
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChromium complexes for luminescence, solar cells, photoredox catalysis, upconversion, and phototriggered NO release
Laura A.
Büldt†
 and
Oliver S.
Wenger
*
and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
First published on 14th September 2017
Abstract
Some complexes of Cr(III) and Cr(0) have long been known to exhibit interesting photophysical and photochemical properties, but in the past few years important conceptual progress was made. This Perspective focuses on the recent developments of Cr(III) complexes as luminophores and dyes for solar cells, their application in photoredox catalysis, their use as sensitizers in upconversion processes, and their performance as photochemical nitric oxide sources. The example of a luminescent Cr(0) isocyanide complex illustrates the possibility of obtaining photoactive analogues of d6 metal complexes that are commonly made from precious metals such as Ru(II) or Ir(III). The studies highlighted herein illustrate the favorable excited-state properties of robust first-row transition metal complexes with broad application potential.
1. Introduction
Luminescence has been observed from materials with chromium in different oxidation states, but Cr(III) is clearly the most important species in this regard. For coordination geometries that can be approximated as octahedral, the Tanabe–Sugano diagram for the d3 electron configuration (Fig. 1a) shows that there is a critical ligand field strength at which the lowest-energetic electronically excited state changes from 4T2 to 2E. The latter is only weakly distorted relative to the ground state because it derives from the same electron configuration (t2g3), and consequently nonradiative relaxation is comparatively inefficient and the 2E state can exhibit lifetimes in the nano- to microsecond regime. Many Cr(III) polypyridine complexes are beyond the 4T2/2E crossing point, and their photophysical and photochemical properties were first explored more than 40 years ago.1,2 Early studies demonstrated that this class of compounds does not only exhibit favorable luminescence properties,3 but that they also have strong oxidizing power in their long-lived excited-states.4–6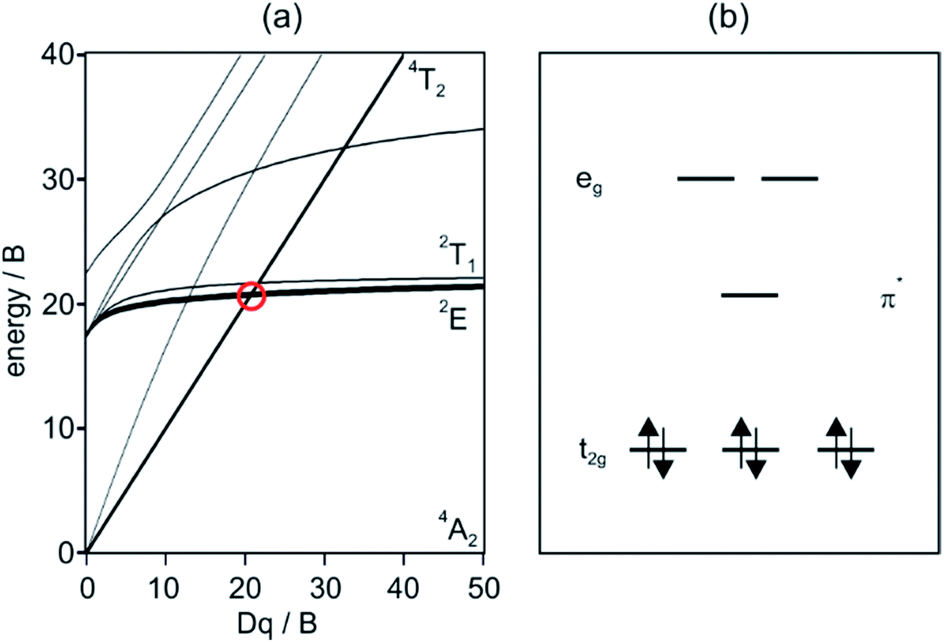 | ||
| Fig. 1 (a) Tanabe–Sugano diagram for the d3 electron configuration. (b) Relevant frontier orbitals for low-spin octahedral d6 metal complexes with low-lying ligand-based π* orbitals. | ||
Cr(0) complexes can be stabilized with π-accepting ligands such as carbonyl or isocyanides. While Cr(CO)6 and related carbonyls were mostly investigated with respect to photoinduced ligand dissociation reactions,7 Cr(0) complexes with arylisocyanide ligands seemed promising for obtaining emissive 3MLCT excited states.8–11 In the low-spin d6 electron configuration, such MLCT states can become the lowest electronically excited states for ligands with accessible empty π* orbitals (Fig. 1b). Ru(II) polypyridines and cyclometalated Ir(III) complexes are currently the most prominent 3MLCT emitters with d6 electron configuration,12 but early studies already indicated that Cr(0) arylisocyanides have the potential to become earth-abundant alternatives to these precious metals.8,9
With both Cr(III) and Cr(0) important progress was made in recent years. Efficient near-infrared photoluminescence from Cr(III) complexes in aqueous solution at room temperature became possible due to advanced ligand design,13 new synthetic approaches gave access to heteroleptic Cr(III) complexes that can be grafted onto semiconductors,14 and Cr(III) complexes were successfully employed for photoredox catalysis of Diels–Alder reactions.15–17 Moreover, when incorporated into helical structures along with Er(III), Cr(III) complexes were able to sensitize near-infrared to visible upconversion for the first time in molecular systems.18 Chelating diisocyanide ligands proved useful for obtaining a luminescent Cr(0) complex that is an emissive analogue of Fe(bpy)32+ (bpy = 2,2′-bipyridine) with a spectacularly long 3MLCT lifetime compared to all previously known isoelectronic Fe(II) complexes.19 All these recent advances are summarized and discussed in the following sections, and they illustrate the broad current interest in chromium as a key constituent of photoactive metal complexes.
2. Near-infrared Cr(III) luminescence in aqueous solution
Early studies demonstrated that photoaquation, i.e., exchange of ligands by H2O upon photoexcitation, occurs in Cr(III) complexes in aqueous solution, particularly under alkaline conditions.20 This is the case for example for Cr(III) polypyridines such as Cr(bpy)33+, Cr(phen)33+ (phen = 1,10-phenanthroline), or Cr(tpy)23+ (tpy = 2,2′:6′,2′′-terpyridine).4 Recently, Heinze, Resch-Genger, and coworkers prepared a new tridentate polypyridine ligand (L1, Fig. 2a) which combines strong σ-donating and π-accepting properties with the capability to chelate to Cr(III) with substantially larger N–Cr–N bite angles than bpy, phen, or tpy.13 In the resulting Cr(L1)23+ complex (Fig. 2b) the ligand field is very strong and given the nearly perfect octahedral coordination, there is an unusually large energy gap of 7100 cm−1 between the relaxed 2E and 4T2 excited states. This is much larger than in common Cr(III) polypyridines, and consequently back intersystem crossing from 2E to 4T2, which normally reduces phosphorescence quantum yields and lifetimes (due to strong distortion in the 4T2 excited state),1 is largely suppressed in Cr(L1)23+. In the form of its tetrafluoroborate salt, this complex is highly soluble in water, and it emits at 775 nm with a quantum yield (ϕ) of 11.0% following excitation at 435 nm into 4A2 → 4T2 and LMCT bands (Fig. 2c). For comparison, Cr(tpy)23+ has ϕ < 0.001% in water, and Cr(bpy)32+ exhibits ϕ = 0.089% under these conditions.13 The luminescence lifetime of Cr(L1)23+ is 898 μs in de-aerated H2O, much longer than for classic Cr(III) polypyridines (Table 1).13 Cr(L1)23+ is stable in aerated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) CH3CN/H2O solution under long-term irradiation at 430 nm while Cr(bpy)33+ undergoes complete photosubstitution within a few hours.
:1 (v/v) CH3CN/H2O solution under long-term irradiation at 430 nm while Cr(bpy)33+ undergoes complete photosubstitution within a few hours.
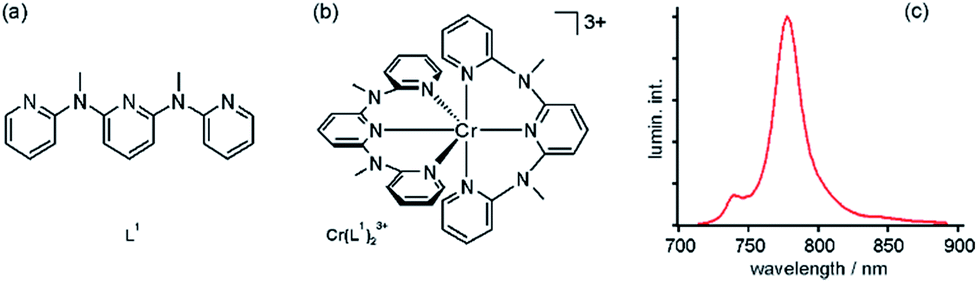 | ||
| Fig. 2 (a) Tridentate ligand that can chelate Cr(III) with a N–Cr–N bite angle close to 90 °C. (b) Resulting bis(terdentate) Cr(III) complex. (c) Luminescence spectrum of this complex in de-aerated H2O at room temperature following excitation at 435 nm. Reproduced with permission from ref. 13. Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
| Complex | τ [μs] | ϕ |
|---|---|---|
| a In aqueous solution at room temperature; from ref. 3,4 and 13. | ||
| Cr(L1)33+ | 898 | 11.0% |
| Cr(bpy)33+ | 63 | 0.089% |
| Cr(phen)33+ | 270 | 0.15% |
| Cr(tpy)23+ | <30 | <0.00089% |
Thus, Cr(L1)23+ is a robust near-infrared emitter with unusually high luminescence quantum yield and excited state lifetime in aqueous solution. In this respect, it is a unique Cr(III) polypyridine complex.13 In a subsequent study the focus was on the temperature dependence of the luminescence emitted by Cr(L1)23+ in aqueous and organic media.21 Emission from both the 2E and 2T1 excited states (Fig. 1a) is observable in the 210–373 K temperature range, and their relative contribution is governed by Boltzmann's law. Thus, the Cr(L1)23+ complex can be used as a contactless molecular thermometer, operating on the basis of ratiometric 2E vs.2T1 luminescence intensity, and this was put in evidence by incorporating it into polystyrene nanoparticles and micelles. It was noted that this could lead to applications in biology, medicine, and material sciences.21
Mn(IV) is isoelectronic with Cr(III), but until very recently luminescence from molecular Mn(IV) compounds had not been reported yet. Now, a bis-tris(carbene)borate complex of Mn(IV) was found to emit from the 2E state at 85 K in the solid state.22
3. Heteroleptic Cr(III) complexes with carboxylate anchor groups for solar cells
While heteroleptic tris(α-diimine) complexes with Ru(II) are synthetically readily accessible, this is less straightforward for Cr(III) due to ligand scrambling when attempting to use substitution-labile Cr(II) intermediates. However, with [Cr(α-diimine)2(OTf)2]+ complexes as precursors, the synthesis of heteroleptic Cr(III) complexes became tractable.23 Building on this strategy, Damrauer, Shores, and coworkers synthesized and explored a series of Cr(III) complexes with the 2,2′-bipyridine-4,4′-carboxylic acid methyl ester ligand (L2) (Fig. 3) and various spectator ligands (L3–L5) with a view to grafting them onto semiconductor surfaces.14 It was hoped that the strongly oxidizing properties of these Cr(III) complexes would render them suitable for photoinduced hole injection into p-type semiconductors (Table 2), and this could be interesting for tandem photovoltaic cells.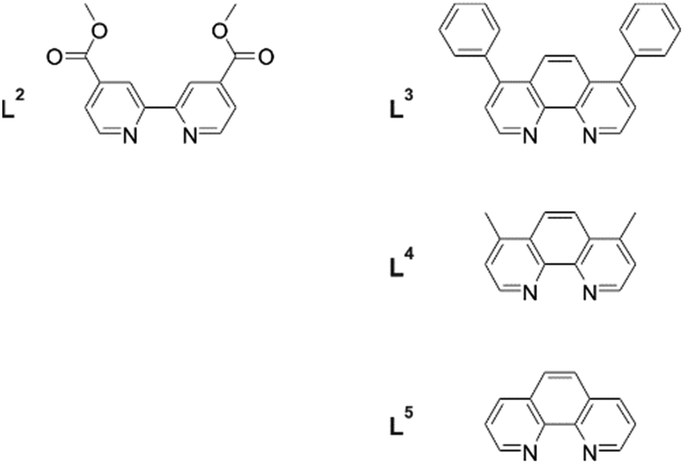 | ||
| Fig. 3 Ligands used for the synthesis of heteroleptic Cr(III) complexes with a view to grafting them onto p-type semiconductor surfaces.14 | ||
| Complex | E 0 (CrL3*3+/2+)/V vs. SCE |
|---|---|
| a From ref. 14, in CH3CN, converted from V vs. Fc+/Fc to V vs. SCE according to ref. 24. b From ref. 15. For the determination of these potentials, the reader is referred to the original literature. | |
| Cr(bpy)33+ | 1.46a |
| Cr(phen)33+ | 1.43a |
| Cr(L2)33+ | 1.82a |
| Cr(L3)33+ | 1.40b |
The investigated complexes exhibited photophysical and electrochemical properties that are in line with prior studies of structurally related homoleptic Cr(III) polypyridines. Ligand L2 seemed to weaken the ligand field in Cr(L2)(L5)23+, thereby decreasing the activation energy for nonradiative relaxation from the emissive 2E state by several kJ mol−1 relative to homoleptic Cr(L5)33+.14 Redox potentials up to 1.82 V vs. SCE were determined for reduction of the photoexcited Cr(III) complexes, owing to the electron-withdrawing nature of L2 (Table 2). Considering these strongly oxidizing properties and the long excited state lifetimes (7.7–108 μs) in solution, the use of these heteroleptic complexes for hole injection into p-type semiconductors is indeed promising. A significant drawback is their relatively weak absorption in the visible spectral range. The most favorable example in this respect was Cr(L2)(L4)23+, which exhibits a molar absorptivity of 1270 M−1 cm−1 at 491 nm.
Damrauer, Shores, and coworkers noted that their study was the first on heteroleptic Cr(III) dipyridyl complexes with at least one carboxylate group for eventual attachment to semiconductors.14 All their studies were performed in presence of 1 M acid, because Cr(III) polypyridine complexes have long been known to hydrolyze in alkaline solution.20
4. Photoredox catalysis with Cr(III)
Shores, Ferreira, and coworkers discovered that under irradiation with visible light, Cr(L2)33+ is able to catalyze the [4 + 2] dimerization of 1,3-cyclohexadiene (Fig. 4a) significantly better than the strongly oxidizing Ru(bpz)32+ complex (bpy = 2,2′-bipyrazine).15 This observation is in line with the higher oxidizing power of photoexcited Cr(L2)33+ relative to Ru(bpz)32+, and it opened the possibility of performing cross-cycloadditions with various other substrates.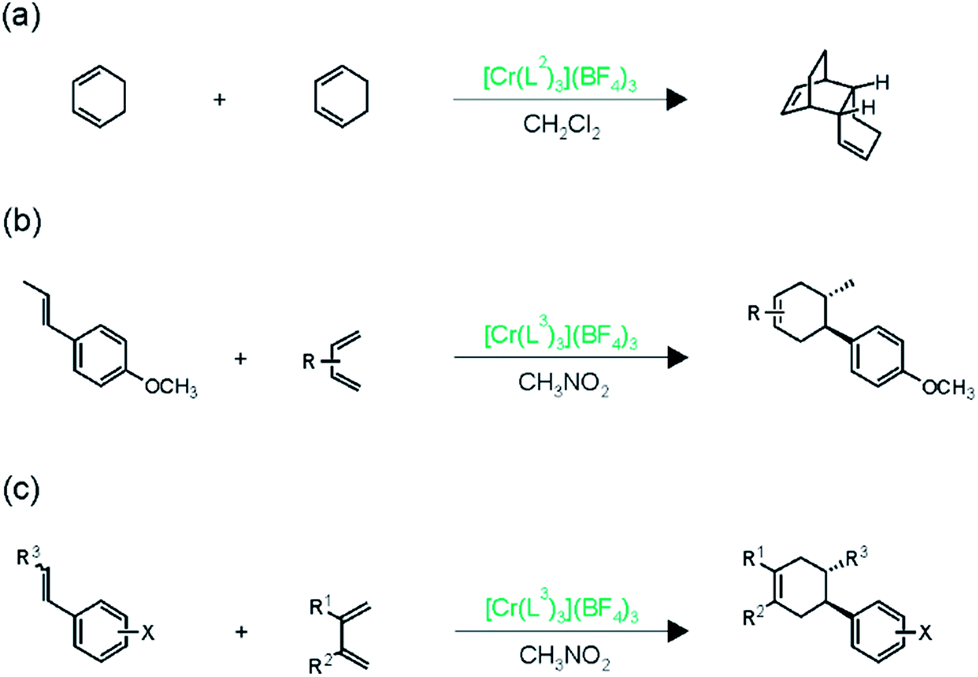 | ||
| Fig. 4 Cycloaddition reactions catalyzed by photoexcited Cr(III) complexes.15 | ||
The combination of electron-rich dienophiles with dienes that are more difficult to oxidize was very successful in this respect, because it allowed the Cr(III) catalysts to discriminate between the two reactants. Cr(L3)33+ was used for this purpose, since its somewhat lower oxidation potential (Table 2) was expected to accentuate selective dienophile oxidation.15 Electron-rich styrenes turned out to be the optimal dienophiles for reaction with isoprene (Fig. 4b) or 2,3-dimethyl-1,3-butadiene (Fig. 4c), yielding more than 15 different Diels–Alder adducts in good to excellent yields under irradiation with 300–419 nm light in CH3NO2. Different functional groups (ethers, esters, and sulfonates) were tolerated and high diastereoselectivities were achieved.
While early studies had already explored the photooxidizing properties of Cr(III) complexes from a fundamental perspective,4–6 the study by Shores, Ferreira, and coworkers was the first to apply Cr(III) complexes as photocatalysts in organic synthesis.15 This represents the key novelty of their work, but conceptually very similar organic transformations had been performed earlier by photoredox catalysis with Ru(bpz)32+.25 Mechanistic aspects of the Cr(III) photoredox catalysis initially remained somewhat unclear, in particular with regard to the role of O2, which was found to be a critically important ingredient for the overall reaction.15
Subsequent investigations revealed that O2 interferes in two ways in the overall catalysis.16 On the one hand, it is able to act as an energy acceptor vis-à-vis photoexcited Cr(III) complexes, thereby forming singlet oxygen. On the other hand, oxygen was also found to act as an electron acceptor in the catalytic cycle, but only in its 1O2 form. Based on detailed electrochemical and spectroscopic investigations, catalysis studies, electronic structure calculations, and a variety of different control experiments, a mechanism taking into account both the energy transfer and the electron transfer pathway was proposed (Fig. 5).16 In this mechanism, photoexcited Cr(III) undergoes competitive reductive quenching by dienophile substrates (1) and energy transfer quenching by 3O2 to form 1O2. For instance, para-anethole (1) is oxidized to its radical cation form (1+), which can then undergo reaction with the diene (2) to result in a cycloadduct radical cation (3+), while the chromium complex is reduced. Meanwhile, other photoexcited Cr(III) complexes undergo energy transfer quenching to result in 1O2, and the latter is then able to re-oxidize CrL32+ to CrL33+, thereby closing the catalytic cycle for the chromium complex and resulting in the formation of O2−. These superoxide anions then reduce the cycloadduct radical cation (3+) to the final Diels–Alder product (3), and with 3O2 as an oxidation product of this reaction, the catalytic cycle for oxygen is closed. Recent computational studies by other researchers complement this mechanistic picture and give insights into the origin of the chemo- and regioselectivity of the overall reaction.26
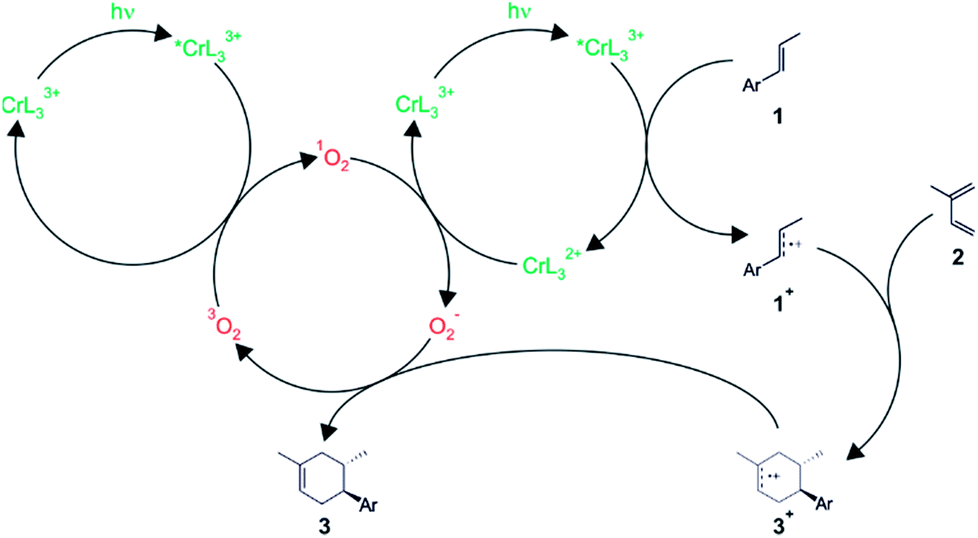 | ||
| Fig. 5 Plausible mechanism for the light-driven [4 + 2] cycloaddition reaction between a dienophile (1) and a diene (2) catalyzed by the Cr(L3)33+ complex in presence of oxygen.16 | ||
This rather complex catalytic cycle relies on the long 2E excited-state lifetime of Cr(III) complexes (13 μs for Cr(L3)33+ under the conditions used here), which permits the build-up of substantial concentrations of 1O2 (τ = 40 μs) and ultimately O2−. By contrast, Ru(bpz)32+ photoredox catalysis of closely related reactions seems to operate mainly through a radical propagation mechanism,27 possibly because Ru(bpz)32+ has a much shorter excited-state lifetime (<0.9 μs), leading to comparatively low 1O2 and O2− concentrations. In the radical chain mechanism (not included in Fig. 5), the hexenyl radical cation (3+) reacts with dienophile 1 to product 3 and radical cation 1+. It was noted that small changes in environmental conditions could allow switching between photocatalytic and radical chain pathways in the chromium systems as well.16
In further studies, the same authors were able to perform chromium-catalyzed [4 + 2] cycloaddition reactions with electron-deficient dienophiles,17 thereby going significantly beyond the substrate scope initially explored with Ru(bpz)32+ (Fig. 6).25 Enone 4 has an oxidation potential (Eox) of 2.0 V vs. SCE and therefore cannot be oxidized by photoexcited Cr(L3)33+, because the latter has an oxidation potential of only 1.33 V vs. SCE in CH3NO2. Nevertheless, in presence of isoprene (2) (Eox = 1.98 V vs. SCE), photoirradiation of Cr(L3)33+ leads to conversion of enone 4 to the cycloaddition product 5 in good yield (Fig. 6a), and this transformation works for several differently substituted chalcone derivatives.17 Interestingly, the regioselectivity of this cycloaddition is always in favor of the reversed Diels–Alder adduct, opposite to what is commonly observed, and the reaction conditions were tolerant of a variety of functional groups on the diene.
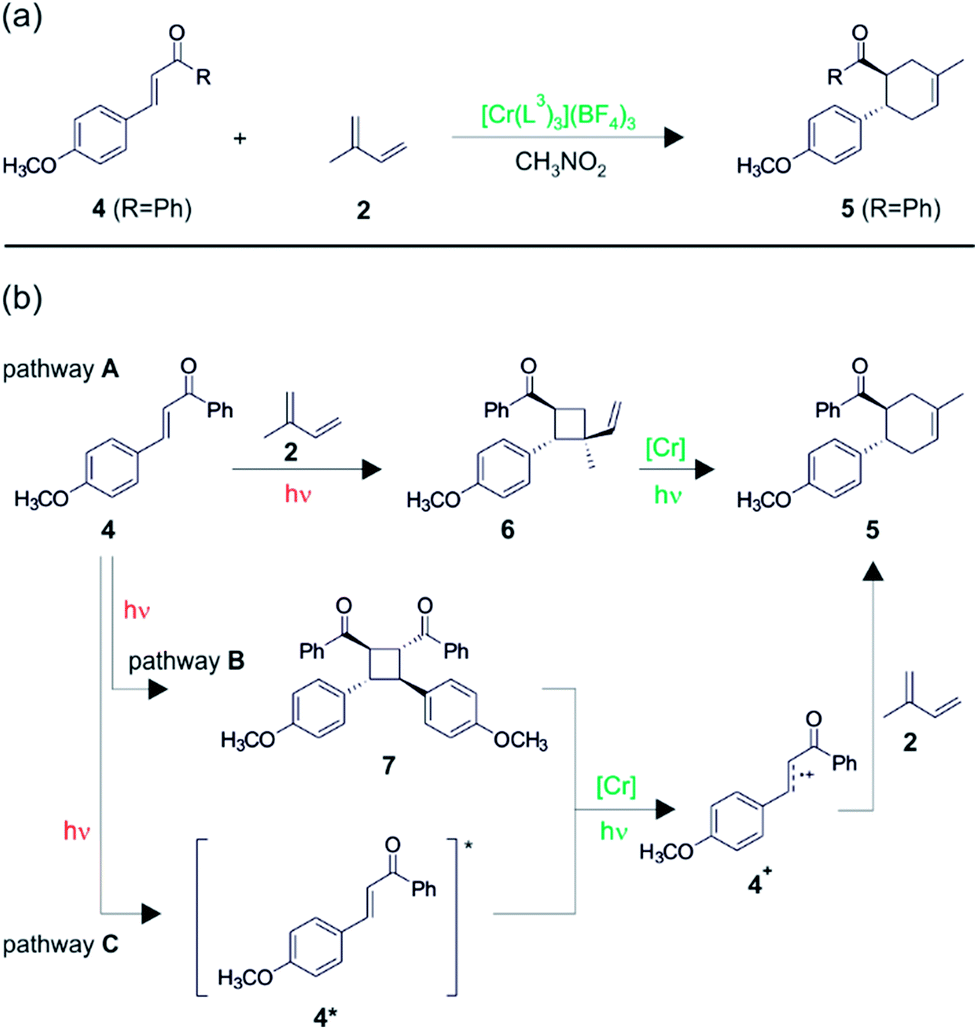 | ||
| Fig. 6 (a) Diels–Alder reaction between chalcone derivative 4 and isoprene 2.17 (b) Possible mechanistic pathways. Reproduced from ref. 17 – Published by The Royal Society of Chemistry. | ||
Three different reaction mechanisms were considered theoretically viable, and two of them were identified as significant contributors. In the first mechanism (pathway A in Fig. 6b), there is an initial organic photochemical step in which [2 + 2] cycloaddition between the two substrates occurs. The resulting vinylcyclobutane (6) species (Eox = 1.68 V vs. SCE) is oxidized much more readily than 2 and 4, and consequently can undergo single-electron oxidative vinylcyclobutane rearrangement to yield 5 after photoexcitation of Cr(L3)33+. In control experiments without Cr catalyst, the vinylcyclobutane 6 was directly observable, confirming that pathway A is indeed viable. Pathway B recognizes that dimerization of enone 4 yields a compound (7) with a relatively low redox potential (Eox = 1.4 V vs. SCE), and after oxidation of the dimer (7) by photoexcited Cr(L3)33+, cycloreversion to radical cation 4+ could occur, followed by reaction of the latter with the diene (2). However, control experiments indicate that pathway B is a minor contributor at most. In pathway C, photoexcited enone (4*) is taken into account. This species was previously reported to have excited-state lifetimes between 20 and 30 ns,28 and the photoexcited Cr(L3)33+ complex is thermodynamically competent to oxidize 4* to 4+. Once formed, radical cation 4+ can then undergo reaction with the diene (2). Pathway C is unusual in that two photoexcited species are proposed to come to reaction with one another, but preliminary experiments did indeed seem to reveal a nonlinear dependency of the overall rate of reactivity on the light intensity. The long 2E excited-state lifetime of the Cr(III) complexes is certainly helpful in this context. Radical chain mechanisms do not seem to be involved, and oxygen is still beneficial to the overall reaction progress, but not to the same extent as in the reactions from Fig. 4 and 5.
Heinze and coworkers recently explored the reaction pathways leading to oxidative C–H bond functionalization in tertiary amines under aerobic photoredox conditions with the Cr(L1)23+ complex from Fig. 2b.29 Since the energy gap between the 2E and 4T2 excited states is very large in this particular case, and because the 2E state is not expected to be particularly redox-active (due to the fact that it derives from the same t2g3 electron configuration as the ground state), it was explored whether energy (EnT) or electron transfer mechanisms are operative in the photo-cyanation of tertiary amines in presence of oxygen. Indeed, the key conclusion is that 1O2 is rapidly generated (ΔGEnT = −0.62 eV; kEnT = 1.2 × 108 M−1 s−1), and 1O2 then initiates the reaction with the α-C–H bonds of amines. The high selectivity for the energy transfer mechanism in this case is very likely due to the thermal inaccessibility of the redox-active 4T2 state in this particular complex, and this is in marked contrast to the other Cr(III) polypyridine complexes discussed in this subsection.
5. Near-infrared to visible upconversion sensitized by Cr(III)
Photon upconversion has long been known to occur in trivalent lanthanide ions doped into solid matrices.31 Typically, near-infrared excitation into weakly absorbing f–f transitions leads to the population of long-lived excited states, which serve as energy reservoirs for the subsequent population of higher excited states emitting in the visible spectral range. The energy conservation law demands the absorption of multiple photons for this purpose, usually leading to nonlinear dependences of the upconversion luminescence intensity on excitation density. Since f–f transitions have inherently low oscillator strengths, the overall upconversion efficiency is often rather limited unless high excitation densities are employed. The use of transition metal sensitizers for upconversion luminescence is an attractive approach to addressing this challenge,32 but until recently success was somewhat limited, except of course for the special case of triplet–triplet annihilation upconversion which is conceptually different from the processes considered in this section.33Building on profound expertise in lanthanide chemistry and helical molecular structures, Piguet in collaboration with Hauser and their coworkers recently took an innovative approach to achieving transition metal sensitized lanthanide upconversion.18,30 The trimetallic compound in Fig. 7a combines a central Er(III) ion with two flanking Cr(α-diimine)33+ complexes in a kinetically inert helical structure made from 3 intertwined ligands (Fig. 7b). The central lanthanide ion (Ln) is in pseudo-tricapped trigonal prismatic coordination, and at 10 K it luminesces from multiple excited states upon UV-excitation. Following laser irradiation at 750 nm into the 2E absorption of the Cr(α-diimine)33+ sensitizers with excitation densities between 195 and 690 mW mm−2, upconversion luminescence at ∼540 nm emitted from the Er(III) 4S3/2 state was observed at 10 K (Fig. 7c).18 This finding is spectacular because it represents the first observation of near-infrared to visible photon upconversion in an isolated molecular system.18,30
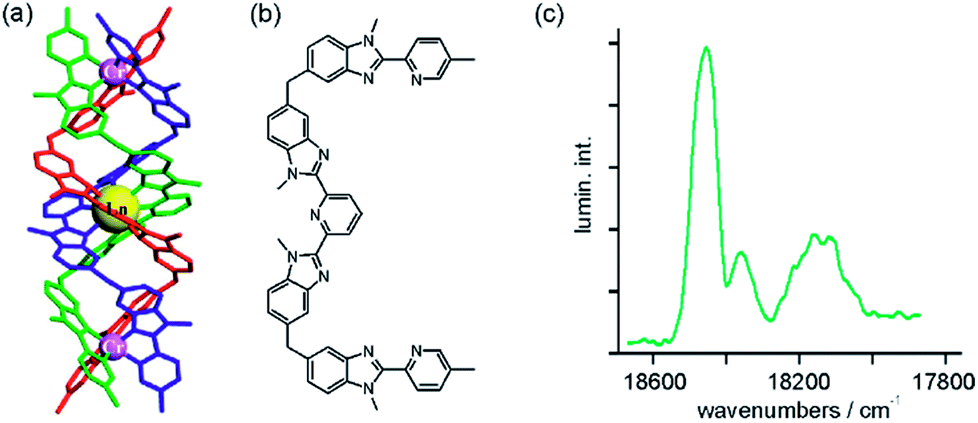 | ||
| Fig. 7 (a) Triple-stranded helix incorporating a central Er(III) ion flanked by two CrN63+ units; the three identical ligands are represented in different colors. Reproduced with permission from ref. 30. Copyright 2012 Elsevier. (b) Ligand used for formation of the helix. (c) Upconverted 4S3/2 → 4I15/2 luminescence emitted from Er(III) after excitation of the CrN63+ units at 13360 cm−1. The helix was present at 10 mM concentration in CH3CN at ∼30 K, and an excitation density of 400 mW mm−2 was used. Reproduced with permission from ref. 18. Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The Cr(α-diimine)33+ sensitizer is exquisitely suited for this purpose not only because it gives access to a kinetically inert helical structure with Er(III), but also because its various excited states are just at the right energies (Fig. 8). The 2E state is slightly above the Er(III) 4I9/2 multiplet, making intramolecular energy transfer (EnT) with a rate constant of ∼500 s−1 possible.18 Nonradiative relaxation of 4I9/2 populates the 4I11/2 and 4I13/2 states (gray dashed downward arrow in Fig. 8). In particular the latter has a long lifetime, and from there the green emissive 4S3/2 state can be reached by a so-called energy transfer upconversion process (right part of Fig. 8).34 In structurally related Cr(III)–Er(III) dimers, the upconversion intensity measured under identical excitation conditions as for a Cr(III)–Er(III)–Cr(III) trimer is 4–10 times weaker, illustrating that the sensitizer to activator ratio is an important factor.35 The observation that the upconversion efficiency enhancement is greater than statistically expected when increasing the number of Cr(III) sensitizers from 1 to 2 (i.e., greater than a factor of 4), might be an indication for an additional upconversion mechanism, in which two excitations are stored on separate Cr(III) sensitizers prior to energy transfer to Er(III).36
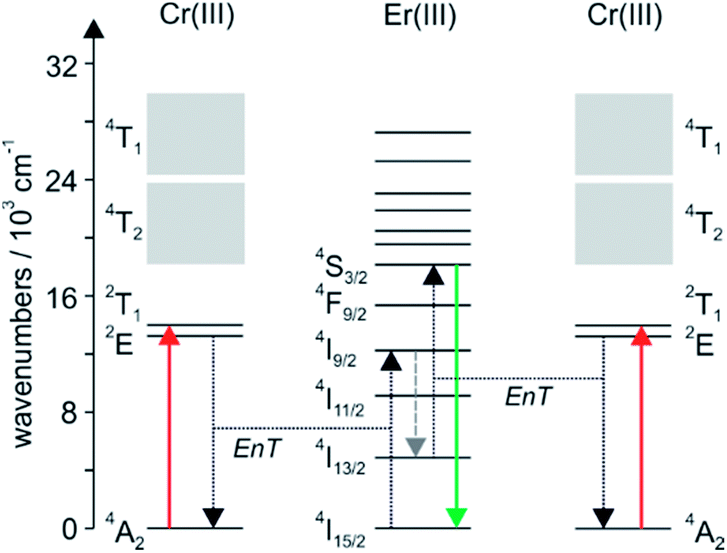 | ||
| Fig. 8 Energy-level scheme for the Cr(III)–Er(III)–Cr(III) helix from Fig. 7 illustrating the near-infrared to visible upconversion process. EnT stands for energy transfer. The dashed gray downward represents nonradiative relaxation. Reproduced with permission from ref. 30. Copyright 2012 Elsevier. | ||
Unfortunately, Cr(α-diimine)33+ complexes do not represent the universal solution to achieving near-infrared to visible upconversion with lanthanides. In a Cr(III)–Tm(III)–Cr(III) trimer, the targeted blue Tm(III) 1G4 → 3H6 emission is quenched by energy transfer to the Cr(III) 4T2 state, and in a Cr(III)–Ho(III)–Cr(III) molecule the desired energy transfer from the Cr(III) 2E state to Ho(III) is too slow (<100 s−1).35 Thus, so far the combination of Cr(III) and Er(III) is unique in these helical systems. Aside from careful matching of excited state energies between sensitizer and activator, the successful protection of the Er(III) ion from high-energy oscillators in the helical structure is a key factor for upconversion. Without this, the 4S3/2 state would simply deactivate nonradiatively.
The combination of Cr(α-diimine)33+ sensitizers with either Nd(III) or Yb(III) in closely related di- and trinuclear helical structures results in an interesting fundamental observation, which could have important practical implications. In these compounds, excitation of the Cr(III) sensitizers in the visible spectral range leads to lanthanide-based emission in the near-infrared with apparent lifetimes in the millisecond regime.37 This is possible because the long-lived 2E state of Cr(III) serves as an energy storage reservoir and feeding level for lanthanide sensitization in the case of Nd(III), Er(III), and Yb(III). For instance, in a Cr(III)–Yb(III)–Cr(III) molecule, the Yb(III) 2F5/2 → 2F7/2 emission at ∼1000 nm decays with τ ≈ 1.3 ms, and in a Cr(III)–Nd(III)–Cr(III) helix the Nd(III) 4F3/2 → 4I11/2 luminescence at ∼1055 nm decays with τ ≈ 0.25 ms at 10 K. If this could be exploited at room temperature in solution, it could become interesting for time-gated luminescence detection in biological tissue.
6. A Cr(III) nitrito complex for photochemical NO release
Nitric oxide is an intracellular regulator of various physiological processes in the cardiovascular and neurological systems of mammals. Photochemical release of NO would be highly desirable for medical applications since this would permit good temporal and spatial control of NO dosage.39 This is important because NO seems to be involved in both tumor growth and suppression depending on local concentrations.In this context, the Ford group explored the water-soluble trans-Cr(cyclam) (ONO)2+ complex (cyclam = 1,4,8,11-tetraazacycltetradecane) (Fig. 9a) which undergoes reversible photochemical NO release.38 The initial photochemical step occurs from a spin-flip (doublet) excited state and leads to homolytic dissociation of NO from a coordinated nitrite and a Cr(IV) intermediate (right part of Fig. 9b). In anaerobic media, this Cr(IV) complex reacts back with NO with a second-order rate constant of 3.06 × 106 M−1 s−1, but under aerobic conditions Cr(IV) is oxidized more rapidly to a Cr(V) complex (Fig. 9b). The quantum yield for NO release following excitation into the spin-allowed (quartet–quartet) d–d transition at 436 nm (λmax = 476 nm, ε = 40 M−1 cm−1) is 0.27 ± 0.03.38 Photoaquation (left part of Fig. 9b) only has a quantum yield of 0.0092 ± 0.0008.
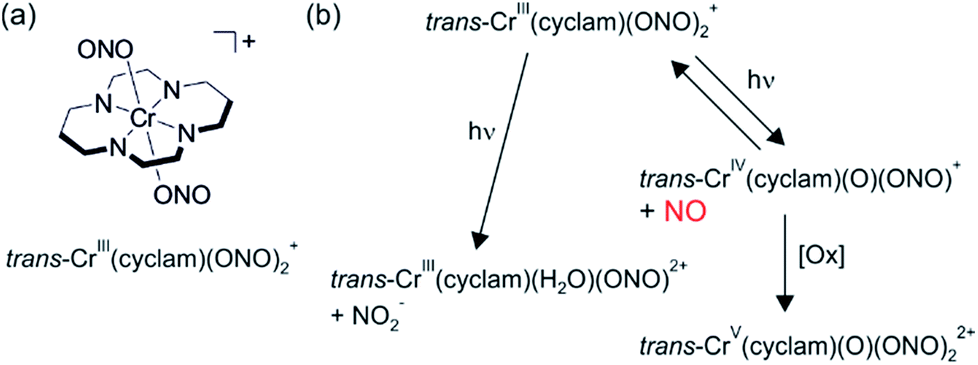 | ||
| Fig. 9 (a) Chemical structure of the trans-Cr(cyclam)(ONO)2+ complex. (b) Photochemistry of this complex. Adapted with permission from ref. 38. Copyright 1999 American Chemical Society. | ||
Oxidative trapping of the Cr(IV) intermediate presumably leads to the formation of superoxide, and this in turn intercepts a fraction of the photochemically released NO through formation of peroxynitrite (ONOO−).40 In presence of 5 mM glutathione in pH 7.4 aqueous buffer solution the Cr(IV) intermediate is reduced sufficiently fast to a Cr(III) product, outcompeting the (undesired) thermal back reaction (NO binding). It was noted that this is ideal for applications where nitric oxide needs to be generated in hypoxic, reducing environments that are typical for tumor tissues.
Lactate dehydrogenase assays indicated that the photochemically released NO and other reactive intermediates do not induce acute toxicity.40 Moreover, it could be demonstrated that low concentrations (3 μM) of the Cr(cyclam)(ONO)2+ complex are sufficient to induce vasorelaxation of porcine coronary arterial rings.41 This occurs through activation of the enzyme guanylyl cyclase by NO.
Although the photoactive doublet excited state is at ca. 14500 cm−1, the Cr(cyclam)(ONO)2+ complex absorbs only very weakly throughout the entire visible spectral range. In the NIR, where human tissue is more transparent, it does not absorb at all.40 Antenna systems were therefore explored for sensitization of NO release. CdSe/ZnS core/shell quantum dots led to a 15-fold enhancement of NO release relative to direct photolysis of aqueous solutions containing only Cr(cyclam)(ONO)2+.39,42,43 It was noted that functionalization of the quantum dots with proteins and antibodies could potentially be employed to target specific tissues for NO release.
7. A Cr(0) isocyanide complex as a luminophore and photosensitizer
Ru(bpy)32+ is the prototype of a d6 metal complex with an emissive 3MLCT excited state, and ligand variation gives access to hundreds of related analogues. Similar excited state structures can readily be obtained with other precious metals such as Re(I), Os(II) and Ir(III).12,44 Early studies already showed that zero-valent group 6 metal complexes with arylisocyanide ligands are promising earth-abundant alternatives. 3MLCT luminescence from Mo(0) and W(0) was readily detectable at room temperature, and Cr(0) emission was observed at 77 K.9 Then, these complexes seemed forgotten for nearly 40 years, until new reports on Mo(0) and W(0) appeared.45–49 We were curious whether it would be possible to obtain a Cr(0) complex that luminesces at room temperature in solution, particularly in view of the recent interest in isoelectronic Fe(II) complexes in the greater context of dye-sensitized solar cells.50–56A chelating diisocyanide ligand with sterically demanding substituents turned out to be well suited for obtaining a Cr(0) complex with a long-lived 3MLCT excited state (Fig. 10a).19,49 In de-aerated THF at 20 °C, the Cr(CNtBuAr3NC)3 complex luminesces with a quantum yield of ∼10−5 and a lifetime of 2.2 ns (inset in Fig. 10b). This is nearly two orders of magnitude longer than the current record 3MLCT lifetime of Fe(II) complexes (37 ps on Al2O3 nanofilms).51,52 UV-Vis absorption and luminescence spectra of Cr(CNtBuAr3NC)3 are reminiscent of those of Ru(bpy)32+ (Fig. 10b). The Cr(0) center can be oxidized up to Cr(III) in quasi-reversible fashion, but photochemically it can of course only act as a donor of single electrons. The excited-state oxidation potential for the Cr(I/*0) couple is −2.4 V vs. Fc+/Fc, making it a very strong photoreductant.19,49 For comparison, the Ru(III/*II) potential for Ru(bpy)32+ is −1.2 V vs. Fc+/Fc and the Ir(IV/*III) potential for fac-Ir(ppy)3 (ppy = 2-phenylpyridine) is −2.1 V vs. Fc+/Fc.57
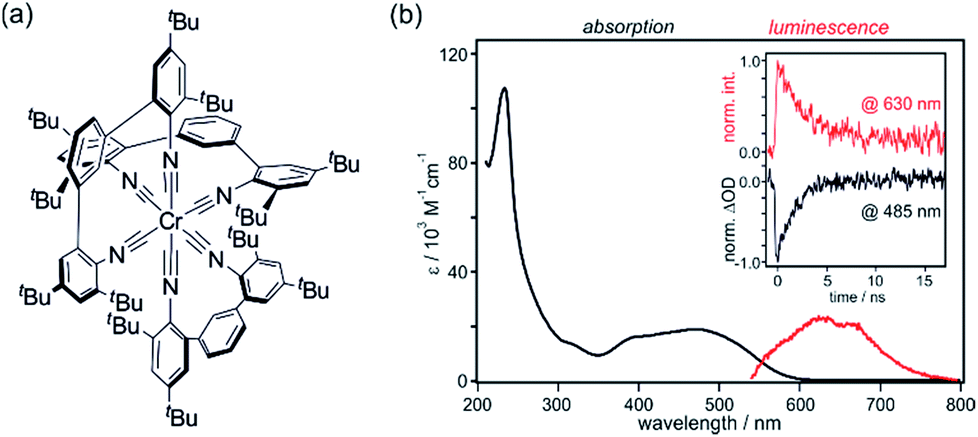 | ||
| Fig. 10 (a) Chemical structure of the Cr(CNtBuAr3NC)3 complex.19 (b) UV-Vis absorption and luminescence spectra of Cr(CNtBuAr3NC)3 in de-aerated THF at 20 °C. Excitation occurred at 450 nm. Inset: luminescence decay at 630 nm (red) and absorption bleach recovery at 485 nm (black) following excitation at 532 nm with pulses of ∼30 ps duration. Adapted with permission from ref. 19. Copyright 2017 American Chemical Society. | ||
An early study of a Cr(0) complex with monodentate arylisocyanide ligands found a luminescence lifetime shorter than 10 ns in a frozen glass at 77 K, from which it was concluded that the emissive excited state could have singlet parentage.9 In Cr(CNtBuAr3NC)3 the emissive MLCT state is quenched efficiently by anthracene, compatible with energy transfer from a triplet excited state.19,49 The triplet-excited anthracene molecules can subsequently undergo triplet–triplet annihilation upconversion, and thus Cr(CNtBuAr3NC)3 is an uncommon photosensitizer for a process that is more commonly sensitized by Ru(α-diimine)32+ complexes, Cu(I) complexes, or porphyrins.33,58 This suggests that the 3MLCT lifetime of Cr(CNtBuAr3NC)3 is sufficiently long for this complex to engage in bimolecular reactions, and this is promising in view of photoredox applications similar to those reported recently for a structurally similar Mo(0) tris(diisocyanide) complex.48,49
Given the long excited-state lifetime of Cr(CNtBuAr3NC)3 compared to Fe(II) complexes, the use of Cr(0) isocyanides as dyes of wide bandgap semiconductor solar cells seems within reach. Considering the high reducing power of photoexcited Cr(CNtBuAr3NC)3, it is possible that semiconductors other than the commonly used TiO2 might lead to optimal energy matching between sensitizer LUMO and semiconductor conduction band.
8. Summary and conclusions
Inorganic photochemistry and photophysics has long focused on complexes made from Ru, Re, Os, Ir, Pt or Au.12,44 There has been a long-standing interest in replacing these precious metals by more earth-abundant elements, but in recent years this has become an area of increasing activity. New photoactive metal complexes made from relatively earth-abundant elements have been developed, including for example the Cr(III) and Cr(0) systems discussed herein, but also complexes with Mn,22 Fe,50–55,59–63 Cu,64–66 Ni,67 Zr,68 Mo,48,49 or W.45–47,49,69 In several cases, it has long been known that the respective transition metal species in the right oxidation states equipped with suitable ligands do indeed exhibit very favorable photophysical properties,3,4,9,70 but the current trend towards performing more sustainable chemistry has led to a revival of sometimes long neglected types of complex classes. Cr(III) polypyridines and Cr(0) arylisocyanides are good examples in this regard, and the new studies discussed above evidently go substantially beyond the early initial investigations. Applications as luminophores, dyes for solar cells, photoredox catalysts, upconversion sensitizers, and photochemical NO sources are either within reach or have now been realized.Given the current interest in the use of photoactive metal complexes for various applications ranging from synthetic organic chemistry to solar energy conversion, it seems likely that this will trigger new fundamental discoveries in the development of non-traditional metal complexes with long-lived excited states. Creative approaches such as those outlined herein are in demand, making sure that a mature field such as coordination chemistry remains a key contributor to chemistry at large.71 Many different options are on the table, as illustrated herein on the specific example of chromium. New discoveries are possible with complexes emitting from ligand-field (typically spin-flip) excited states or from charge transfer excited states. Cr(III) with its d3 electron configuration or Cr(0) d6 complexes are only two possibilities among many others. There are several dn configurations in which the ligand field should be tunable to such an extent that emissive d–d excited states result, and charge transfer excited states are in principle accessible with many different metal–ligand combinations beyond the well-beaten path of d6, d8 or d10 metal diimines with precious metals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swiss National Science Foundation through grant number 200021_156063/1, through the NCCR Molecular Systems Engineering, and through R'Equip grant number 206021_157687 is gratefully acknowledged.Notes and references
- M. Maestri, F. Bolletta, L. Moggi, V. Balzani, M. S. Henry and M. Z. Hoffman, J. Am. Chem. Soc., 1978, 100, 2694–2701 CrossRef CAS
.
- N. A. P. Kane-Maguire, J. Conway and C. H. Langford, J. Chem. Soc., Chem. Commun., 1974, 801–802 RSC
- A. D. Kirk and G. B. Porter, J. Phys. Chem., 1980, 84, 887–891 CrossRef CAS
- N. Serpone, M. A. Jamieson, M. S. Henry, M. Z. Hoffman, F. Bolletta and M. Maestri, J. Am. Chem. Soc., 1979, 101, 2907–2916 CrossRef CAS
- R. Ballardini, G. Varani, F. Scandola and V. Balzani, J. Am. Chem. Soc., 1976, 98, 7432–7433 CrossRef CAS
- F. Bolletta, M. Maestri, L. Moggi and V. Balzani, J. Chem. Soc., Chem. Commun., 1975, 901–902 RSC
- E. R. Davidson, K. L. Kunze, F. B. C. Machado and S. J. Chakravorty, Acc. Chem. Res., 1993, 26, 628–635 CrossRef CAS
- K. R. Mann, M. Cimolino, G. L. Geoffroy, G. S. Hammond, A. A. Orio, G. Albertin and H. B. Gray, Inorg. Chim. Acta, 1976, 16, 97–101 CrossRef CAS
- K. R. Mann, H. B. Gray and G. S. Hammond, J. Am. Chem. Soc., 1977, 99, 306–307 CrossRef CAS
- E. Maskova and A. Vlcek, Inorg. Chim. Acta, 1996, 242, 17–23 CrossRef CAS
- L. E. Shaw and C. H. Langford, Inorg. Chem., 2000, 39, 541–546 CrossRef CAS PubMed
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed
- A. M. McDaniel, H. W. Tseng, N. H. Damrauer and M. P. Shores, Inorg. Chem., 2010, 49, 7981–7991 CrossRef CAS PubMed
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS PubMed
- R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappe and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451–5464 CrossRef CAS PubMed
- S. M. Stevenson, R. F. Higgins, M. P. Shores and E. M. Ferreira, Chem. Sci., 2017, 8, 654–660 RSC
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J. C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108–4112 CrossRef CAS PubMed
- L. A. Büldt, X. Guo, R. Vogel, A. Prescimone and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 985–992 CrossRef PubMed
- M. A. Jamieson, N. Serpone, M. S. Henry and M. Z. Hoffman, Inorg. Chem., 1979, 18, 214–216 CrossRef CAS
- S. Otto, N. Scholz, T. Behnke, U. Resch-Genger and K. Heinze, Chem.–Eur. J., 2017, 23, 12131–12135 CrossRef CAS PubMed
- V. Baslon, J. P. Harris, C. Reber, H. E. Colmer, T. A. Jackson, A. P. Forshaw, J. M. Smith, R. A. Kinney and J. Telser, Can. J. Chem., 2017, 95, 547–552 CrossRef CAS
- K. D. Barker, K. A. Barnett, S. M. Connell, J. W. Glaeser, A. J. Wallace, J. Wildsmith, B. J. Herbert, J. F. Wheeler and N. A. P. Kane-Maguire, Inorg. Chim. Acta, 2001, 316, 41–49 CrossRef CAS
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS
- S. S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed
- Y. Yang, Q. Liu, L. Zhang, H. Yu and Z. Dang, Organometallics, 2017, 36, 687–698 CrossRef CAS
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC
- R. A. Caldwell and M. Singh, J. Am. Chem. Soc., 1983, 105, 5139–5140 CrossRef CAS
- S. Otto, A. M. Nauth, E. Emilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef
- L. Aboshyan-Sorgho, M. Cantuel, S. Petoud, A. Hauser and C. Piguet, Coord. Chem. Rev., 2012, 256, 1644–1663 CrossRef CAS
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed
- O. S. Wenger and H. U. Güdel, J. Phys. Chem. B, 2002, 106, 10011–10019 CrossRef CAS
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS
- Y. Suffren, B. Golesorkhi, D. Zare, L. Guénée, H. Nozary, S. V. Eliseeva, S. Petoud, A. Hauser and C. Piguet, Inorg. Chem., 2016, 55, 9964–9972 CrossRef CAS PubMed
- D. Zare, Y. Suffren, L. Guénée, S. V. Eliseeva, H. Nozary, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, Dalton Trans., 2015, 44, 2529–2540 RSC
- Y. Suffren, D. Zare, S. V. Eliseeva, L. Guénée, H. Nozary, T. Lathion, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, J. Phys. Chem. C, 2013, 117, 26957–26963 CAS
- L. Aboshyan-Sorgho, H. Nozary, A. Aebischer, J. C. G. Bünzli, P. Y. Morgantini, K. R. Kittilstved, A. Hauser, S. V. Eliseeva, S. Petoud and C. Piguet, J. Am. Chem. Soc., 2012, 134, 12675–12684 CrossRef CAS PubMed
- M. De Leo and P. C. Ford, J. Am. Chem. Soc., 1999, 121, 1980–1981 CrossRef CAS
- A. D. Ostrowski and P. C. Ford, Dalton Trans., 2009, 10660–10669 RSC
- A. D. Ostrowski, R. O. Absalonson, M. A. De Leo, G. Wu, J. G. Pavlovich, J. Adamson, B. Azhar, A. V. Iretskii, I. L. Megson and P. C. Ford, Inorg. Chem., 2011, 50, 4453–4462 CrossRef CAS PubMed
- A. D. Ostrowski, S. J. Deakin, B. Azhar, T.
W. Miller, N. Franco, M. M. Cherney, A. J. Lee, J. N. Burstyn, J. M. Fukuto, I. L. Megson and P. C. Ford, J. Med. Chem., 2010, 53, 715–722 CrossRef CAS PubMed
- D. Neuman, A. D. Ostrowski, R. O. Absalonson, G. F. Strouse and P. C. Ford, J. Am. Chem. Soc., 2007, 129, 4146–4147 CrossRef CAS PubMed
- D. Neuman, A. D. Ostrowski, A. A. Mikhailovsky, R. O. Absalonson, G. F. Strouse and P. C. Ford, J. Am. Chem. Soc., 2008, 130, 168–175 CrossRef CAS PubMed
- K. S. Schanze, D. B. MacQueen, T. A. Perkins and L. A. Cabana, Coord. Chem. Rev., 1993, 122, 63–89 CrossRef CAS
- W. Sattler, M. E. Ener, J. D. Blakemore, A. A. Rachford, P. J. LaBeaume, J. W. Thackeray, J. F. Cameron, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 10614–10617 CrossRef CAS PubMed
- W. Sattler, L. M. Henling, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2015, 137, 1198–1205 CrossRef CAS PubMed
- H. Kvapilova, W. Sattler, A. Sattler, I. V. Sazanovich, I. P. Clark, M. Towrie, H. B. Gray, S. Zalis and A. Vlcek, Inorg. Chem., 2015, 54, 8518–8528 CrossRef CAS PubMed
- L. A. Büldt, X. Guo, A. Prescimone and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 11247–11250 CrossRef PubMed
- L. A. Büldt and O. S. Wenger, Angew. Chem., Int. Ed., 2017, 129, 5770–5776 CrossRef
- L. L. Jamula, A. M. Brown, D. Guo and J. K. McCusker, Inorg. Chem., 2014, 53, 15–17 CrossRef CAS PubMed
- T. C. B. Harlang, Y. Z. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chabera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed
- Y. Z. Liu, P. Persson, V. Sundström and K. Warnmark, Acc. Chem. Res., 2016, 49, 1477–1485 CrossRef CAS PubMed
- T. Duchanois, T. Etienne, C. Cebrian, L. Liu, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, Eur. J. Inorg. Chem., 2015, 2469–2477 CrossRef CAS
- L. Liu, T. Duchanois, T. Etienne, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, Phys. Chem. Chem. Phys., 2016, 18, 12550–12556 RSC
- S. G. Shepard, S. M. Fatur, A. K. Rappe and N. H. Damrauer, J. Am. Chem. Soc., 2016, 138, 2949–2952 CrossRef CAS PubMed
- D. C. Ashley and E. Jakubikova, Coord. Chem. Rev., 2017, 337, 97–111 CrossRef CAS
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed
- V. Gray, K. Borjesson, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Phys. Chem. C, 2016, 120, 19018–19026 CAS
- P. Chabera, Y. Liu, O. Prakash, E. Thyrhaug, A. El Nahhas, A. Honarfar, S. Essén, L. A. Fredin, T. C. B. Harlang, K. S. Kjaer, K. Handrup, F. Ericsson, Y. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695–699 CrossRef CAS
- P. Zimmer, P. Müller, L. Burkhardt, R. Schepper, A. Neuba, J. Steube, F. Dietrich, U. Flörke, S. Mangold, M. Gerhards and M. Bauer, Eur. J. Inorg. Chem., 2017, 1504–1509 CrossRef CAS
- A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS
- S. Parisien-Collette, A. C. Hernandez-Perez and S. K. Collins, Org. Lett., 2016, 18, 4994–4997 CrossRef CAS PubMed
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed
- O. Reiser, Acc. Chem. Res., 2016, 49, 1990–1996 CrossRef CAS PubMed
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386–8398 RSC
- M. Gernert, U. Müller, M. Haehnel, J. Pflaum and A. Steffen, Chem.–Eur. J., 2017, 23, 2206–2216 CrossRef CAS PubMed
- L. A. Büldt, C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2017, 23, 8577–8580 CrossRef PubMed
- Y. Zhang, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2016, 138, 13115–13118 CrossRef CAS PubMed
- K.-T. Yeung, W.-P. To, C. Sun, G. Cheng, C. Ma, G. S. O. Tong, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 133–137 CrossRef CAS PubMed
- C. O. Dietrich-Buchecker, P. A. Marnot, J. P. Sauvage, J. R. Kirchhoff and D. R. McMillin, J. Chem. Soc., Chem. Commun., 1983, 513–515 RSC
- P. C. Ford, Chem. Sci., 2016, 7, 2964–2986 RSC
Footnote |
| † Current address: Institute of Inorganic Chemistry, University of Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. |
| This journal is © The Royal Society of Chemistry 2017 |