3,† shows isolated silanols at 3727 cm−1, as well as the characteristic vibrations of the bipyridyl units in the 1500–1350 cm−1 region. Characterisation of the material by solid-state NMR spectroscopy revealed five main resonances in the 13C Cross Polarisation Magic Angle Spinning (CP MAS) spectrum of bpy-PMO at 158, 153, 142, 124 and 121 ppm, characteristic for the bipyridyl fragments (Fig. S5†). The 29Si CPMAS spectrum also shows the expected signals of the T2 and T3-sites of the material −69.0 and −77.6 ppm, respectively (Fig. S6†), in agreement with the previously reported bpy-based PMO materials.24A series of PMO-supported Mn catalysts were synthesized by reaction of bpy-PMO with manganese pentacarbonyl bromide in diethyl ether at room temperature, affording coral-coloured materials (Scheme 1).25 Materials with different loadings of Mn were obtained by varying the ratio between the amount of Mn precursor introduced and the amount of bpy units of the material determined by elemental analysis. We report here the synthesis of materials using nMn/nbpy ratio of 1/2, 1/10 and 1/50, named 1, 2 and 3 respectively. However, while the elemental analyses of materials 2 and 3 were in agreement with the expected Mn loading of 1.3% and 0.4% respectively, material 1 revealed a much lower Mn weight loading than expected if the post-synthetic immobilization was quantitative (ca. 1.4% vs. 6.6% expected). This loading, comparable to that obtained for material 2, corresponds to a functionalisation of ca. 10% of the bipyridine moieties in the material, which appears to be the maximum loading achievable under these conditions. This result is consistent with previous observations with analogous phenylpyridine-based PMO materials, for which a maximum of 15% of the phenylpyridine units could be functionalized.17 Such a value was shown to correspond to the functionalisation of one third of the surface phenylpyridine ligands, the walls of the material being composed of four layers of phenylpyridine building blocks, out of which only the two outer layers are available for functionalisation.26
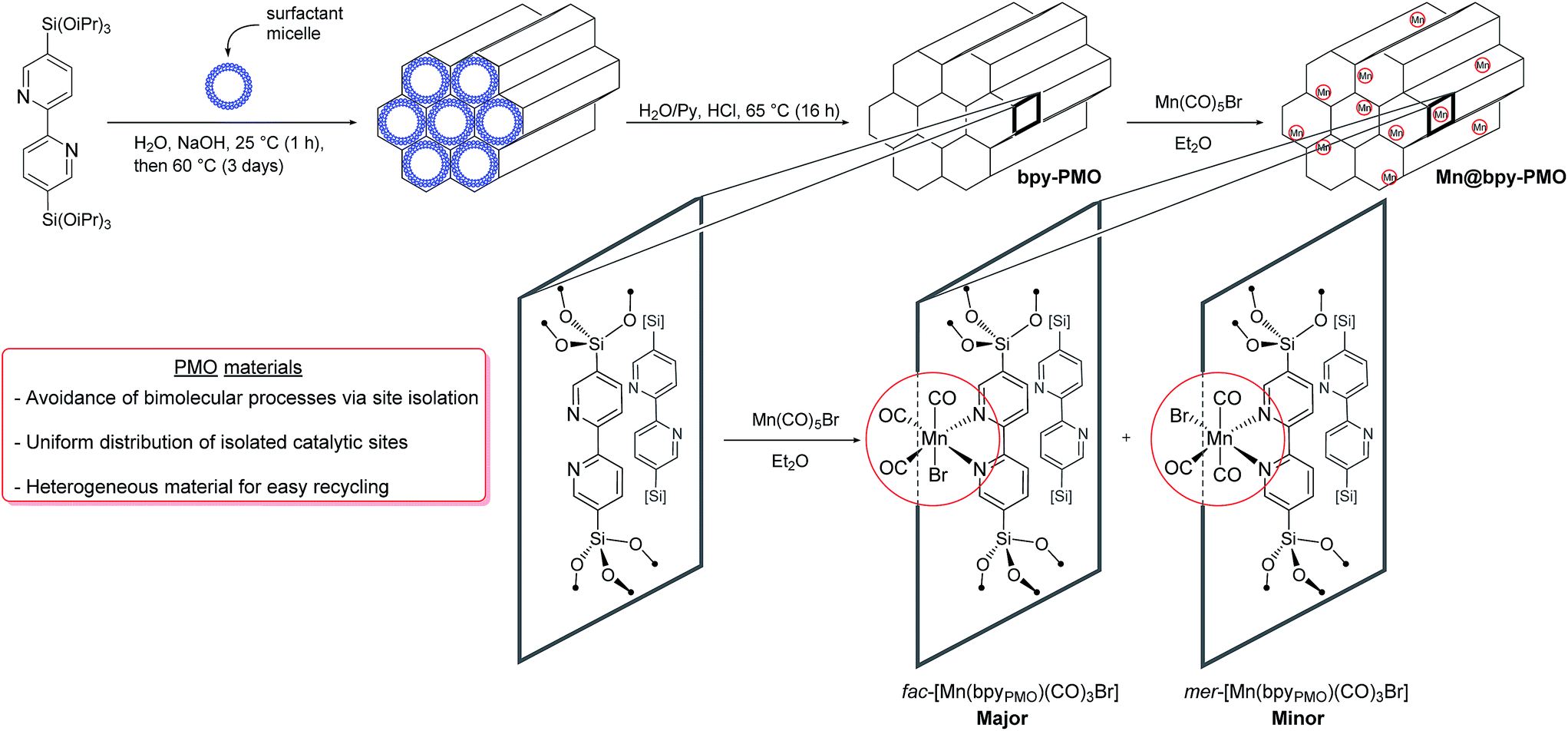 | ||
| Scheme 1 Synthetic pathway to [Mn(bpyPMO)(CO)3Br]. | ||
As a representative example, we will describe only characterisation of material 2 in detail here. The porous structure of bpy-PMO and material 2 was essentially the same, as suggested by the similar TEM images and pore size distribution. Scanning Transmission Electron Microscopy – High-Angle Annular Dark-Field and X-ray Energy-Dispersive Spectroscopy (STEM-HAADF and EDXS) analyses of 2 confirmed the homogeneous distribution of Mn over the whole material (Fig. S7†). The FT-IR spectrum of 2 revealed bands at 2051, 2036, 1951 and 1936 cm−1 (black curve in Fig. 1). These signals cannot be assigned to a single species but are indicative of a mixture of the fac- and mer-isomers of [Mn(bpyPMO)(CO)3Br] (νCO stretches at 2036, 1936 cm−1 and 2051, 1951 cm−1 respectively), with the fac-isomer representing the major species in the material.27 The presence of these two isomers was further confirmed by UV-diffuse reflectance spectroscopy (UV-DRS). The spectrum of 2 (black curve in Fig. 2) displays a broad absorption band from 450 to 600 nm, in agreement with the presence of both fac- and mer-species. Indeed, molecular fac- and mer-[Mn(bpy)(CO)3Br] complexes show absorption maxima at 460 and 510 nm, respectively.27,28
 | ||
| Fig. 1 Infrared spectra of 2 in CO atmosphere (300 mbar) before (black) and after 1 min (red), 15 min (green) or 30 min (blue) irradiation with a 300 W Xe arc lamp equipped with a 400 nm filter at 20 °C. Symbols refer to the species described in Scheme 2. | ||
 | ||
| Fig. 2 UV-diffuse reflectance spectra of 2 in Ar before (black) and after 1 min (Red) or 30 min (blue) irradiation with a 300 W Xe arc lamp equipped with a 400 nm filter at 20 °C. Symbols refer to the species described in Scheme 2. | ||
This finding is surprising since the presence of the mer-isomer at room temperature is not expected. Note, however, that similar behaviour was previously observed upon immobilisation of Mn tris-carbonyl species into a MOF (the mer-species presenting νCO stretches at 2049, and 1946 cm−1, in good agreement with the stretches observed for 2 at 2051 and 1951 cm−1).29 Adventitious light exposure during synthesis of the material could be responsible for this isomerisation. This is in line with the high light-sensitivity of these materials, turning quickly to a pink colour upon light exposure; this behaviour is even more pronounced under vacuum. In order to further investigate processes occurring under light irradiation, we characterized material 2 using FTIR and UV-DRS during irradiation. Proposed photolysis pathway based on these experiments is presented in Scheme 2 and described in details below.
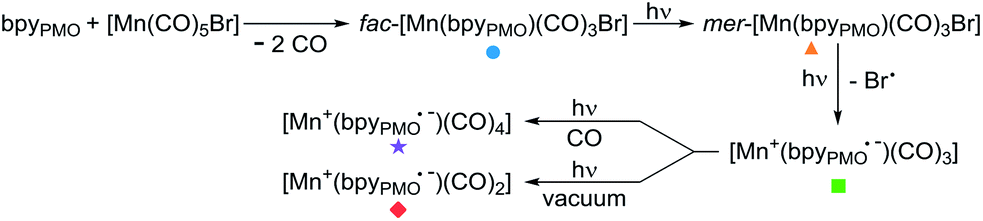 | ||
| Scheme 2 Proposed photolysis pathway of [Mn(bpyPMO)(CO)3Br] species. | ||
The UV-DRS spectrum of 2 (Fig. 2) significantly changed upon light exposure, with the appearance of two new bands at 395 nm and 510 nm attributed to mer-[Mn(bpyPMO)(CO)3Br],27,28 as well as a new band at 540 nm that becomes more pronounced after prolonged irradiation. This feature is reminiscent of the Laporte-forbidden π*–π* transition of bpy˙− ligands and could be tentatively assigned to [Mn+(bpyPMO˙−)(CO)3].30,31
The FTIR spectrum of a pellet of material 2 mixed with KBr was recorded before and after light exposure, under a CO atmosphere or dynamic vacuum. Under a CO atmosphere, at short exposure time (1 min) the νCO bands assigned to the mer-isomer increased in intensity while those assigned to the fac-isomer decreased (Fig. 1). At longer exposure times (15 and 30 min), in addition to a further increase of mer-/fac-ratio, new νCO bands at 2007, 1993, 1975, 1880 (broad), 1857 (broad) and 1820 cm−1 (broad) were observed. The bands at 1975 and 1857 cm−1 are likely due to a singly-reduced Mn complex [Mn+(bpyPMO˙−)(CO)3].8 The features at 2007 and 1880 cm−1 can be assigned to the formation of an octahedral Mn(I) carbonyl species resulting from the coordination of a fifth ligand to the complex [Mn+(bpyPMO˙−)(CO)3]. This ligand originates from a neighbouring bpy moiety in the PMO material or a fourth CO ligand, as both species are known to have similar stretches.8,10,32 Since these bands rapidly disappeared when a sample was exposed to high vacuum (Fig. S9†), the presence of [Mn+(bpyPMO˙−)(CO)4] species is more likely. The weak νCO bands at 1993 and 1820 cm−1 will be discussed below.
As fac- to mer-isomerisation is known to occur in a two-step mechanism via de-coordination/coordination of a CO ligand triggered by light,33 we reasoned that irradiation in dynamic vacuum could aid observing other reaction intermediates. Indeed, when a pellet of material 2 mixed with KBr was exposed to light for 30 minutes under dynamic vacuum, a colour change to dark pink was quickly observed, concomitantly with significant changes in the FTIR spectrum. The intensity of the νCO stretches of both isomers of [Mn(bpyPMO)(CO)3Br] decreased, and new νCO stretches at 1993, 1927, 1876 and 1820 cm−1 appeared (Fig. 3). The shifts of νCO stretching peaks to lower wavenumbers as well as their decreased intensity are likely reflecting a partial loss of CO, consistent with the formation of coordinatively unsaturated bis- and mono-CO Mn(I) complexes.
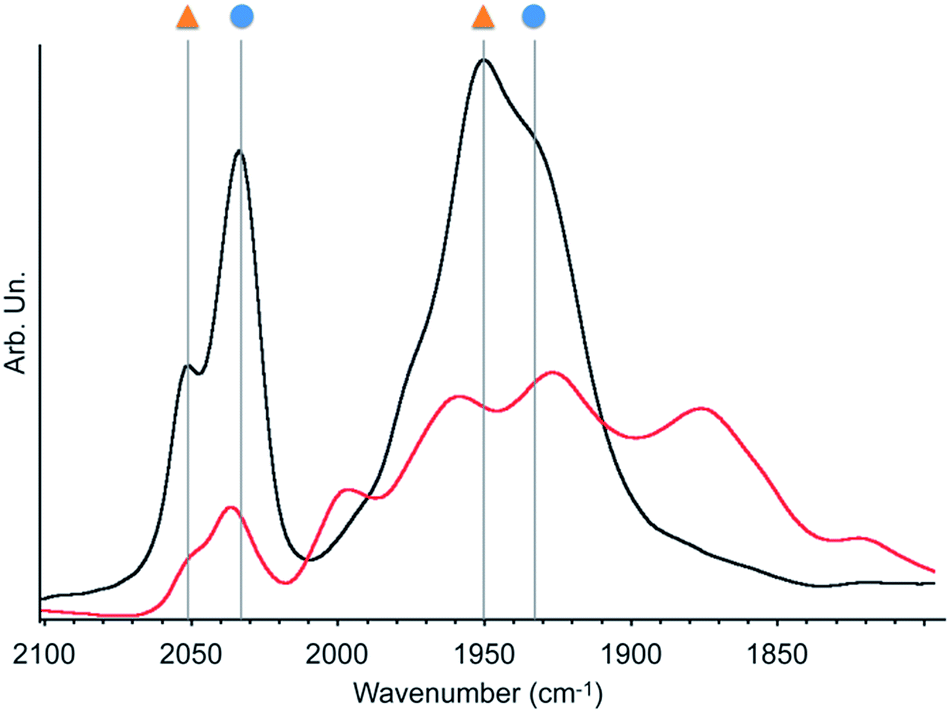 | ||
| Fig. 3 Infrared spectra of 2 under dynamic vacuum (10−5 mbar) before (black) and after (red) 30 minutes irradiation with a 300 W Xe arc lamp. Symbols refer to the species described in Scheme 2. | ||
After 24 h of light exposure under high vacuum the CO stretching peaks in the 2100–1800 cm−1 region have almost fully disappeared, indicating that most of the Mn centers have lost all their initial CO ligands (Fig. S10†). Such CO decoordination has been previously observed with analogous homogeneous Mn(I) complexes bearing diimine ligands. It is likely due to the MLCT into the CO π* orbital, which reduces the metal-to-CO back-bonding and weakens the M–CO bond.34
EPR measurements of the material revealed a clear isotropic signal at giso = 2.007 (linewidth 50 G) (Fig. S11†). This signal is close to the g-factor value of the free electron and is different from the g-factors reported for Mn centred radicals at g = 2.03,35,36 further supporting the presence of a bpy˙− ligand and the assignment of [Mn+(bpyPMO˙−)(CO)3].27,33,37
The spectroscopic results above demonstrate the considerably increased stability of the mer-isomer of [Mn(bpyPMO)(CO)3Br] and of the 16 e− radical [Mn+(bpyPMO˙−)(CO)3] species when immobilised in the PMO material, allowing their characterisation at room temperature. EPR, UV-DRS and FTIR studies confirmed the presence of a reduced bpy˙− ligand in the [Mn+(bpyPMO˙−)(CO)3] radical species generated upon light irradiation. As such radical plays a crucial role in the proposed mechanisms of CO2 reduction using Mn bpy complexes,8–11 its formation in the absence of external photosensitizer highlights the potential of using bpy-PMO materials for photocatalytic CO2 reduction.
Photocatalytic CO2 reduction
The catalytic activity in photochemical CO2 reduction of materials 2 and 3 was assayed in the presence of a photosensitizer and a sacrificial reductant. The system was optimized through varying the electron donors, the photosensitizers and the concentrations of the reactants and catalysts.:1, v/v) solvent mixture, no CO2 reduction activity could be observed (Table S1,† entry 1). No activity was observed upon addition of BNAH either (Table S1,† entry 2). Addition of 0.1 M of BIH gave 49 TONs of formate, 70 TONs of CO and 33 TONs of H2 after 16 h reaction (Table S1,† entry 3). The lack of catalytic activity in the presence of BNAH is intriguing, as a MeCN:TEOA:BNAH mixture has been used6,13 for promoting the photocatalytic activity of Mn(bpy)(CO)3X complexes using ruthenium-based photosensitizers. Consequently, BIH was used as the sacrificial reductant in the following photocatalytic tests.
| Entry | Solvent | Cat. (μmol) | PS (μmol) | Ratio PS/Cat. | Products (μmol) | TON | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CO | Formate | H2 | CO | Formate | H2 | |||||
|
a Samples containing different amounts of catalyst (2, 3 or molecular Mn(bpy)(CO)3Br) and [Ru(bpy)3]Cl2 as PS, 0.1 M of BIH in 1 mL CO2-saturated MeCN/TEOA (5:1, v/v) or N,N-DMF/TEOA (5:1, v/v) solvent mixture upon 16 hours irradiation with a 300 W Xe arc lamp equipped with a 400 nm filter at 20 °C.
|
||||||||||
| 1 | MeCN | 2 (0.1) | 0.1 | 1:1 |
0.2 | 0.5 | 0.1 | 2 | 5 | 1 |
| 2 | MeCN | 2 (1) | 10 | 10:1 |
49 | 70 | 33 | 49 | 70 | 33 |
| 3 | MeCN | 2 (0.1) | 1 | 10:1 |
5.3 | 10.6 | 2.2 | 53 | 106 | 22 |
| 4 | MeCN | 2 (0.01) | 0.1 | 10:1 |
0.29 | 1.42 | 0.39 | 29 | 142 | 39 |
| 5 | MeCN | 3 (0.01) | 0.1 | 10:1 |
1.68 | 2.92 | 0.72 | 168 | 292 | 72 |
| 6 | N,N-DMF | 2 (0.1) | 1 | 10:1 |
0.9 | 1.8 | 0.1 | 9 | 18 | 1 |
| 7 | MeCN | Mn(bpy)(CO)3Br (0.01) | 0.1 | 10:1 |
0.15 | 0.65 | 0.14 | 15 | 65 | 14 |
We investigated the effect of dilution of the metal centres in the materials by comparing the activity of material 2 and material 3 (Table 1, entries 4–5). As mentioned above, the Mn catalyst in 3 is ca. 5 times more diluted than in 2. Catalyst 3 yielded higher TONs than 2 while being slightly less selective for formate. The higher TON values obtained with 3 likely indicate that only a part of the catalytic sites is accessible to the substrate when the loading of the Mn catalyst in the material is too high. The TONs obtained with material 3 were the highest observed with all materials described here, and are ca. 6 times greater than those observed with the [Mn(bpy)(CO)3]Br molecular analogue (Table 1, entries 5 and 7).
| Entry | PS | TON | ||
|---|---|---|---|---|
| CO | Formate | H2 | ||
|
a Samples containing 0.01 μmol of catalyst 3 and 0.1 mM of different photosensitizers, 0.1 M of BIH in 1 mL CO2-saturated ACN/TEOA (5:1, v/v) solvent mixture upon 5 hours irradiation with a 300 W Xe arc lamp equipped with a 400 nm filter at 20 °C.
|
||||
| 1 | [Ru(bpy)3]Cl2 | 153 | 243 | 25 |
| 2 | Fluorescein | 11 | 65 | 0 |
| 3 | ZnTPP | 17 | 52 | 0 |
Kinetic and recyclability studies
The time-dependent formation of formate, CO and H2 during irradiation of a sample containing 0.01 μmol mL−1 of catalyst 3, 0.1 mM of [Ru(bpy)3]Cl2 photosensitizer and 0.1 M of BIH in MeCN/TEOA solvent mixture is presented in Fig. 4. No induction period could be observed for the formation of CO or formate. The latter product forms with a slightly higher initial rate (TOF = 38 min−1) with respect to that for CO formation (TOF = 30 min−1), which is consistent with the results presented in Table 1 as a higher selectivity for formate is observed under these conditions. The decreased rate occurring after 2 hours of irradiation is likely due to the deactivation of the Ru photosensitizer upon illumination.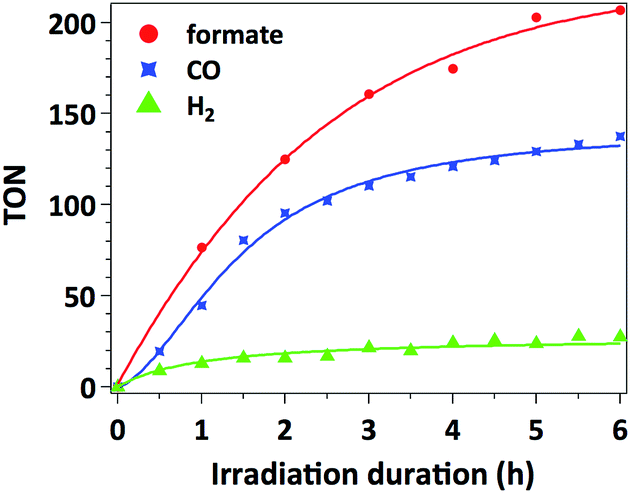 | ||
| Fig. 4 Formate (red circle), CO (blue quadrangle) and H2 (green triangle) evolution catalyzed by 0.01 μmol of catalyst 3 in the presence of 0.1 mM of [Ru(bpy)3]Cl2 and 0.1 M of BIH in 1 mL CO2-saturated MeCN/TEOA (5:1, v/v) solvent mixture upon irradiation with a 300 W Xe arc lamp equipped with a 400 nm filter at 20 °C. TONs are defined relative to Mn weight loading in the catalyst. Lines are sketched through the data points to guide the eye. | ||
Accordingly, separating and washing the catalyst and finally adding a second portion of 10 equiv. of [Ru(bpy)3]Cl2 restores more than 60% of the initial catalytic activity. When such treatment is repeated three times (5 hours for each run), catalyst 3 yields overall formate and CO TONs of 484 and 239 respectively (Fig. 5). However, decreasing CO2 conversion was observed at each run. This behaviour is typically observed with such heterogenized catalysts and could be attributed to (I) catalyst mass loss during separation/washing steps, (II) irreversible deactivation of the catalyst when the material was exposed to air during the rinsing procedure, (III) Mn leaching from the catalyst.13 In the latter case, Mn leaching in solution could lead to in situ formation of Mn–Mn dimers, by analogy with what was observed with the molecular analogue [Mn(bpy)(CO)3Br]. However, characterization of the catalysts by UV-DRS and FTIR immediately after catalytic tests does not show the appearance of the characteristic spectroscopic signatures of the Mn–Mn dimer such as intense absorption bands at λ = 620 and 830 nm in the UV-DRS spectrum and νCO bands at 1975, 1936 and 1886 cm−1 (Fig. S12 and S13†),27 proving that dimeric Mn species do not form under catalytic conditions and thereby illustrating the advantage of site isolation obtained by anchoring Mn onto the bpy-PMO material.
 | ||
| Fig. 5 TONs of CO (blue), formate (red) and H2 (green) production over 0.01 μmol catalyst 3 as a recyclability test. Each run was performed on samples containing recycled 3, a fresh solution of 0.1 mM of [Ru(bpy)3]Cl2 and 0.1 M BIH in 1 mL CO2-saturated MeCN/TEOA (5:1, v/v) solvent mixture. Samples were illuminated for 5 hours with a 300 W Xe arc lamp equipped with a 400 nm filter at 20 °C. | ||
Reaction mechanism
As reported in the photocatalytic activity section, the main differences between the Mn PMO materials reported here and the previously described Mn–bipyridine molecular catalyst6,10 are (i) the need of using BIH instead of BNAH to observe photocatalytic activity, (ii) the increased formate selectivity at higher BIH/Cat. ratio and (iii) the ease of formation of the one-electron reduced radical species in the material, the latter being observed even in the absence of photosensitizers. We propose here a reaction mechanism comprising two parts, a photoexcitation cycle and a catalytic cycle (Schemes 3 and 4 respectively), taking these observations into account.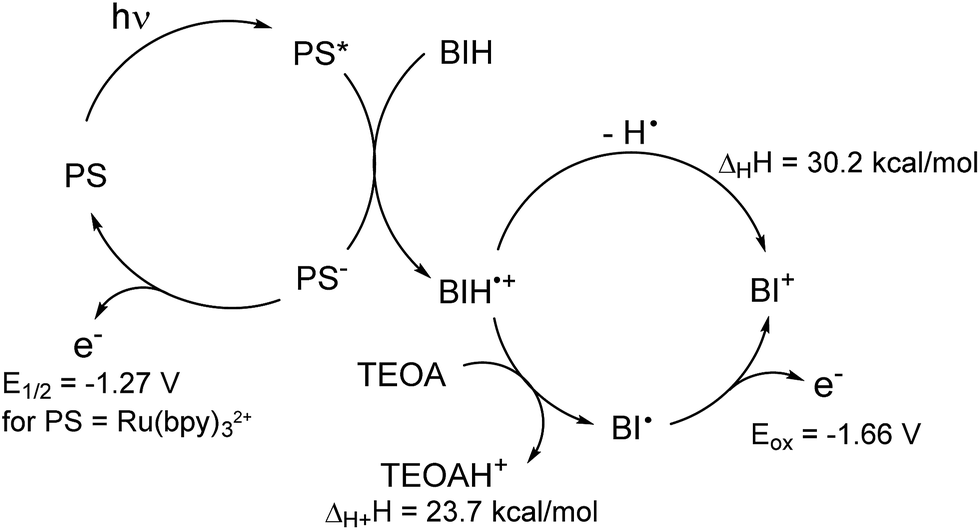 | ||
| Scheme 3 Proposed photoexcitation scheme involving a BIH sacrificial donor. Enthalpy changes in hydrogen atom and proton transfer, as well as Eox(BI˙), were determined in ref. 44, E1/2(Ru(bpy)2+/+) value is given in ref. 45. Redox potentials are given vs. SCE. | ||
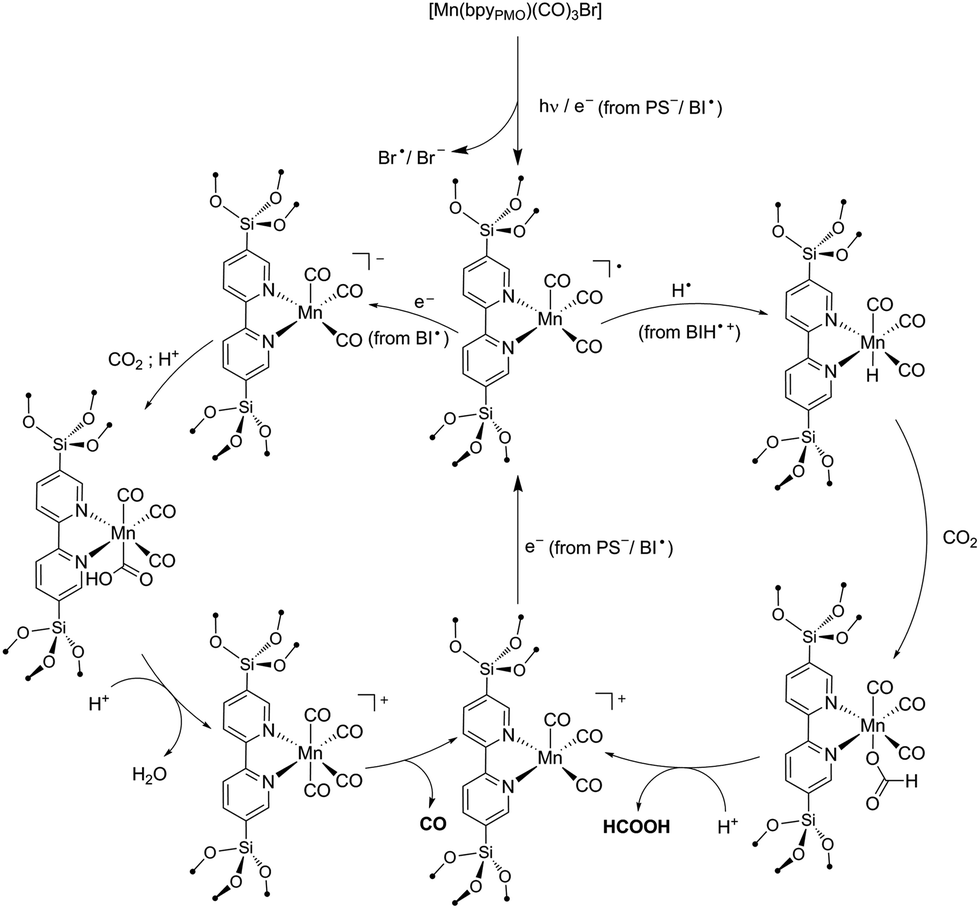 | ||
| Scheme 4 Proposed Mechanisms for the formation of HCO2H and CO from the photocatalytic reaction with [Mn(bpyPMO)(CO)3Br]. | ||
The lack of catalytic activity in the absence of PS (Table S2† entry 2) confirmed that the photoexcitation cycle is initiated by the photoexcitation of the PS. The excited PS is then reductively quenched by BIH to afford the one-electron reduced PS and BIH˙+ (Scheme 3). BIH˙+ can further react through two reaction paths. It can be deprotonated by TEOA to afford BI˙ or undergo hydrogen atom transfer generating the two-electron reduced species BI+.41 These two paths were determined to have a rather small difference in enthalpy change and both are likely to occur in the reaction conditions.48 In addition, the radical species BI˙ was shown to have a higher reducing power (Eox = −1.66 V vs. SCE in MeCN)48 than that of the one-electron reduced photosensitizer [Ru(bpy)3]+ itself (E1/2 = −1.27 V vs. SCE in MeCN)49 and can potentially reduce the Mn active site directly as well as the PS. BNAH could follow the same photoexcitation cycle forming PS− species as well.41 However, BNAH differs from BIH in two aspects: proton transfer is ca. 20 kcal mol−1 more favourable than hydrogen atom transfer in the case of BNAH,48 rendering hydrogen atom transfer much less likely, and the radical species BNA˙ formed from BNAH is not a strong reductant like BI˙, since BNA˙ rapidly dimerizes (BNAH only functions as a one electron donor of Eox = 0.57 V vs. SCE in MeCN).41 This difference in behaviour between BIH and BNAH explains the lack of activity with the latter, and will be developed in more details below.
Previous studies on photochemical CO2 reduction using molecular6,10 and heterogenized [Mn(bpy)(CO)3X]13 catalysts have proposed the formation of CO and formate products through two distinct reaction pathways. One involves the reaction of a photo-generated radical species [Mn(bpy)(CO)3]˙ (from [Mn(bpy)(CO)3]2) with a hydrogen atom donor to afford a Mn–H hydride intermediate which converts CO2 into formate. The second one relies on the direct reduction of CO2 by a doubly reduced [Mn(bpy)(CO)3]− species to afford CO.6,8–10 The ability of N,N-DMF in contrast to MeCN to stabilize the singly reduced radical species [Mn(bpy)(CO)3]˙ was proposed to explain the higher selectivity for formate in N,N-DMF.10
Building on these studies, we interpret our data in terms of the proposed catalytic mechanism shown in Scheme 4. In the photosensitization step, the complex is singly reduced by PS−/BI˙ or via homolysis of the Mn–Br bond. As the singly reduced species [Mn+(bpyPMO˙−)(CO)3] was observed under light by FTIR even in the absence of PS or SD, this initial step likely occurs independently of the SD used. In contrast to the reactions using analogous Mn complexes under homogeneous conditions, in the case of reactions using Mn-containing bpy-PMO materials both formation of dimeric species and disproportionation of singly-reduced radical intermediates are highly unlikely events due to the immobilization and high dilution of the Mn centers in the materials. This is consistent with the surprising stability of the radical [Mn+(bpyPMO˙−)(CO)3] species which can be observed by FTIR spectroscopy during light irradiation at room temperature. This species could be further stabilized by coordination of solvent molecules (omitted in Scheme 4 for brevity). In particular, DMF proved to strongly stabilize such a radical intermediate.10 The higher stability of the radical species in the catalytic cycle in the presence of DMF could also explain the slower kinetics here observed with this solvent.
The singly reduced [Mn+(bpyPMO˙−)(CO)3] species could then follow two parallel paths. Abstraction of a hydrogen atom from BIH˙+ generates a hydride species.13 This hydride species reacts with CO2 to give formate. The larger selectivity for formate observed at lower catalyst concentration (i.e. at higher relative BIH concentration) is consistent with this hydrogen atom abstraction step. Hydrogen atom transfer could be potentially observed as well with BNAH and TEOA SDs. However, these are much weaker hydrogen atom donors than BIH, explaining the absence of formate product when using these SDs.
Alternatively, [Mn+(bpyPMO˙−)(CO)3] can be further reduced to a [Mn(bpy)(CO)3]− species. Such doubly-reduced species have been shown to reduce CO2 to CO.6,8–10 As no CO production was observed in the absence of BIH, we reasoned that the second reduction of the complex cannot be realized by PS− but requires the presence of a stronger reducing agent. This is in agreement with the highly negative potential required for the second reduction of Mn bpy complexes when dimerization is prevented.9 As highlighted above, the radical species BI˙ has very strong reducing power and is the most likely reducing agent for the direct reduction of [Mn+(bpyPMO˙−)(CO)3] species. The lack of activity observed when BNAH is used instead of BIH corroborates this hypothesis. Finally, after protonation of formate or CO release, both reaction paths lead to a [Mn+(bpyPMO)(CO)3] species that can be further reduced by PS− or BI˙ to regenerate the active [Mn+(bpyPMO˙−)(CO)3] species.
In summary, the main difference of the mechanism proposed here and those proposed for molecular species originates from the restraint to dimerization or disproportionation provided by the site isolation in PMO. Since bimolecular interactions (formation of Mn–Mn dimers or disproportionation reaction) are known to ease the two electron reduction of molecular Mn bpy complexes,5,10 stronger reducing agents such as BI˙ are required to doubly reduce the Mn sites when immobilized in PMO materials. In addition, the PMO material provides a higher stability to [Mn+(bpyPMO˙−)(CO)3] intermediate, independently of the solvent used. This permits the observation of different product selectivity than for its homogeneous counterparts.
Conclusions
Well-defined supported Mn carbonyl bipyridine-PMO catalysts have been developed; they show high photo-reduction activity of CO2 in the presence of a photosensitizer, reaching cumulated TONs of 484 and 239 in formate and CO with [Ru(bpy)3]Cl2 photosensitizer. The catalytic activity is preserved using simple earth-abundant photosensitizers such as organic dyes and Zn-porphyrin. A large set of reaction conditions have been explored providing mechanistic insight on the reaction and deactivation mechanism. Preventing dimerization and disproportionation reactions through site isolation allow the rare observation of reaction intermediates, such as mer-[Mn(bpyPMO)(CO)3Br], and radical anion species, such as [Mn+(bpyPMO˙−)(CO)3]. Using this principle, we are currently further developing efficient CO2 photoreduction catalysts.Experimental section
General considerations
Elemental analyses were performed by Mikrolabor Pascher, Remagen, Germany. All infrared (IR) spectra were recorded using a Bruker FT-IR Alpha spectrometer placed in the glovebox. Alternatively, a thin pellet of the sample was pressed in a glovebox and loaded to the reactor using a sample holder into glass reactors with IR-transparent CaF2 windows. The spectra were then recorded in a transmission mode on a Nicolet 6700 or a Shimadzu Prestige 21 FTIR spectrophotometer. The samples were measured in vacuo (10−5 mbar) or in argon or carbon monoxide atmospheres. UV-vis diffuse reflectance spectra were recorded on a Cary 5000 UV-vis-NIR spectrophotometer from Agilent Technologies. Solid-state NMR spectra were recorded under MAS conditions on Bruker Advance III 400 or 700 spectrometers with conventional triple resonance 2.5 and 4 mm CP-MAS probe. Samples were introduced in zirconia rotors in the glovebox. Electron microscopy was done using the ScopeM facilities, ETH Zürich. Nitrogen adsorption/desorption experiments were performed on a BELsorp-mini II instrument, and the specific surface area of the samples was determined using Brunauer–Emmett–Teller (BET) analysis.50 The pore size distributions were calculated at the limit of Barrett–Joyner–Halenda analysis. Experiments under photoirradiation were performed at 20 °C using 300 W Xe arc lamp equipped with a 400 nm filter.All reagents were purchased from Sigma Aldrich except for 1-benzyl-1,4-dihydronicotinamide (TCI), and were used without further purification. [Mn(bpy)(CO)3]Br,5 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzoimidazole,48 zinc tetraphenylporphyrin51 and 2-bromo-5-(triisopropoxysilyl)pyridine24 were synthesized according to published procedures. 5,5′-Bis(triisopropoxysilyl)-2,2′-bipyridine was synthesized according to the general literature procedure24 except that a 125 mL Parr bomb reactor was used (140 °C, 3 days), bis(hexabutyl)tin and toluene in place of bis(tributyltin) and reflux in m-xylene (yield: 1.66 g, 53%).
Synthesis of bpyPMO
Trimethylstearylammonium iodide (0.91 g) was dissolved in dist. water (49 mL) and NaOH(aq) (10 M, 0.17 mL) and stirred for 1 h at room temperature. 5,5′-Bis(triisopropoxysilyl)-2,2′-bipyridine (1.11 g, 2 mmol) was dissolved in ethanol (4 mL) and added via syringe pump (3.0 mL h−1) to the surfactant solution at 60 °C. After complete addition the suspension was sonicated at 50 °C for 1 h, before it was stirred at 60 °C for 3 days, followed by static conditions at 60 °C for 3 days. The solid was then filtered and washed with water (2 × 100 mL) and acetone (2 × 100 mL). The white powder was then suspended in a premixed solution of 2 M HCl, pyridine and water (50 mL:50 mL:17 mL) and stirred at 65 °C for 16 h. The product was filtered and washed with deionized water (3 × 100 mL) and acetone (3 × 100 mL). Drying under high vacuum (10−5 mbar) at 135 °C for 16 h yielded bpy-PMO as a white powder (yield: 350 mg).
EA found: C, 40.62%; H, 2.54%; N, 8.94%.
BET surface area (N2, 77 K) 596 m2 g−1; pore size: 2.4 nm.
EA found: C, 40.86%; H, 2.60%; N, 8.37%; Mn 1.22% corresponding to 0.04 Mn/N (0.08 Mn/bpy). BET surface area (N2, 77 K) 240 m2 g−1; pore size: 2.4 nm.
EA found: C, 42.58%; H, 2.70%; N, 8.93%, Mn 0.41% corresponding to 0.01 Mn/N (0.02 Mn/bpy).
Photolysis procedure
Photochemical reactions were performed using a 300 W, high pressure Xe arc lamp (Oriel Instruments). The beam was passed through an infrared filter, a collimating lens, a filter holder equipped with a 400 nm band pass filter. Samples were prepared in a 1 cm path length quartz cuvette (Starna) which was placed in a temperature controlled cuvette holder (Quantum Northwest) maintained at 20 °C with a circulated water bath.For the entirety of the study, a 5:1 (v/v) mixture of ACN/TEOA or N,N-DMF/TEOA were used as a solvent mixture. The electron donor utilized was a 100 mM solution of BIH or a 100 mM solution of BNAH. The photosensitizer used a solution of [Ru(bpy)3]Cl2, ZnTPP or fluorescein. Samples were saturated with CO2via directly bubbling CO2 through the solution mixture for 10 minutes.
Product detection
H2 measurements were performed by gas chromatography on a Shimadzu GC-2014 equipped with a Quadrex column, a Thermal Conductivity Detector with N2 as a carrier gas. CO was measured using a Shimadzu GC-2010 Plus gas chromatography, fitted with a S9 Restek Shin Carbon column, helium carrier gas, a methanizer and a Flame Ionization Detector. H2 and CO were also analyzed by gas chromatography (SRI Instruments), Multi-Gas Analyzer #5 equipped with a Haysep D column and MoleSieve 5A column. Thermal Conductivity Detector (TCD) and Flame Ionization Detector (FID) with methanizer using argon as a carrier gas. The typical volume of gas injected was 50 μL.Formate concentration was determined using a Metrohm 883 Basic IC plus ionic exchange chromatography instrument, using a Metrosep A Supp 5 column and a conductivity detector. A typical measurement requires the sampling of 200 μL of solution (except in kinetic studies where 15 μL aliquots were sampled), followed by a 100 times dilution in deionised 18 MΩ water and injection of 20 μL into the instrument.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Huan Ngoc Tran for assistance with GC characterization of the reaction products, and Dr Reinhard Kissner and Christopher P. Gordon for assistance with EPR spectroscopy. I. T. and A. F. thank the German Academic Exchange Service (DAAD) and the Holcim Stiftung for a postdoctoral and a Habilitation fellowship, respectively. I. T. and A. F. were also partially funded by the SCCER Heat and Energy storage. The authors acknowledge ScopeM of the ETH Zürich for the use of their electron microscopy facilities.Notes and references
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- H. Ishida, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 905–911 CrossRef CAS.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- C. W. Machan, C. J. Stanton, J. E. Vandezande, G. F. Majetich, H. F. Schaefer, C. P. Kubiak and J. Agarwal, Inorg. Chem., 2015, 54, 8849–8856 CrossRef CAS PubMed.
- P. L. Cheung, C. W. Machan, A. Y. S. Malkhasian, J. Agarwal and C. P. Kubiak, Inorg. Chem., 2016, 55, 3192–3198 CrossRef CAS PubMed.
- M. V. Vollmer, C. W. Machan, M. L. Clark, W. E. Antholine, J. Agarwal, H. F. S. Iii, C. P. Kubiak and J. R. Walensky, Organometallics, 2015, 34, 3–12 CrossRef CAS PubMed.
- T. E. Rosser, C. D. Windle and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 7388–7392 CrossRef CAS PubMed.
- H. Fei, M. D. Sampson, Y. Lee, C. P. Kubiak and S. M. Cohen, Inorg. Chem., 2015, 54, 6821–6828 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608–620 RSC.
- A. Mehdi, C. Reye and R. Corriu, Chem. Soc. Rev., 2011, 40, 563–574 RSC.
- W. R. Grüning, G. Siddiqi, O. V. Safonova and C. Copéret, Adv. Synth. Catal., 2014, 356, 673–679 CrossRef.
- Y. Maegawa and S. Inagaki, Dalton Trans., 2015, 44, 13007–13016 RSC.
- H. Takeda, M. Ohashi, T. Tani, O. Ishitani and S. Inagaki, Inorg. Chem., 2010, 49, 4554–4559 CrossRef CAS PubMed.
- Y. Kuramochi, M. Sekine, K. Kitamura, Y. Maegawa, Y. Goto, S. Shirai, S. Inagaki and H. Ishida, Chem.–Eur. J., 2017, 23, 10301–10309 CrossRef CAS PubMed.
- I. Thiel, A. Fedorov, R. Verel, S. Yakunin, M. V. Kovalenko and C. Copéret, Phys. Chem. Chem. Phys., 2016, 18, 13746–13749 RSC.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- C. Copéret, A. Fedorov and P. A. Zhizhko, Catal. Lett., 2017, 147, 2247–2259 CrossRef.
- M. Waki, Y. Maegawa, K. Hara, Y. Goto, S. Shirai, Y. Yamada, N. Mizoshita, T. Tani, W.-J. Chun, S. Muratsugu, M. Tada, A. Fukuoka and S. Inagaki, J. Am. Chem. Soc., 2014, 136, 4003–4011 CrossRef CAS PubMed.
- A control reaction of [Mn(CO)5Br] with bisphenyl-PMO, a material where bpy units are replaced by bisphenyl ones, did not result in any immobilization of Mn in this PMO material, as evidenced by the absence of νCO bands in the FTIR spectra (Fig. S14†) and catalytic activity of the resulting material (see ESI† for details).
- W. R. Grüning, A. J. Rossini, A. Zagdoun, D. Gajan, A. Lesage, L. Emsley and C. Copéret, Phys. Chem. Chem. Phys., 2013, 15, 13270 RSC.
- G. J. Stor, S. L. Morrison, D. J. Stufkens and A. Oskam, Organometallics, 1994, 13, 2641–2650 CrossRef CAS.
- C. J. Kleverlaan, F. Hartl and D. J. Stufkens, J. Organomet. Chem., 1998, 561, 57–65 CrossRef CAS.
- A. J. Blake, N. R. Champness, T. L. Easun, D. R. Allan, H. Nowell, M. W. George, J. Jia and X.-Z. Sun, Nat. Chem., 2010, 2, 688–694 CrossRef CAS PubMed.
- C. Creutz, Comments Inorg. Chem., 1982, 1, 293–311 CrossRef CAS.
- C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, Organometallics, 1985, 4, 2161–2166 CrossRef CAS.
- J. Tory, B. Setterfield-Price, R. A. W. Dryfe and F. Hartl, ChemElectroChem, 2015, 2, 213–217 CrossRef CAS.
- F. Hartl, B. D. Rossenaar, G. J. Stor and D. J. Stufkens, Recl. Trav. Chim. Pays-Bas, 1995, 114, 565–570 CrossRef CAS.
- C. Daniel, D. Guillaumont, C. Ribbing and B. Minaev, J. Phys. Chem. A, 1999, 103, 5766–5772 CrossRef CAS.
- D. R. Kidd, C. P. Cheng and T. L. Brown, J. Am. Chem. Soc., 1978, 100, 4103–4107 CrossRef CAS.
- R. W. Saalfrank and W. Rost, Angew. Chem., Int. Ed., 1985, 24, 855–856 CrossRef.
- R. R. Andréa, W. G. J. de Lange, T. van der Graaf, M. Rijkhoff and D. J. Stufkens, Organometallics, 1988, 7, 1100–1106 CrossRef.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146–15151 CAS.
- M. B. Chambers, X. Wang, N. Elgrishi, C. H. Hendon, A. Walsh, J. Bonnefoy, J. Canivet, E. A. Quadrelli, D. Farrusseng, C. Mellot-Draznieks and M. Fontecave, ChemSusChem, 2015, 8, 603–608 CrossRef CAS PubMed.
- K. Kalyanasundaram, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2401–2415 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- While the concentration of the catalyst in the sample cannot be rigorously defined as the reaction is heterogeneous, we describe here by convenience the amount of of Mn sites present in the 1 mL sample in μmol mL−1.
- H. Takeda, K. Ohashi, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 4354–4357 CrossRef CAS PubMed.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S.-M. Ng, C.-C. Ko, G. Liu, L. Chen, M. Robert and T.-C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed.
- J.-X. Zhang, C.-Y. Hu, W. Wang, H. Wang and Z.-Y. Bian, Appl. Catal., A, 2016, 522, 145–151 CrossRef CAS.
- X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed.
- C. M. Elliott and E. J. Hershenhart, J. Am. Chem. Soc., 1982, 104, 7519–7526 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. G. Azenha, A. C. Serra, M. Pineiro, M. M. Pereira, S. de Melo, L. G. Arnaut, S. J. Formosinho and A. M. d'. A. Rocha Gonsalves, Chem. Phys., 2002, 280, 177–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed characterization of the material and photolytic experiments. See DOI: 10.1039/c7sc03512h |
| This journal is © The Royal Society of Chemistry 2017 |