DOI:
10.1039/C7SC02738A
(Edge Article)
Chem. Sci., 2017,
8, 6400-6408
Potent mechanism-based sirtuin-2-selective inhibition by an in situ-generated occupant of the substrate-binding site, “selectivity pocket” and NAD+-binding site†
Received
20th June 2017
, Accepted 14th July 2017
First published on 21st July 2017
Abstract
Sirtuin 2 (SIRT2), a member of the NAD+-dependent histone deacetylase family, has recently received increasing attention due to its potential involvement in neurodegenerative diseases and the progression of cancer. Potent and selective SIRT2 inhibitors thus represent desirable biological probes. Based on the X-ray crystal structure of SIRT2 in complex with a previously reported weak inhibitor (6), we identified in this study the potent mechanism-based inactivator KPM-2 (36), which is selective toward SIRT2. Compound 36 engages in a nucleophilic attack toward NAD+ at the active site of SIRT2, which affords a stable 36-ADP-ribose conjugate that simultaneously occupies the substrate-binding site, the “selectivity pocket” and the NAD+-binding site. Moreover, 36 exhibits antiproliferative activity in cancer cells and remarkable neurite outgrowth activity. This strategy for the selective inhibition of SIRT2 should allow further probing of the biology of SIRT2, and promote the development of new disease treatment strategies.
Introduction
Human sirtuins (SIRT1–7) are currently the only known class of histone deacetylases that require NAD+ to catalyze the selective deacetylation of lysine in histone and non-histone proteins.1 Recently, a growing body of evidence has emerged, which suggests that sirtuins can also efficiently catalyze the removal of long chain fatty acids (SIRT1–3, 6 and 7) and 4-oxononanonyl to ε-amino lysine residues (SIRT2).2–5 SIRT5 exhibits desuccinylase enzymatic activity.6 The catalytic region of sirtuins consists of a Rossmann-fold domain, a zinc-binding domain and the loops connecting them.7 The interface between the loops and the two domains form both NAD+ (subdivided by the pockets A–C) and Ac-Lys substrate-binding sites. This region is highly conserved in SIRT1–7 and mutations can drastically reduce the enzymatic activity. Even though the sirtuin subcellular localization is strongly connected to the cell type, stress conditions and interactions with other proteins, SIRT1, 6 and 7 are mainly localized in the nuclei, SIRT3–5 are mitochondrial sirtuins, and SIRT2 is classified as a cytoplasmic isoform.8 The ability of sirtuins to deacetylate histones, transcription factors and nuclear receptors reflects their involvement in many physiopathological processes. The activity of sirtuins has been related to metabolic disorders,9 cancer progression10 (in which they play a Janus-faced role) and neurodegenerative diseases.11
Of the seven isoforms, SIRT1 is considered the genome “guardian angel” in normal cells, i.e., it can regulate genomic stability through DNA repair. In cancer cells, its overexpression leads to the negative regulation of p53,12,13 FOXO3a,14 HIF1-α15 and a blockage of Bax translocation to the mitochondria.16 Thus, SIRT1 induces cancer cell survival, accumulation of DNA mutations and drug resistance. While it is undisputed that the activity of SIRT1 is correlated to the growth of several kinds of cancer cells, the exact function of SIRT2 within cancer cells was the subject of strong controversy until recently. SIRT2 has been reported to act as a tumor suppressor and as an oncogene.17 SIRT2 levels are reduced in various types of cancer such as glioma,18 liver cancer,19 esophageal adenocarcinoma20 and breast cancer,21 while they are increased in neuroblastoma,21 acute myeloid leukemia22 and prostate cancer.23 However, recent reports strongly suggest that SIRT2 plays a key role in the invasion and metastasis formation of malignancy by increasing the cell motility of cancer cells.23 On the other hand, the function of SIRT2 in neurons remains the subject of controversy. While it has been reported that SIRT2 inhibition impairs neurogenesis and results in depression-like behaviors,24 a SIRT2 inhibitor has been reported to induce antidepressant-like action.25,26 Thus, potent and selective SIRT2 inhibitors are desirable as biological probes.
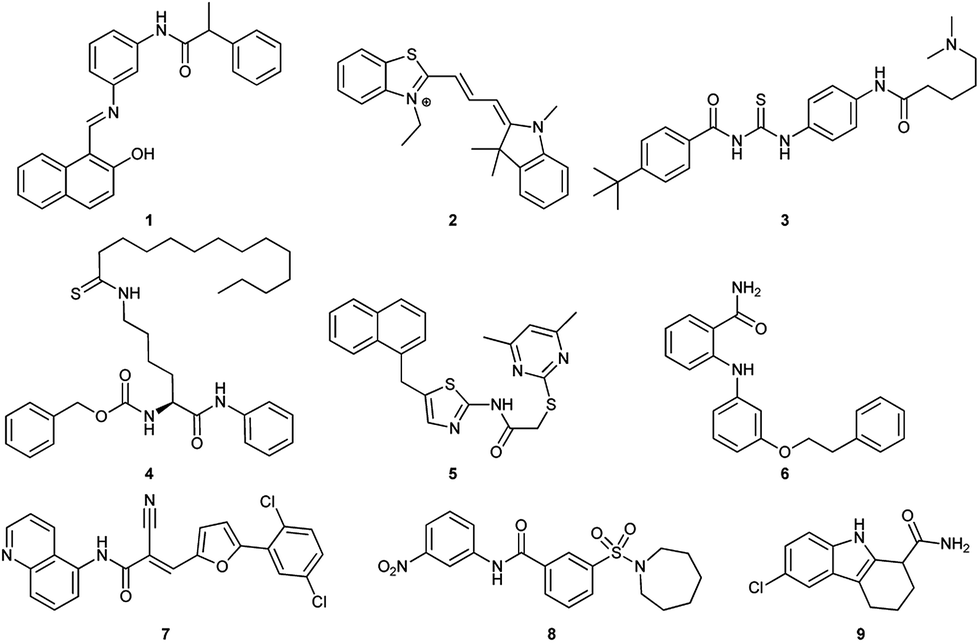
So far, several SIRT1 and SIRT2 inhibitors have been developed (Fig. 1), including e.g. salermide (1),27 AC93253 (2),28 tenovin-6 (3),29,30 TM (4),31 SirReal2 (5),32,33 3′-phenethyloxy-2-anilinobenzamide (6),34 AGK2 (7),35 AK-1 (8)36 and EX-527 (9).37 These compounds have shown antiproliferative activity when tested in cancer cells and some of these have shown beneficial effects in neurological disorder models, but in many cases, the lack of potency and isoform specificity suggests their effect is not solely related to SIRT1/2 inhibition. Therefore, the development of more potent and specific compounds is currently of high interest.
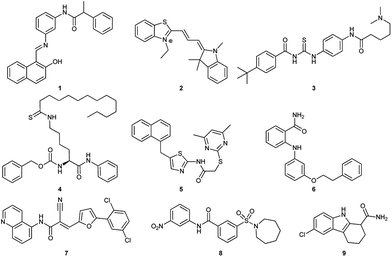 |
| | Fig. 1 Examples of SIRT1 and SIRT2 inhibitors (1–9). | |
In the field of SIRT2 inhibitor design, a milestone on the elucidation of the mechanism behind the isotype selectivity of SIRT2 inhibitors has recently been reported by Rumpf et al., who discovered that the novel drug-like small molecule SirReal2 (5)32,33 is able to induce a rearrangement of the SIRT2 active site by creating a binding pocket, called the “selectivity pocket”, which is responsible for isoform specificity, suggesting a potential common mode of action for selective SIRT2 inhibitors. In fact, the “selectivity pocket” is the pocket that accommodates the long chain fatty acyl groups.3,38,39
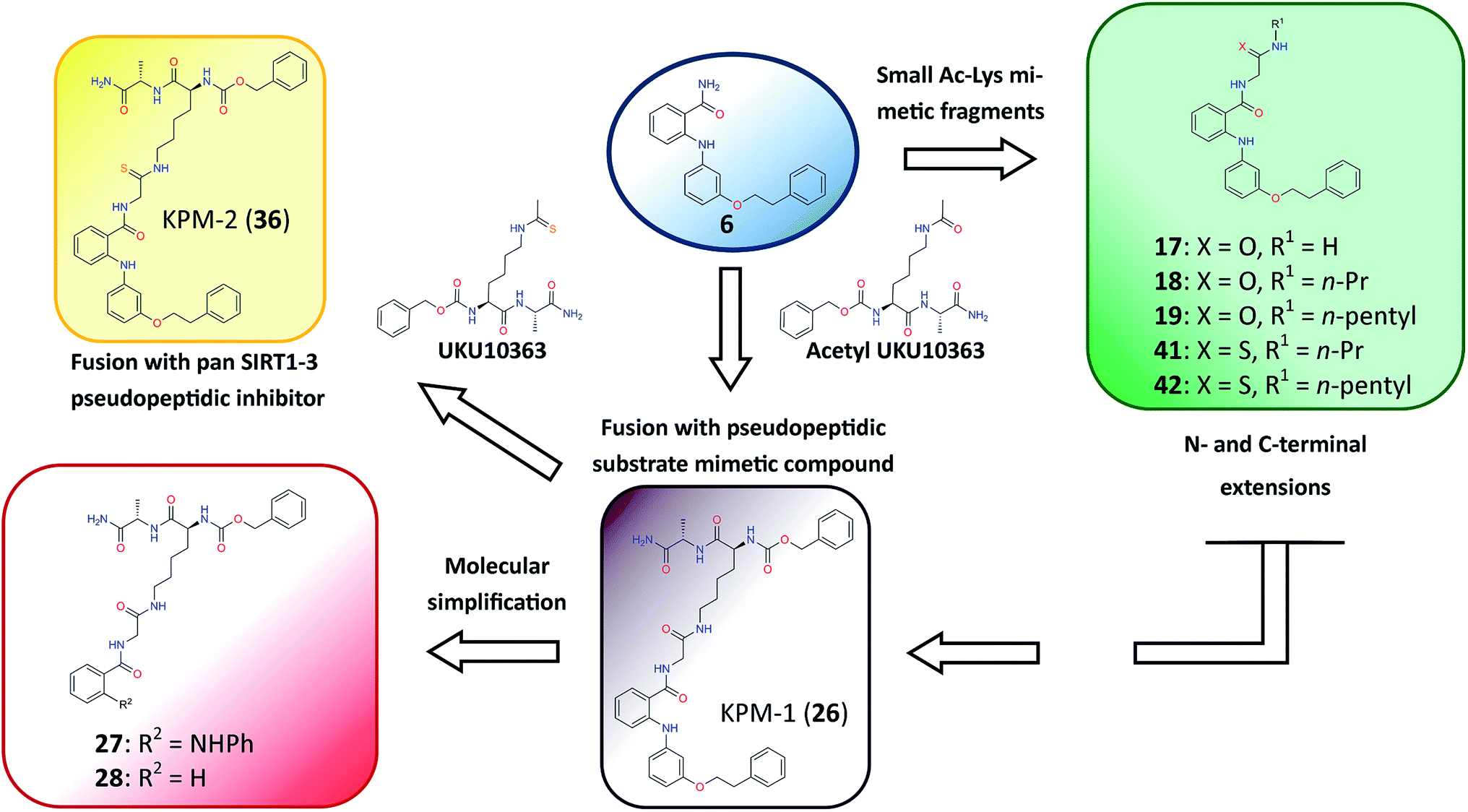
Previously, we proposed a novel class of 2-anilinobenzamides34 as selective SIRT2 inhibitors, in which a phenylethyl ether moiety acted as the optimal fragment to improve both potency and isoform selectivity. Herein, with the aim to elucidate the binding mode of this prototype and to further improve the inhibitory activity, we initially solved the single-crystal X-ray diffraction structure of SIRT2 in complex with 3′-phenethyloxy-2-anilinobenzamide (6),34 which revealed that 6 is located in the selectivity pocket. Then, inspired by this result, we applied structural modifications to the amide moiety in order to target the acetyl–lysine-substrate-binding site and NAD+-binding pockets, which finally led to the identification of KPM-2 (36) as a novel class of mechanism-based SIRT2 inactivators. Moreover, 36 shows highly potent and selective SIRT2 inhibition with antiproliferative activity in breast cancer cells and potent neurite outgrowth activity in neuro-2a (N2a) cells.
Results and discussion
Binding mode analysis of 2-anilinobenzamide 6 in complex with SIRT2
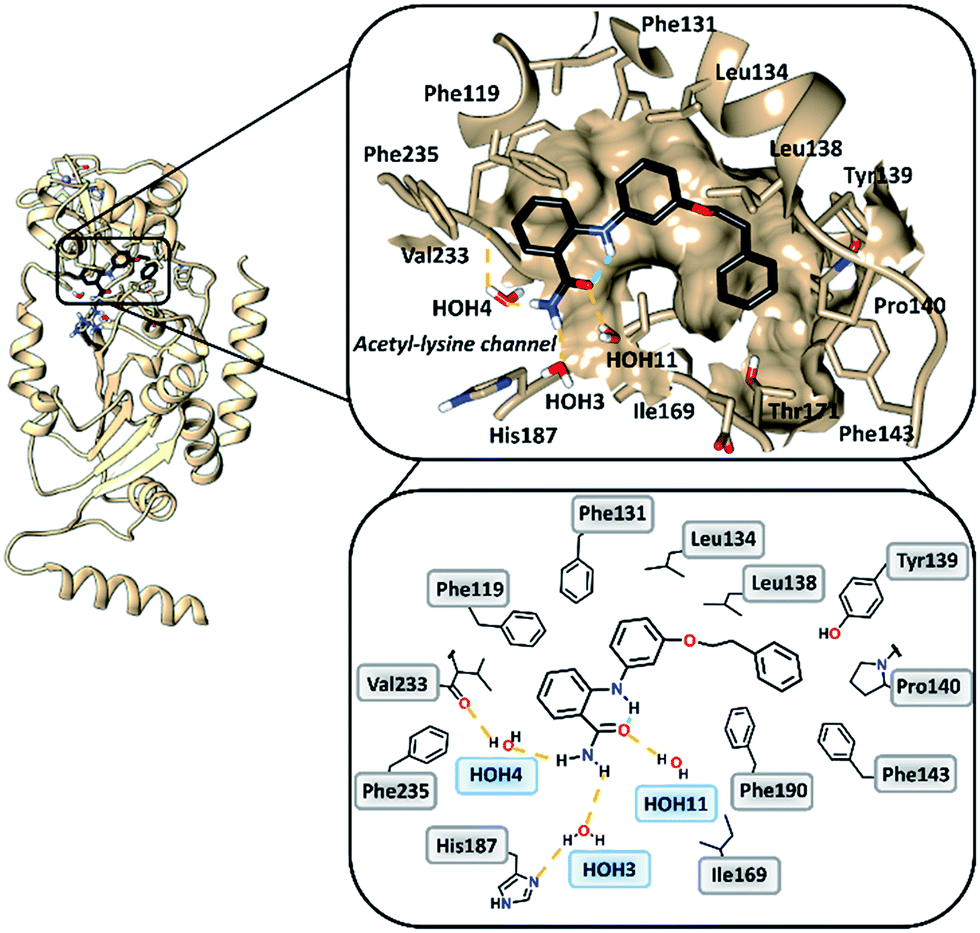
To gain insight into the binding mode of 2-anilinobenzamides, we determined the single-crystal X-ray diffraction structure of SIRT2 in complex with 6 (Table S1†). In general, the overall fold of complex SIRT2/6 is similar to the previously reported SIRT2 structure in complex with the SIRT2-selective SirReal inhibitors (PDB code: 5DY5, 5DY4, 4RMG and 4RMH), whereby the main divergences occur at the cofactor binding loop (Gly92-Leu112), which is not well defined, and at the downward-shifted connection loop at β8–α10 (ref. 40) (Thr262-Phe269). The inhibitor binds to a highly lipophilic pocket localized at the interface between the Rossmann-fold domain and a small domain adjacent to the C-pocket of the NAD+-binding site. The superimposition of the SIRT2/6 crystal structure with that of the SIRT2 apo structure (PDB: 3ZGO, Fig. S1†) demonstrates that 6 induces a shift of the helix α5 (ref. 40) and the connection loop to α6 (Glu129-Thr146), which is responsible for the accommodation of the inhibitor. The binding mode analysis of 6 (Fig. 2) revealed that its phenoxyethylphenyl moiety is surrounded by Phe131, Leu134, Leu138, Tyr139, Pro140, Phe143 and Ile169, which leads to the establishment of π–π and H–π interactions. The 2-aminobenzamide moiety oriented towards the acetyl–lysine tunnel exhibits hydrogen bonds with water molecules HOH3, HOH4 and HOH11. Of these three water molecules, HOH3 and HOH4 are stabilized by a hydrogen bond network with the side chain of the catalytically active His187 and the main chain of Val233, i.e., the key amino acid residues that stabilize the binding of the acetyl–lysine substrate during the deacetylation reaction.40 Furthermore, 6 shows an intramolecular hydrogen bond between the amide (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and aromatic amine (–NH) moieties. This partially locked conformation renders the speculation feasible that there might be an additional structural requirement to achieve SIRT2 inhibition, as previously observed for SIRT1.41
O) and aromatic amine (–NH) moieties. This partially locked conformation renders the speculation feasible that there might be an additional structural requirement to achieve SIRT2 inhibition, as previously observed for SIRT1.41
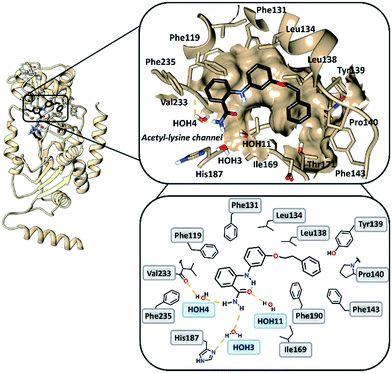 |
| | Fig. 2 Overall structure of SIRT2/6 (light brown ribbon; left). The windows on the right show magnifications of the inhibitor-binding mode, wherein 6 (black) occupies the SIRT2 selectivity pocket delineated by the molecular surface. Intramolecular (green) and intermolecular (yellow) hydrogen bonds are illustrated by dashed lines. | |
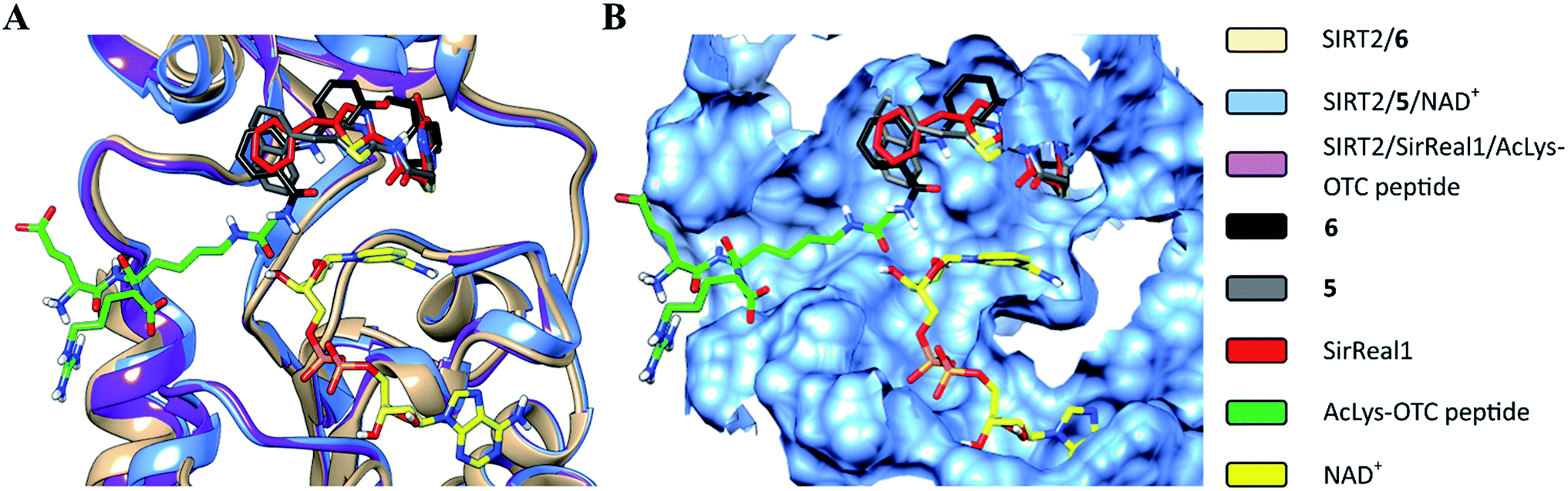
The structural overlay of the SIRT2/6, SIRT2/5/NAD+ (PDB: 4RMG) and SIRT2/SirReal1/AcLys-OTC peptide (PDB: 4RMI) crystal structures demonstrate that inhibitors 6, SirReal1 and 5 occupy the same binding site (Fig. 3 and S2†).
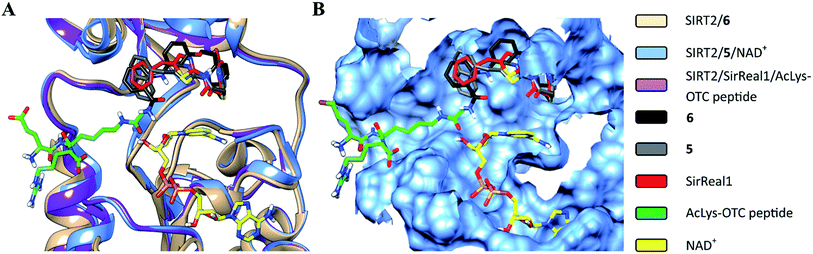 |
| | Fig. 3 Superimposition of SIRT2/6 with the complex structures of 5/NAD+ (PDB: 4RMG) and the SirReal1/AcLys OTC peptide substrate (PDB: 4RMI). (A) Colored ribbons; (B) molecular surface; the binding sites of the acetylated substrate, NAD+, 6, 5 and SirReal1 are shown in cyan. The carbon atoms of 6, SirReal1, 5, AcLys-OTC peptide and NAD+ are colored in black, grey, red, green and yellow, respectively. | |
Design of novel SIRT2-selective inhibitors and their in vitro biological evaluation
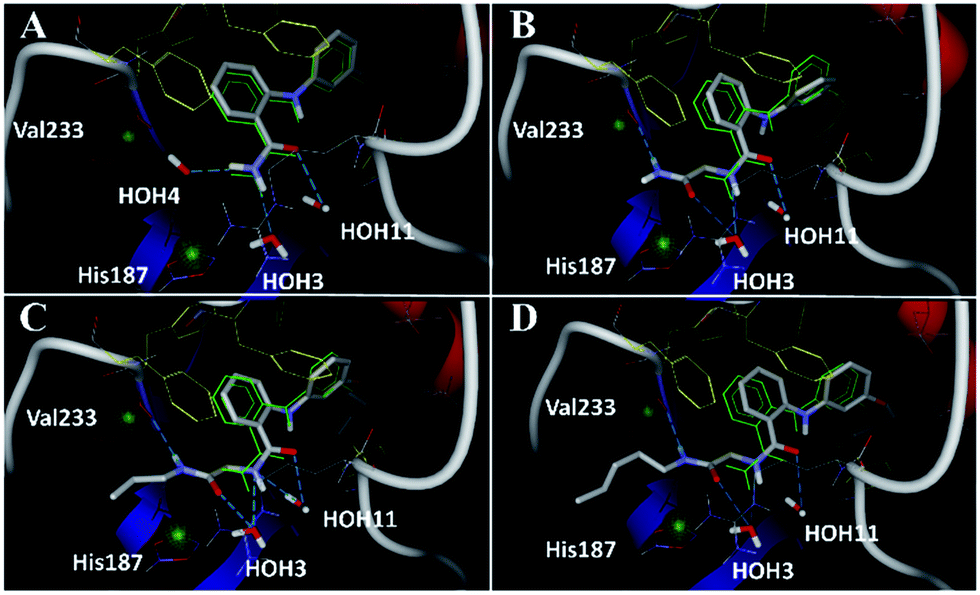
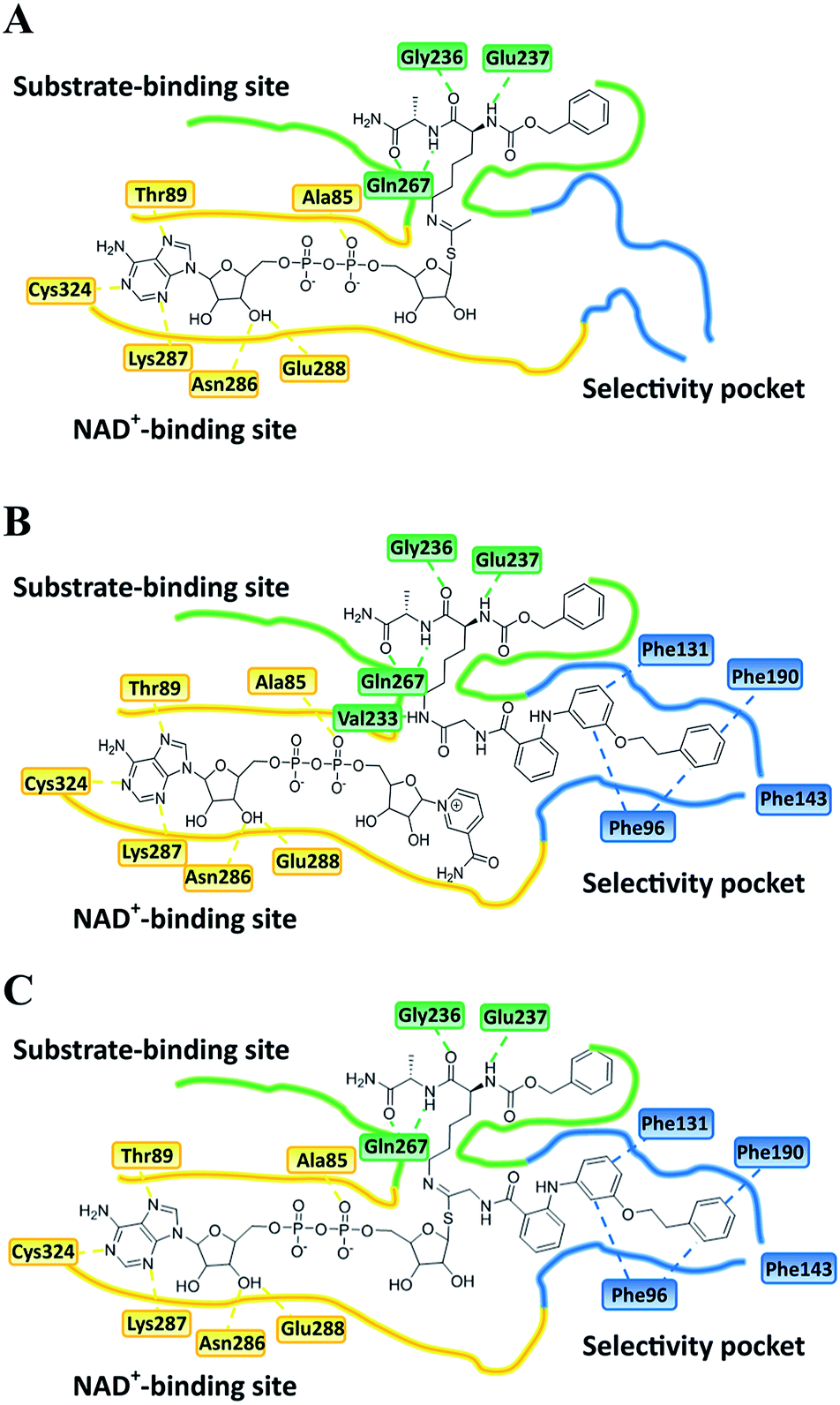
Closer evaluation of the superimposed SIRT2 structures revealed that the amide group of 6 might occupy the same space as the methyl group of the Nε-acetyl–lysine substrate (Fig. 3). This result prompted us to take a new perspective toward targeting the substrate-binding site through the functionalization of the benzamide moiety in 6 to mimic the acetyl–lysine interactions, which afforded compounds 17–19, 26–28, 36, 41 and 42 (Fig. 4). All compounds prepared in this study (Schemes S1–4†) were screened in vitro against SIRT1–3 and SIRT5 (concentration: 10 μM; Table 1), while 6, UKU10363 (Fig. 4) and 5 were used as reference compounds. In a first attempt to mimic the lysine acetyl amide, we explored small fragments using a glycinamide linker (17–19). The screening of SIRT1–3 and 5 (Table 1) revealed that both isotype selectivity and good SIRT2 inhibitory activity were retained upon amide functionalization of 6, although the inhibitory potency was not improved. In order to rationalize the results, the putative binding mode of 17–19 was studied via docking simulations (Fig. 5). The glycinamide handle exhibits hydrogen bonding with HOH3 and HOH11 (Fig. 5B–D), located in proximity of the amide groups of 17–19, and the water molecules can be displaced by NAD+ once it binds to the active site. The glycinamide substituents in 17–19 displace HOH4, thus mimicking the behavior of acetyl–lysine. This may lead to the direct interaction of 17–19 with Val233 (Fig. 5B–D) and a subsequent loss of hydrogen bonding with HOH4, resulting in an “abated” inhibitory activity. In order to test whether SIRT2 inhibition could be improved by introducing N-, C-terminal pseudopeptidic extensions on 19, we designed KPM-1 (26). A methylene bridge was used to link 6 with the acetyl derivative of UKU10363,42 which is a previously identified pan-SIRT1–3 inhibitor (Fig. 4). UKU10363 has been proposed to inhibit SIRT1–3 through the attack of its thioacetyl group to NAD+ to afford a UKU10363-ADP-ribose conjugate (Fig. S3A, S3B† and 6A). The occupation of the SIRT2 selectivity pocket by a single molecule through the 2-anilinobenzamide core and the substrate-binding site through the pseudopeptidic backbone should lead to potent inhibition (Fig. 6B). The screening output revealed a strong and selective SIRT2 inhibitory effect (IC50 = 0.37 μM) by 26 (Table 1), which was fourfold more active than lead compound 6 and equipotent to UKU10363 and 5. This result supports the hypothesis that targeting both the substrate-binding site and the selectivity pocket by linking two distinct inhibitor scaffolds represents a useful strategy to develop novel SIRT2 inhibitors. In order to obtain additional information on the structure–activity relationship around 26, the 2-anilinobenzamide core was simplified in 27 and 28 (Fig. 4). As expected, the removal of the phenoxyethyl fragment in 27 reduced the SIRT2 inhibitory activity by ∼20% (Table 1), while further simplification of the core in 28 led to no SIRT2 inhibition. These results are consistent with those of our previous 2-anilinobenzamide SAR study.34 Then, we turned our efforts toward finding a suitable strategy to boost the inhibitory potency of 26. Thioacetyl–lysine peptide/pseudopeptide inhibitors interact stronger with the substrate-binding site than their acetylated counterparts through the formation of a stalled intermediate with NAD+, inducing a retardation of the enzymatic turnover rate (Fig. 6A and S3B†).43,44 Following this lead, we designed KPM-2 (36) to test whether replacement of the acyl group in 26 with a thioacyl group could lead to an improvement of the SIRT2 inhibitory activity by generating a novel trapped intermediate (Fig. S3C†) and, consequently, the full occupation of the substrate/NAD+-binding site and the selectivity pocket (Fig. 6C). At 1 μM concentration, 36 potently inhibited SIRT2, even though it did not show strong inhibition towards SIRT1 and SIRT3. As indicated in Table 1, 36 is a moderate SIRT1 and SIRT3 inhibitor, while it potently inhibits SIRT2 (SIRT1 [IC50]/SIRT2 [IC50] = 28.4; SIRT3 [IC50]/SIRT2 [IC50] = 172.5) with an IC50 of 55 nM, which is by a factor of ∼32 lower than that of 6, and by a factor of ∼11 lower than UKU10363. Notably, the SIRT2-inhibitory activity of 36 was much greater than representative previously reported SIRT2 inhibitors 3 and 5–9. The moderate SIRT1 and SIRT3 inhibition by 36 is not surprising as SIRT1 and SIRT3 are also known to be a demyristoylase and have a pocket similar to the SIRT2 selectivity pocket that can accommodate the long chain fatty acyl group.3
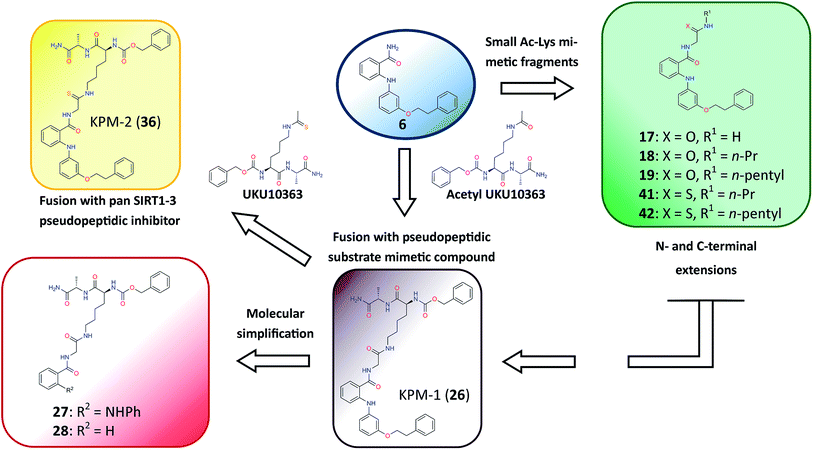 |
| | Fig. 4 Molecular design applied to 6 in order to mimic acetyl–lysine substrate interactions. | |
Table 1 Screening of derivatives of 6, UKU10363, 3, 5, 7–9, 17–19, 26–28, 36, 41 and 42
| Cpd. |
% inhibition at 10 μMa |
IC50 ± SDc [μM] or % inhibition at 50 μM |
| SIRT1 |
SIRT2 |
SIRT3 |
SIRT5 |
SIRT1 |
SIRT2 |
SIRT3 |
SIRT5 |
|
Fluor de Lys assay; values represent mean values ± standard deviation of at least two experiments.
% inhibition at 1 μM.
Fluor de Lys assay; values are calculated from three independent determinations, which afford a total of 21 data points (Fig. S4).
Data from ref. 34.
Data from ref. 32.
|
|
6
|
3 ± 3.6 |
90 ± 1.5 |
1 ± 2.6 |
10 ± 2.6 |
14% at 50 μM, >300d |
1.74 ± 0.26 |
12% at 50 μM, >300d |
12% at 50 μM |
| 32 ± 4.1b |
1.0 ± 0.12d |
| UKU 10363 |
97 ± 0.04 |
99 ± 3.2 |
65 ± 0.23 |
3 ± 3.9 |
0.18 ± 0.013 |
0.59 ± 0.13 |
4.39 ± 0.28 |
6% at 50 μM |
| 83 ± 0.4b |
56 ± 1.9b |
19 ± 0.78b |
|
17
|
10 ± 1.9 |
85 ± 0.47 |
10 ± 0.95 |
0 ± 1.1 |
18% at 50 μM |
3.60 ± 0.03 |
13% at 50 μM |
4% at 50 μM |
| 19 ± 0.63b |
|
18
|
2 ± 1 |
81 ± 0.43 |
0 ± 1.1 |
0 ± 0.2 |
12% at 50 μM |
4.04 ± 0.65 |
3% at 50 μM |
3% at 50 μM |
|
19
|
4.5 ± 3.4 |
69 ± 2.2 |
3 ± 3.2 |
6 ± 3.2 |
4% at 50 μM |
6.71 ± 0.42 |
2% at 50 μM |
6% at 50 μM |
|
26
|
16 ± 0.1 |
99 ± 0.15 |
14 ± 4.9 |
0 ± 3.5 |
25% at 50 μM |
0.37 ± 0.03 |
56% at 50 μM |
6% at 50 μM |
| 74 ± 2b |
|
27
|
24 ± 2.1 |
80 ± 0.12 |
9 ± 2.2 |
0 ± 2.5 |
68% at 50 μM |
1.84 ± 0.21 |
16% at 50 μM |
3% at 50 μM |
|
28
|
3.5 ± 1 |
8 ± 3.3 |
1 ± 1.8 |
0 ± 0.6 |
1% at 50 μM |
18% at 50 μM |
0% at 50 μM |
0% at 50 μM |
|
36
|
29.5 ± 2.1b |
100 ± 3.4b |
2 ± 1.8b |
4 ± 2.6 |
1.56 ± 0.06 |
0.055 ± 0.0035 |
9.49 ± 1.7 |
10% at 50 μM |
|
5
|
3 ± 0.65 |
98 ± 3.1 |
5 ± 0.6 |
3 ± 1.5 |
6% at 50 μM, >100e |
0.30 ± 0.06, 0.4e |
1% at 50 μM, >100e |
1% at 50 μM |
| 80 ± 1.5b |
|
3
|
0.9 ± 0.17 |
45 ± 2.5 |
0 ± 1.0 |
0 ± 1.4 |
64% at 50 μM |
9.66 ± 2.3 |
9% at 50 μM |
2% at 50 μM |
|
7
|
0.9 ± 0.62 |
45 ± 2.9 |
0 ± 1.0 |
0 ± 1.1 |
14% at 50 μM |
11.5 ± 3.8 |
14% at 50 μM |
3% at 50 μM |
|
8
|
6.2 ± 0.85 |
31 ± 0.42 |
0 ± 0.89 |
0 ± 0.05 |
25% at 50 μM |
30.1 ± 1.3 |
0% at 50 μM |
0% at 50 μM |
|
9
|
97 ± 0.13 |
79 ± 1.3 |
20 ± 1.2 |
0 ± 1.1 |
0.24 ± 0.01 |
2.53 ± 0.04 |
54% at 50 μM |
0% at 50 μM |
|
41
|
0 ± 0.84 |
54 ± 1.5 |
0 ± 0.50 |
1 ± 2.2 |
10% at 50 μM |
8.47 ± 0.30 |
0% at 50 μM |
3% at 50 μM |
|
42
|
0 ± 1.1 |
40 ± 3.4 |
0 ± 1.1 |
0 ± 0.23 |
11% at 50 μM |
19.3 ± 3.1 |
2% at 50 μM |
3% at 50 μM |
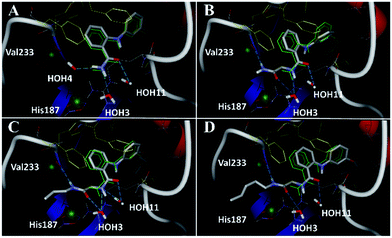 |
| | Fig. 5 Docking poses for 6 ((A), redocked ligand RMSD 0.70 Å), 17 (B), 18 (C), and 19 (D) in the SIRT2/6 crystal structure. Crystallized ligand 6 and docking poses are colored in green and gray, respectively. Val233 and His187 are colored in red and delimited by green spheres. The Phe residues surrounding selectivity pocket are colored in yellow; hydrogen bonds are represented by dashed lines. | |
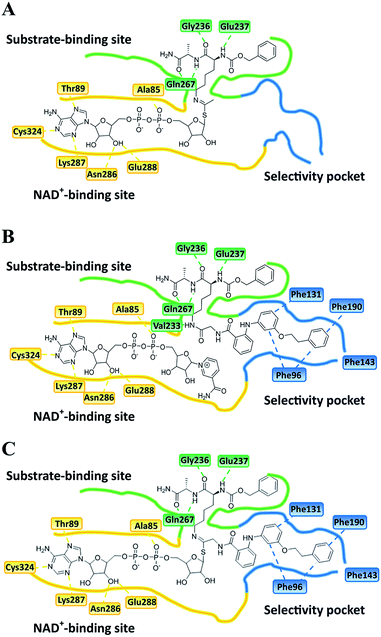 |
| | Fig. 6 Plausible binding mode for (A) UKU10363, (B) 26 and (C) 36. The substrate-binding site, NAD+-binding site, and selectivity pocket are outlined by green, yellow and blue lines, respectively. | |
In addition, we examined the SIRT-inhibitory activity of compounds 41 and 42, thioamide analogues of 18 and 19, respectively. As shown in Table 1, even though SIRT2 inhibition was observed with 41, no improvement was achieved as compared with 18. Furthermore, the elongation of aliphatic chain (19) resulted in a loss of potency. These results suggest that compounds 41 and 42 do not react with NAD+ because the short alkyl chain of 41 and 42 is too flexible to fix the thioamide group in proximity to the nicotinamide moiety of NAD+.
Mechanistic analysis of SIRT2 inhibition by 36
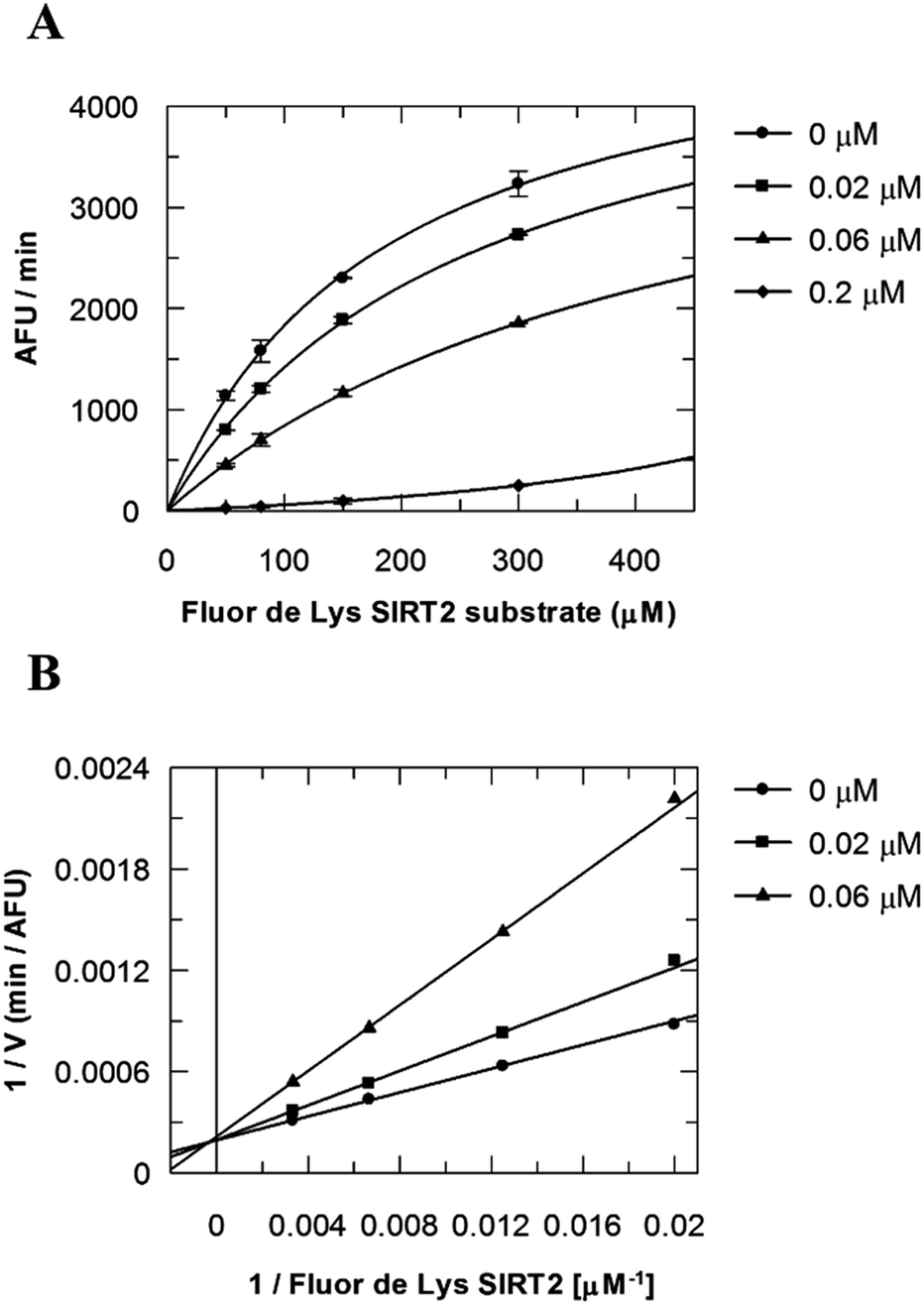
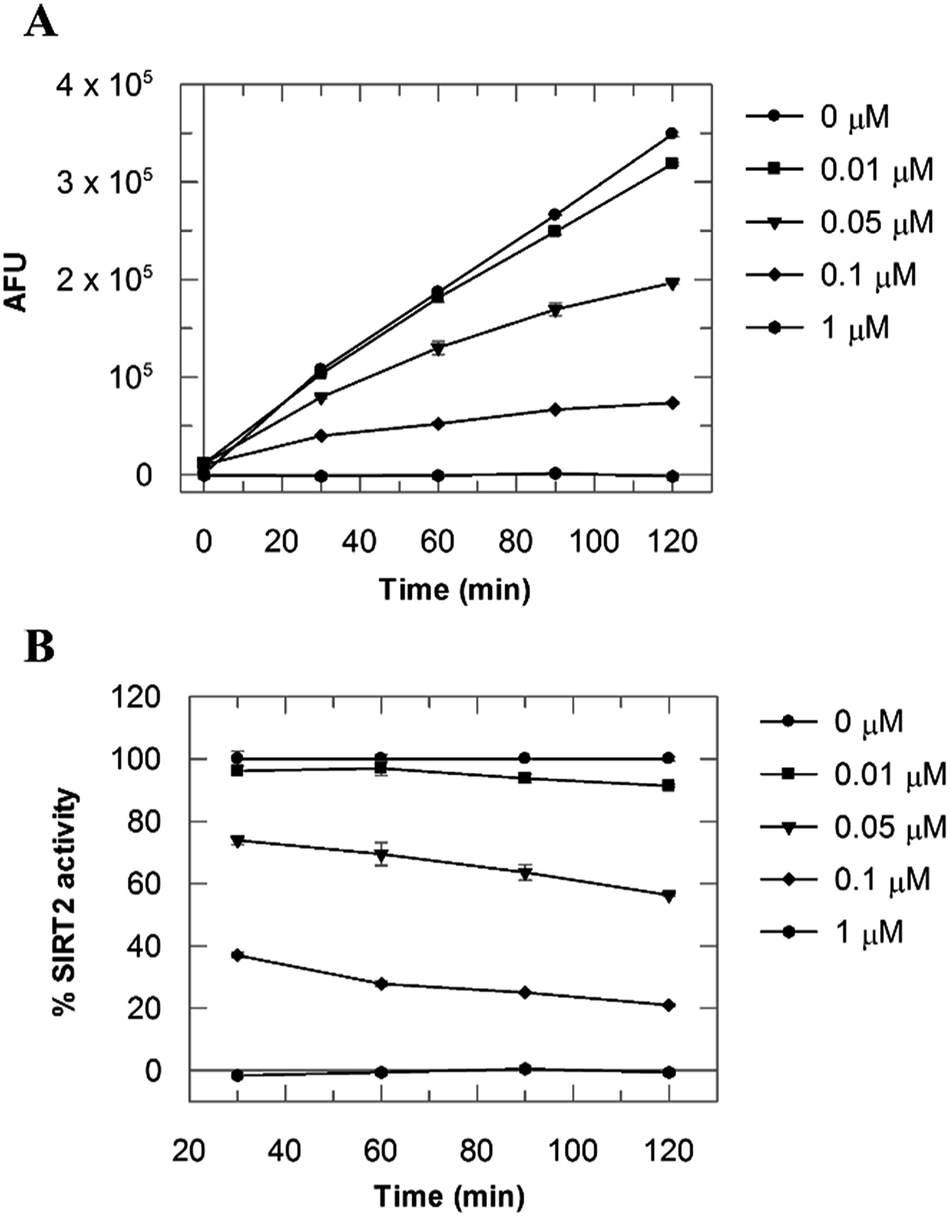
Due to the peculiar structure of the novel SIRT2 inhibitor, further investigations seemed pertinent. To elucidate the 36-induced SIRT2 inhibition mechanism, a substrate competition analysis was carried out (Fig. 7A). The double reciprocal plot of initial velocity as a function of substrate concentration 1/V against 1/[S] revealed a series of regression lines intersecting the 1/V axis, suggesting competitive inhibition with the acetylated lysine substrate (Fig. 7B). Furthermore, the apparent Michaelis constant Km of the substrate increased with increasing concentration of 36 under NAD+ saturation conditions. Subsequently, we monitored the SIRT2 reaction course in the presence and absence of 36 (Fig. 8A). The inhibitory effect was time-dependent and exhibited a pronounced effect after 120 min of simultaneous incubation of SIRT2, the acetylated substrate, NAD+ and 36 (Fig. 8B). These results support a potential SIRT2 mechanism-based inhibition. To confirm that the time-dependent SIRT2 inhibition by 36 was not due to an assay artifact or chemical instability, a SIRT deacetylated standard assay and an HPLC stability test were carried out (Fig. S5 and S6†). Under the applied conditions, 36 was stable and did not inhibit the developer reaction of the SIRT2 assay used in this study.
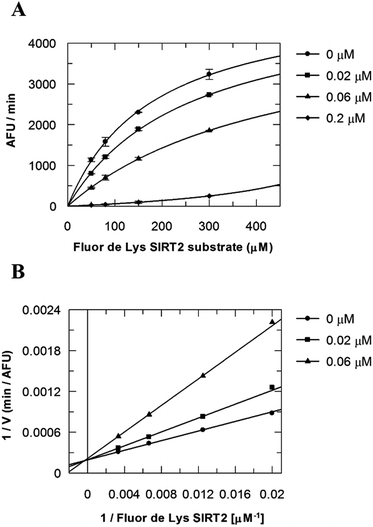 |
| | Fig. 7 Competition analysis for 36 with the acetylated lysine substrate. (A) Michaelis–Menten plot showing the Fluor de Lys SIRT2 substrate (μM) competition analysis for of 36 at 0, 0.02, 0.06 and 0.2 μM. (B) Lineweaver–Burk plot: 1/V as a function of the reciprocal acetylated lysine substrate in the presence of 0, 0.02 and 0.06 μM of 36. Values were calculated from two independent determinations. | |
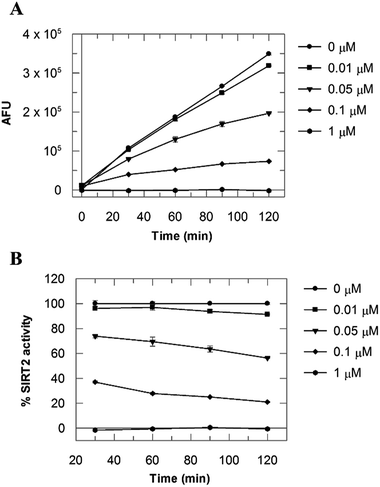 |
| | Fig. 8 (A) SIRT2 time reaction course at 0, 0.01, 0.05, 0.1 and 1 μM of 36. (B) SIRT2 activity expressed as % of the control value. Values were calculated from two independent determinations. | |
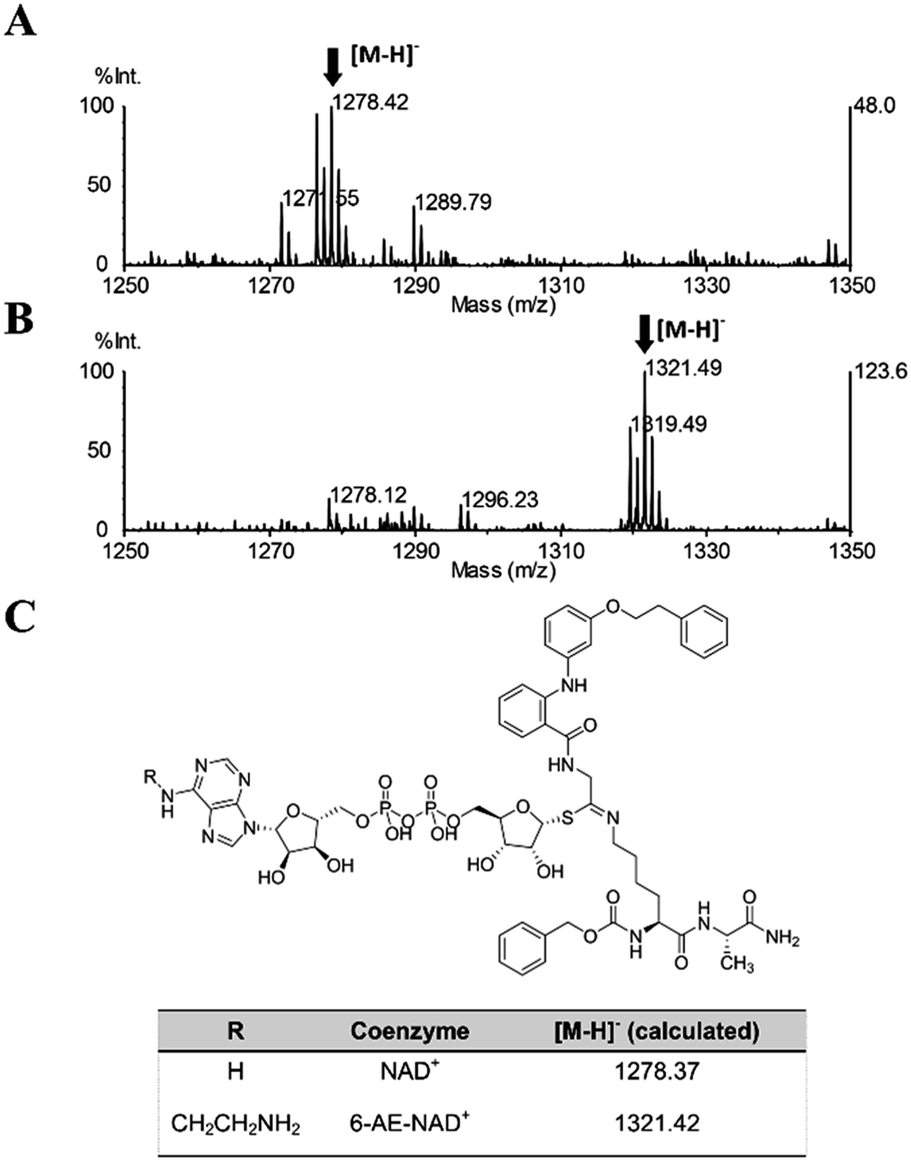
Once proven that 36 is a substrate-competitive and time-dependent inhibitor, a MALDI-TOF (matrix-assisted laser desorption/ionization time-of-flight) mass spectrometric analysis was conducted, in order to determine whether the 36-ADP-ribose conjugate could be detected after simultaneous incubation of 36 with SIRT2 and NAD+ (Fig. S3C† and 6C). As shown in Fig. 9A, a peak with m/z 1278.42 corresponding to [M − H]− of 36-ADP-ribose conjugate was observed only in the presence of SIRT2 (Fig. S7†). In order to provide additional insight, a similar experiment was carried out using 6-aminoethyl (AE)-NAD+. Upon generation of the trapped intermediate, a mass shift of +43 Da from that of the 36-ADP-ribose conjugate should be observed. As shown in Fig. 9B, a mass peak of m/z 1321.49 confirmed the ability of 36 to specifically react with 6-(AE)-NAD+, which is catalyzed by SIRT2. In their entirety, these results suggest that 36 is a substrate-competitive inhibitor. Moreover, they indicate that 36 specifically reacts in a time-dependent manner with NAD+ exclusively in the presence of SIRT2, which affords a Michaelis adduct intermediate, supporting the hypothesis that 36 is a SIRT2-mechanism-based inhibitor.
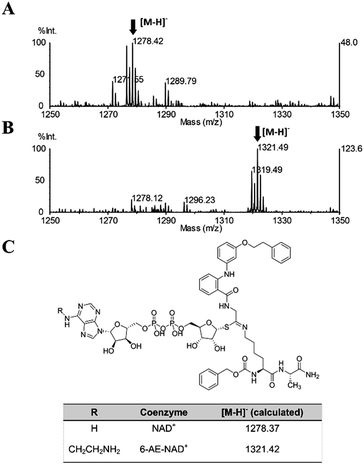 |
| | Fig. 9 MALDI-TOF mass spectrometric detection of the ADP-ribose conjugate formed between 36 and (A) NAD+ or (B) 6-AE-NAD+. (C) Chemical structure of the conjugate and the calculated m/z value for the [M − H]− ion. | |
The effect of 26 and 36 in breast cancer cells (MDA-MB-231 and MCF-7)
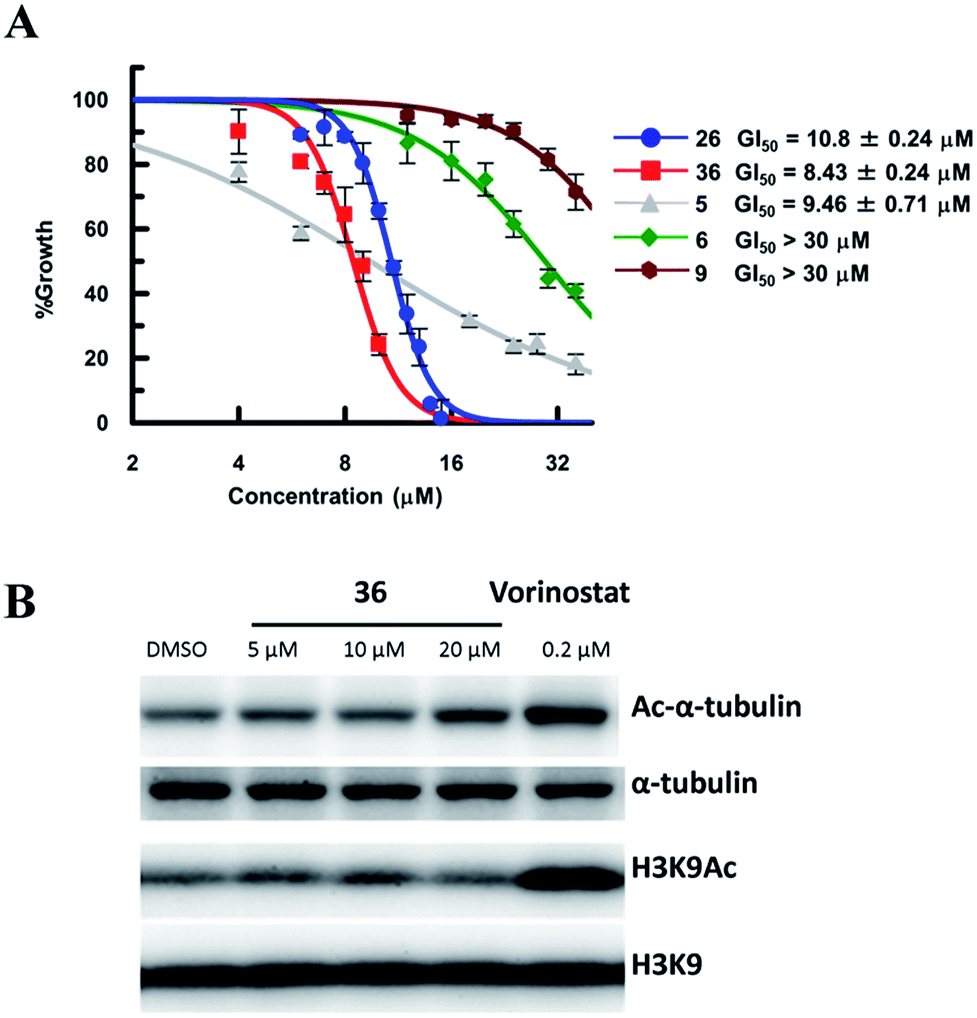
So far, valuable antiproliferative activity in cancer cells has been established for several SIRT1/2 inhibitors. However, on account of their questionable potency of action and selectivity profile, a possible off-target activity could not be ruled out. In this study, the ability of the highly potent and selective SIRT2 inactivators 26 and 36 toward the reduction of MDA-MB-231 and MCF-7 cancer cell proliferation and the selective intracellular inhibition of SIRT2 was examined. As reference compounds, 5, 6 and EX-527 (9)37 were included in the assay as a potent SIRT2-selective inhibitor, a weak SIRT2-selective inhibitor and a potent SIRT1-selective inhibitor, respectively. After 72 h of incubation, GI50 values were determined. Neither the potent and selective SIRT1 inhibitor 9, nor the weak SIRT2-selective inhibitor 6 showed significant antiproliferative activity against MDA-MB-231 cells (GI50 > 30 μM; Fig. 10A). However, under the applied conditions, 5 and 26 efficiently reduced the proliferation of MDA-MB-231 cells (GI50 = 9.46 μM and 10.8 μM, respectively). 36, i.e., the most potent SIRT2 inhibitor, showed the highest activity (GI50 = 8.3 μM). We also examined the effect of 6, 9, 26 and 36 in MCF-7 cells (Fig. S8†). The antiproliferative activity of 6, 9, 26 and 36 in MCF-7 cells was similar to that in MDA-MB-231 cells, with a strong correlation between the antiproliferative activity and the SIRT2-inhibitory activity of the inhibitors. These results suggest that SIRT2-selective inhibition is responsible for the observed cancer cell growth inhibition. Subsequently, 36 was used for further examination of its cellular “isotype selectivity” using western blotting (Fig. 10B). Treatment of MDA-MB-231 breast cancer cells with 20 μM 36 induced the accumulation of acetylated α-tubulin, which is a known SIRT2 substrate,47 in the cells. On the other hand, 36 did not upregulate the levels of acetylated lysine 9 on histone H3 (H3K9Ac), which is a SIRT1 substrate,48–50 in MDA-MB-231 cells. In their entirety, these results suggest that 36 selectively inhibits SIRT2 over SIRT1 in the cellular context.
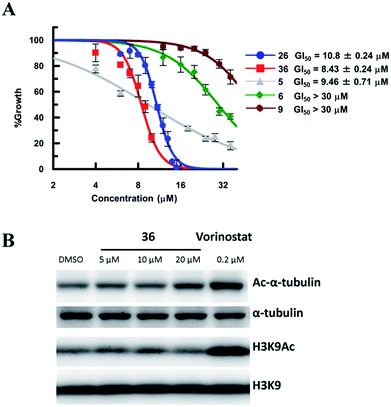 |
| | Fig. 10 (A) Antiproliferative activity of 5, 6, 9, 26 and 36 in MDA-MB-231 cells after 72 h of treatment. Values were calculated from three independent determinations. (B) Western blot detection of α-tubulin or H3K9 acetylation levels in MDA-MB-231 cells after 6 h treatment with 36 or vorinostat, which is a pan-HDAC inhibitor. The latter was used as a positive control that induces acetylation of both α-tubulin45 and H3K9.46 | |
Effect of 36 on the neurite outgrowth of neuro-2a cells
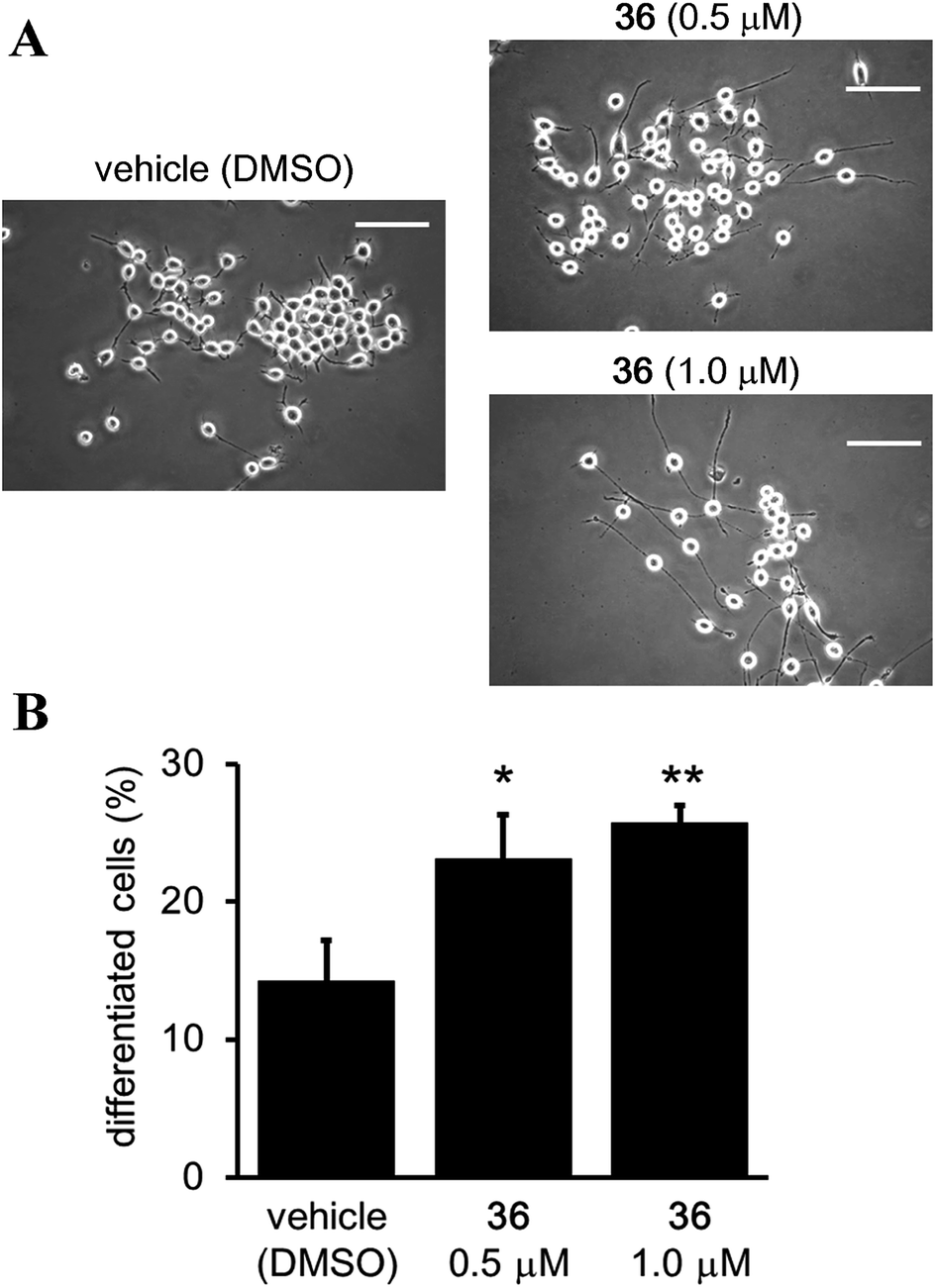
Although it has been suggested that SIRT2 inhibition by small molecules might be beneficial in the context of neurological disorders,25,26,34,51,52 the function of the catalytic activity of SIRT2 in neurogenesis is still unclear.24 Therefore, we investigated the effect of the SIRT2-selective inhibitor 36 on neurite outgrowth. Mouse neuroblastoma neuro-2a (N2a) cells, which have previously been validated for the study of neurite morphology in neuronal differentiation, were used in this study.53,54 Treatment of N2a cells with 36 (0.5 and 1 μM) for 72 h induced neurite outgrowth (Fig. 11A) and significantly increased the percentage of differentiated cells relative to the vehicle control group (0.5 μM and 1 μM of 36vs. vehicle; 23.09 ± 3.21% and 25.73 ± 1.3% vs. 14.22 ± 2.9%; 36 0.5 μM vs. vehicle, p < 0.05, 36 1 μM vs. vehicle, p < 0.01; Fig. 11B). These results suggest that the catalytic activity of SIRT2 is associated with neurite outgrowth and that SIRT2-selective inhibitors may be useful therapeutic agents for neurological disorders.
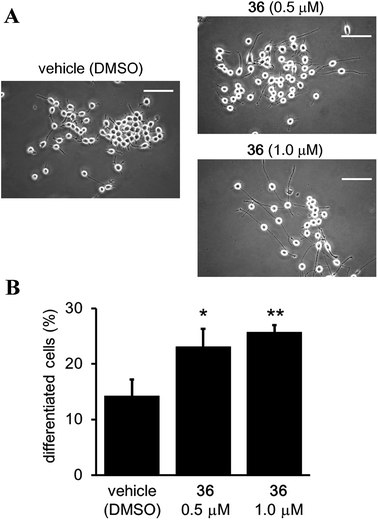 |
| | Fig. 11 Effect of 36 on neurite outgrowth. (A) Representative images showing N2a cells treated with 36 (0.5 and 1 μM) or vehicle (0.1% DMSO) for 72 h. Scale bar: 100 μm. (B) Effect of 36 on N2a differentiation, represented as the % of differentiated cells relative to the total counted cells (at least 100 cells for each condition). Bars represent the mean values ± SD from three independent experiments; *p < 0.05, **p < 0.01, compared to the vehicle group. | |
Conclusions
Since 2012, 2-anilinobenzamides have been considered as one of the most promising classes of drug-like SIRT2 inhibitors. In this study, the binding mode of 6 on SIRT2 was elucidated using X-ray crystallography. On the basis of these results, a new molecular design was developed around the scaffold of this compound. Herein, we demonstrated that targeting of both the SIRT2 selectivity pocket and the substrate-binding site with 26 represents an efficient strategy to achieve potent and selective inhibition. Furthermore, the molecular conjugation of two distinct sirtuin inhibitors, UKU10363 and 6, through a simple methylene bridge led to the new potent SIRT2 inhibitor 36. Kinetic and mass spectroscopic analyses supported the hypothesis that 36 is a mechanism-based inhibitor that affords in situ occupation of the substrate-binding site, the selectivity pocket, and the NAD+-binding site. Thus, 36 should be a valuable tool for the detailed investigation of the effect of SIRT2 inhibition in the cellular context. In MDA-MB-231 and MCF-7 breast cancer cell cultures, 36 showed antiproliferative activity with selective acetylation the SIRT2 substrate α-tubulin. Moreover, 36 showed neurite outgrowth activity in N2a cells, suggesting the possibility of SIRT2-selective inhibitors as therapeutic agents for neurological disorders.
Our strategy to simultaneously target the substrate-binding site, the selectivity pocket and the NAD+-binding site provides a new approach to the design of mechanism-based selective sirtuin inhibitors, which could be potentially extended to explore and probe the limits of the SIRT1–7 active sites.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
The authors would like to thank Yasunao Hattori, Kenichi Akaji, Naoya Ieda, Mitsuyasu Kawaguchi and Hidehiko Nakagawa for their technical support. This work was supported in part by the JST CREST program (T. S.; JPMJCR14L2) and a JSPS fellowship for foreign researchers (P. M.).
Notes and references
- J. S. Smith, C. B. Brachmann, I. Celic, M. A. Kenna, S. Muhammad, V. J. Starai, J. L. Avalos, J. C. Escalante-Semerena, C. Grubmeyer, C. Wolberger and J. D. Boeke, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6658–6663 CrossRef CAS.
- J. L. Feldman, J. Baeza and J. M. Denu, J. Biol. Chem., 2013, 288, 31350–31356 CrossRef CAS PubMed.
- Y.-B. Teng, H. Jing, P. Aramsangtienchai, B. He, S. Khan, J. Hu, H. Lin and Q. Hao, Sci. Rep., 2015, 5, 8529 CrossRef CAS PubMed.
- J. Jin, B. He, X. Zhang, H. Lin and Y. Wang, J. Am. Chem. Soc., 2016, 138, 12304–12307 CrossRef CAS PubMed.
- Z. Tong, M. Wang, Y. Wang, D. D. Kim, J. K. Grenier, J. Cao, S. Sadhukhan, Q. Hao and H. Lin, ACS Chem. Biol., 2017, 12, 300–310 CrossRef CAS PubMed.
- J. Du, Y. Zhou, X. Su, J. J. Yu, S. Khan, H. Jiang, J. Kim, J. Woo, J. H. Kim, B. H. Choi, B. He, W. Chen, S. Zhang, R. A. Cerione, J. Auwerx, Q. Hao and H. Lin, Science, 2011, 3, 806–809 CrossRef PubMed.
- K. G. Hoff, J. L. Avalos, K. Sens and C. Wolberger, Structure, 2006, 14, 1231–1240 CrossRef CAS PubMed.
- E. Michishita, J. Y. Park, J. M. Burneskis, J. C. Barrett and I. Horikawa, Mol. Biol. Cell, 2005, 16, 4623–4635 CrossRef CAS PubMed.
- L. Guarente, Nature, 2006, 444, 868–874 CrossRef CAS PubMed.
- A. Chalkiadaki and L. Guarente, Nat. Rev. Cancer, 2015, 15, 608–624 CrossRef CAS PubMed.
- G. Donmez and T. F. Outeiro, EMBO Mol. Med., 2013, 5, 344–352 CrossRef CAS PubMed.
- J. Luo, A. Y. Nikolaev, S. Imai, D. Chen, F. Su, A. Shiloh, L. Guarente and W. Gu, Cell, 2001, 107, 137–148 CrossRef CAS PubMed.
- H. Vaziri, S. K. Dessain, E. Ng Eaton, S. I. Imai, R. A. Frye, T. K. Pandita, L. Guarente and R. A. Weinberg, Cell, 2001, 107, 149–159 CrossRef CAS PubMed.
- F. Wang, C.-H. Chan, K. Chen, X. Guan, H.-K. Lin and Q. Tong, Oncogene, 2012, 31, 1546–1557 CrossRef CAS PubMed.
- A. Laemmle, A. Lechleiter, V. Roh, C. Schwarz, S. Portmann, C. Furer, A. Keogh, M. P. Tschan, D. Candinas, S. A. Vorburger and D. Stroka, PLoS One, 2012, 7, e33433 CAS.
- H. Y. Cohen, C. Miller, K. J. Bitterman, N. R. Wall, B. Hekking, B. Kessler, K. T. Howitz, M. Gorospe, R. de Cabo and D. A. Sinclair, Science, 2004, 305, 390–392 CrossRef CAS PubMed.
- L. M. McGlynn, S. Zino, A. MacDonald, J. Curle, J. E. Reilly, Z. M. A. Mohammed, D. C. McMillan, E. Mallon, A. P. Payne, J. Edwards and P. G. Shiels, Eur. J. Cancer, 2014, 50, 290–301 CrossRef CAS PubMed.
- M. Hiratsuka, T. Inoue, T. Toda, N. Kimura, Y. Shirayoshi, H. Kamitani, T. Watanabe, E. Ohama, C. G. T. Tahimic, A. Kurimasa and M. Oshimura, Biochem. Biophys. Res. Commun., 2003, 309, 558–566 CrossRef CAS PubMed.
- H.-S. Kim, A. Vassilopoulos, R.-H. Wang, T. Lahusen, Z. Xiao, X. Xu, C. Li, T. D. Veenstra, B. Li, H. Yu, J. Ji, X. W. Wang, S.-H. Park, Y. I. Cha, D. Gius and C.-X. Deng, Cancer Cell, 2011, 20, 487–499 CrossRef CAS PubMed.
- C.-A. J. Ong, J. Shapiro, K. S. Nason, J. M. Davison, X. Liu, C. Ross-Innes, M. O'Donovan, W. N. M. Dinjens, K. Biermann, N. Shannon, S. Worster, L. K. E. Schulz, J. D. Luketich, B. P. L. Wijnhoven, R. H. Hardwick and R. C. Fitzgerald, J. Clin. Oncol., 2013, 31, 1576–1582 CrossRef PubMed.
- P. Y. Liu, N. Xu, A. Malyukova, C. J. Scarlett, Y. T. Sun, X. D. Zhang, D. Ling, S.-P. Su, C. Nelson, D. K. Chang, J. Koach, A. E. Tee, M. Haber, M. D. Norris, C. Toon, I. Rooman, C. Xue, B. B. Cheung, S. Kumar, G. M. Marshall, A. V. Biankin and T. Liu, Cell Death Differ., 2013, 20, 503–514 CrossRef CAS PubMed.
- A. Deng, Q. Ning, L. Zhou and Y. Liang, Sci. Rep., 2016, 6, 27694 CrossRef CAS PubMed.
- H. Hou, W. Chen, L. Zhao, Q. Zuo, G. Zhang, X. Zhang, H. Wang, H. Gong, X. Li, M. Wang, Y. Wang and X. Li, J. Clin. Pathol., 2012, 65, 1088–1096 CrossRef PubMed.
- R. Liu, W. Dang, Y. Du, Q. Zhou, K. Jiao and Z. Liu, Sci. Rep., 2015, 5, 8415 CrossRef CAS PubMed.
- M. Erburu, I. Muñoz-Cobo, J. Domínguez-Andrés, E. Beltran, T. Suzuki, A. Mai, S. Valente, E. Puerta and R. M. Tordera, Eur. Neuropsychopharmacol., 2015, 25, 2036–2048 CrossRef CAS PubMed.
- M. Erburu, I. Muñoz-Cobo, T. Díaz-Perdigon, P. Mellini, T. Suzuki, E. Puerta and R. M. Tordera, Neuropharmacology, 2017, 117, 195–208 CrossRef CAS PubMed.
- D. Rotili, D. Tarantino, A. Nebbioso, C. Paolini, C. Huidobro, E. Lara, P. Mellini, A. Lenoci, R. Pezzi, G. Botta, M. Lahtela-Kakkonen, A. Poso, C. Steinkühler, P. Gallinari, R. De Maria, M. Fraga, M. Esteller, L. Altucci and A. Mai, J. Med. Chem., 2012, 55, 10937–10947 CrossRef CAS PubMed.
- Y. Zhang, Q. Au, M. Zhang, J. R. Barber, S. C. Ng and B. Zhang, Biochem. Biophys. Res. Commun., 2009, 386, 729–733 CrossRef CAS PubMed.
- A. R. McCarthy, L. Pirrie, J. J. Hollick, S. Ronseaux, J. Campbell, M. Higgins, O. D. Staples, F. Tran, A. M. Z. Slawin, S. Lain and N. J. Westwood, Bioorg. Med. Chem., 2012, 20, 1779–1793 CrossRef CAS PubMed.
- L. Li, L. Wang, L. Li, Z. Wang, Y. Ho, T. McDonald, T. L. Holyoake, W. Chen and R. Bhatia, Cancer Cell, 2012, 21, 266–281 CrossRef CAS PubMed.
- H. Jing, J. Hu, B. He, Y. L. Negrón Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y.-L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, Cancer Cell, 2016, 29, 297–310 CrossRef CAS PubMed.
- T. Rumpf, M. Schiedel, B. Karaman, C. Roessler, B. J. North, A. Lehotzky, J. Oláh, K. I. Ladwein, K. Schmidtkunz, M. Gajer, M. Pannek, C. Steegborn, D. A. Sinclair, S. Gerhardt, J. Ovádi, M. Schutkowski, W. Sippl, O. Einsle and M. Jung, Nat. Commun., 2015, 6, 6263 CrossRef CAS PubMed.
- M. Schiedel, T. Rumpf, B. Karaman, A. Lehotzky, S. Gerhardt, J. Ovádi, W. Sippl, O. Einsle and M. Jung, Angew. Chem., Int. Ed., 2016, 55, 2252–2256 CrossRef CAS PubMed.
- T. Suzuki, M. N. A. Khan, H. Sawada, E. Imai, Y. Itoh, K. Yamatsuta, N. Tokuda, J. Takeuchi, T. Seko, H. Nakagawa and N. Miyata, J. Med. Chem., 2012, 55, 5760–5773 CrossRef CAS PubMed.
- T. F. Outeiro, E. Kontopoulos, S. M. Altmann, I. Kufareva, K. E. Strathearn, A. M. Amore, C. B. Volk, M. M. Maxwell, J. C. Rochet, P. J. McLean, A. B. Young, R. Abagyan, M. B. Feany, B. T. Hyman and A. G. Kazantsev, Science, 2007, 317, 516–519 CrossRef CAS PubMed.
- M. G. Cheon, W. Kim, M. Choi and J. E. Kim, Cancer Lett., 2015, 356, 637–645 CrossRef CAS PubMed.
- A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J. F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg, E. Hamelin, R. J. Thomas, M. Kates, S. Jones, M. A. Navia, J. O. Saunders, P. S. DiStefano and R. Curtis, J. Med. Chem., 2005, 48, 8045–8054 CrossRef CAS PubMed.
- J. L. Feldman, K. E. Dittenhafer-Reed, N. Kudo, J. N. Thelen, A. Ito, M. Yoshida and J. M. Denu, Biochemistry, 2015, 54, 3037–3050 CrossRef CAS PubMed.
- Y. Wang, Y. M. Fung, W. Zhang, B. He, M. W. Chung, J. Jin, J. Hu, H. Lin and Q. Hao, Cell Chem. Biol., 2017, 24, 339–345 CrossRef CAS PubMed.
- J. L. Avalos, I. Celic, S. Muhammad, M. S. Cosgrove, J. D. Boeke and C. Wolberger, Mol. Cell, 2002, 10, 523–535 CrossRef CAS PubMed.
- T. Suzuki, K. Imai, H. Nakagawa and N. Miyata, ChemMedChem, 2006, 1, 1059–1062 CrossRef CAS PubMed.
- P. Mellini, T. Kokkola, T. Suuronen, H. S. Salo, L. Tolvanen, A. Mai, M. Lahtela-Kakkonen and E. M. Jarho, J. Med. Chem., 2013, 56, 6681–6695 CrossRef CAS PubMed.
- B. C. Smith and J. M. Denu, Biochemistry, 2007, 46, 14478–14486 CrossRef CAS PubMed.
- T. Suzuki, T. Asaba, E. Imai, H. Tsumoto, H. Nakagawa and N. Miyata, Bioorg. Med. Chem. Lett., 2009, 19, 5670–5672 CrossRef CAS PubMed.
- Y. Itoh, T. Suzuki, A. Kouketsu, N. Suzuki, S. Maeda, M. Yoshida, H. Nakagawa and N. Miyata, J. Med. Chem., 2007, 50, 5425–5438 CrossRef CAS PubMed.
- Y. Itoh, M. Suzuki, T. Matsui, Y. Ota, Z. Hui, K. Tsubaki and T. Suzuki, Chem. Pharm. Bull., 2016, 64, 1124–1128 CrossRef CAS PubMed.
- B. J. North, B. L. Marshall, M. T. Borra, J. M. Denu and E. Verdin, Mol. Cell, 2003, 11, 437–444 CrossRef CAS PubMed.
- A. M. Kalle, A. Mallika, J. Badiger, Alinakhi, P. Talukdar and Sachchidanand, Biochem. Biophys. Res. Commun., 2010, 401, 13–19 CrossRef CAS PubMed.
- A. Vaquero, M. Scher, D. Lee, H. Erdjument-Bromage, P. Tempst and D. Reinberg, Mol. Cell, 2004, 16, 93–105 CrossRef CAS PubMed.
- Y. Feng, T. Liu, S.-Y. Dong, Y.-J. Guo, J. Jankovic, H. Xu and Y.-C. Wu, J. Neurochem., 2015, 134, 668–676 CrossRef CAS PubMed.
- L. Quinti, M. Casale, S. Moniot, T. F. Pais, M. J. Van Kanegan, L. S. Kaltenbach, J. Pallos, R. G. Lim, S. D. Naidu, H. Runne, L. Meisel, N. A. Rauf, D. Leyfer, M. M. Maxwell, E. Saiah, J. E. Landers, R. Luthi-Carter, R. Abagyan, A. T. Dinkova-Kostova, C. Steegborn, J. L. Marsh, D. C. Lo, L. M. Thompson and A. G. Kazantsev, Cell Chem. Biol., 2016, 23, 849–861 CrossRef CAS PubMed.
- R. Luthi-Carter, D. M. Taylor, J. Pallos, E. Lambert, A. Amore, A. Parker, H. Moffitt, D. L. Smith, H. Runne, O. Gokce, A. Kuhn, Z. Xiang, M. M. Maxwell, S. A. Reeves, G. P. Bates, C. Neri, L. M. Thompson, J. L. Marsh and A. G. Kazantsev, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7927–7932 CrossRef CAS PubMed.
- S.-Y. Tsai, T. Hayashi, B. K. Harvey, Y. Wang, W. W. Wu, R.-F. Shen, Y. Zhang, K. G. Becker, B. J. Hoffer and T.-P. Su, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22468–22473 CrossRef CAS PubMed.
- A. Ma'ayan, S. L. Jenkins, A. Barash and R. Iyengar, Sci. Signaling, 2009, 2, cm1 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details, crystallographic data collection and refinement, details of chemical synthesis, additional figures and tables. See DOI: 10.1039/c7sc02738a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yukihiro
Itoh
a,
Hiroki
Tsumoto
b,
Ying
Li
a,
Miki
Suzuki
a,
Natsuko
Tokuda
c,
Taeko
Kakizawa
d,
Yuri
Miura
b,
Jun
Takeuchi
c,
Maija
Lahtela-Kakkonen
e and
Takayoshi
Suzuki
a,
Yukihiro
Itoh
a,
Hiroki
Tsumoto
b,
Ying
Li
a,
Miki
Suzuki
a,
Natsuko
Tokuda
c,
Taeko
Kakizawa
d,
Yuri
Miura
b,
Jun
Takeuchi
c,
Maija
Lahtela-Kakkonen
e and
Takayoshi
Suzuki