
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trapping intermediate MLCT states in low-symmetry {Ru(bpy)} complexes†
Alejandro
Cadranel
 *a,
Paola S.
Oviedo
b,
German E.
Pieslinger
b,
Shiori
Yamazaki
c,
Valeria D.
Kleiman
c,
Luis M.
Baraldo
b and
Dirk M.
Guldi
*a
*a,
Paola S.
Oviedo
b,
German E.
Pieslinger
b,
Shiori
Yamazaki
c,
Valeria D.
Kleiman
c,
Luis M.
Baraldo
b and
Dirk M.
Guldi
*a
aDepartment of Chemistry and Pharmacy, Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany. E-mail: ale.cadranel@fau.de; dirk.guldi@fau.de
bDepartamento de Química Analítica, Inorgánica y Química Física, INQUIMAE, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2, C1428EHA, Buenos Aires, Argentina
cDepartment of Chemistry, University of Florida, PO BOX 117200, Gainesville, FL 32611-7200, USA
First published on 29th August 2017
Abstract
The picosecond excited state dynamics of [Ru(tpm)(bpy)(NCS)]+ (RubNCS+) and [Ru(tpm)(bpy)(CN)]+ (RubCN+) (tpm = tris(1-pyrazolyl)methane, bpy = 2,2′-bipyridine) have been analyzed by means of transient absorption measurements and spectroelectrochemistry. Emissive 3MLCTs with (GS)HOMO(h+)–(GS)LUMO(e−) configurations are the lowest triplet excited states regardless of whether 387 or 505 nm photoexcitation is used. 387 nm photoexcitation yields, after a few picoseconds, the emissive 3MLCTs. In contrast, 505 nm photoexcitation populates an intermediate excited state that we assign as a 3MLCT state, in which the hole sits in a metal-centered orbital of different symmetry, prior to its conversion to the emissive 3MLCTs. The disparities in terms of electronic configuration between the intermediate and the emissive 3MLCTs have two important consequences. On one hand, both states feature very different fingerprint absorptions in transient absorption measurements. On the other hand, the reconfiguration is impeded by a kinetic barrier. As such, the conversion is followed spectroscopically and kinetically on the 300 ps timescale.
Introduction
The MLCT excited state manifold of ruthenium polypyridines constitutes a unique playground for extraordinary photophysics and photochemistry. Their high versatility in terms of both structural modifications and ligand substitutions allows for the fine tuning of their excited state properties.1–4 Many attempts have been made to exploit them as integrative components of supramolecular architectures for catalysis and energy conversion.5–13 MLCT manifolds are typically populated upon visible light absorption. Such MLCT excited states involve a hole in the parent octahedral t2g ruthenium orbitals and an excited electron in a π* orbital of the polypyridinic ligand. One of the greatest challenges in the field is to gain full control over these excited states. If successful, this would assist in promoting charge separation and ultimately using these redox equivalents to catalyze specific reactions or to collect them at electrodes.In a general scenario, the guiding of the excited state energy or charges requires unidirectional energy and/or electron transfer processes. Depending on the precise reaction mechanism, a myriad of energy or symmetry requirements will need optimization. One of the more conventional approaches relies on tuning the energetics of heteroleptic or mixed-ligand complexes. Upon populating the Franck–Condon excited states, differences in the reduction potentials between the non-equivalent ligands promote efficient inter ligand charge transfer (ILCT) events on multiple timescales.14 It is implicit that an electron potentially hops from one ligand to another and, in turn, is directed to the energetically most favorable orbital.15–19 Once the electron reaches the energetically lowest orbital it becomes accessible as a reductive equivalent for catalysis or for injection into electrodes.20
We envision an alternative strategy based on the symmetry of MLCT excited states, rather than on their energetics, and a judicious choice of ancillary ligands. The excited metal ion features three t2g orbitals on which the hole usually sits. An unsymmetrical coordination splits these orbitals in energy affording energy or electron donors of vastly different symmetry. To the best of our knowledge, the only evidence for MLCT states of different symmetry has been documented for [Os(phen)3]2+.21 In this complex, the lowest MLCT state presents a transient absorption band in the near-infrared region which was ascribed to an interconfigurational dπ → dπ transition. This photoinduced absorption transition results in a MLCT state of different symmetry than the lowest one. However, it has never been directly observed. The lack of clear-cut cases demonstrates the need for a better understanding of the factors that determine the population of MLCT states of different symmetry.
In this study, we establish the means by which light absorption in a particular region of the visible spectrum results in the population of an intermediate excited state, that we assign as a high energy 3MLCT state. There, the hole is likely to sit in a metal-based orbital of different symmetry than those in the four lowest-lying 3MLCT states described by Kober and Meyer.22 We show for the first time that high-energy {Ru(bpy)} MLCT excited states are trappable, potentially allowing for the utilization of their energy before dissipation. To shed light on this phenomenon we take advantage of the synergistic combination of transient absorption and spectroelectrochemistry,23,24 investigating the ultrafast dynamics of mixed-ligand ruthenium polypyridines. [Ru(tpm)(bpy)(NCS)]+ (RubNCS+) and [Ru(tpm)(bpy)(CN)]+ (RubCN+) were selected as models because of their overall low symmetry (Cs point group), and because they contain ligands with very different donor properties, which should result in a split of the energy of the dπ orbitals of the ruthenium ions (Fig. 1). The selected ligands are prone to participate in LMCT transitions and hence could provide information on the hole dynamics in these compounds.
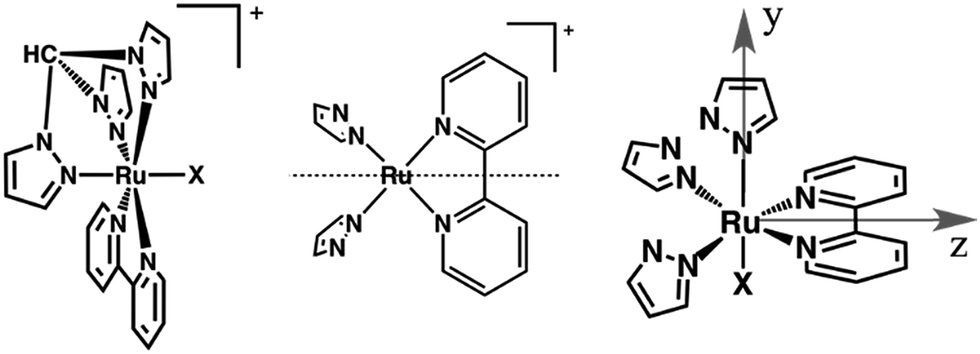 | ||
| Fig. 1 Left: Sketches of the [Ru(tpm)(bpy)X]+ complexes studied in this work, RubNCS+ (X = NCS−) and RubCN+ (X = CN−). Center: The dotted line indicates the reflection plane that bisects the bpy ligand, determining the Cs symmetry. Right: Axis denomination for RubNCS+ and RubCN+. The x axis is normal to the page. | ||
Results
Spectroelectrochemistry
The UV-vis absorption spectra of RubNCS+ and RubCN+ were previously reported.25Fig. 2 shows the spectroelectrochemical evolution throughout the visible region upon one electron oxidation and one electron reduction. As the oxidations are metal-centered,25 the electrolysis of RubNCS+ and RubCN+ at anodic potentials produces a decrease in the MLCT absorptions due to Ru(II) depletion. In the oxidized forms, the absorption features peaks at 391 nm (RubNCS2+) and 425 nm (RubCN2+), which are assigned to LMCT dπ(Ru) ← π(heterocycle) transitions.26–29 A strong LMCT transition is observed at 730 nm for RubNCS2+. Similar fingerprints are observed for related complexes23,30–32 and, in turn, we ascribe them to dπ(Ru) ← π(NCS) charge transfer transitions. RubCN2+ shows weaker transitions at 590 nm, which are also assigned to LMCT transitions. These are likely dπ(Ru) ← π(tpm), since tpm is a stronger electron donor than bpy and CN−. | ||
| Fig. 2 Upper panel: Spectral evolution during the Ru(II) → Ru(III) oxidation of RubNCS+ and RubCN+ in acetonitrile (0.1 M [TBA]PF6); initial: solid black line, final: dashed red line, intermediate: grey lines. Middle panel: Spectral evolution during the one-electron reduction of RubNCS+ and RubCN+ in acetonitrile (0.1 M [TBA]PF6); initial: solid black line, final: dashed blue line, intermediate: grey lines. Bottom panel: Sum of the oxidative and reductive differential spectra of RubNCS+ and RubCN+ in acetonitrile. | ||
As previously reported,25 the first reduction of RubNCS+ and RubCN+ is bpy-centered. This is simply understood in terms of the more extended conjugation found in bpy relative to tpm. Support for this assumption is based on the spectral changes for RubNCS and RubCN, which are in sound agreement with those seen for the reduction of [Ru(bpy)3]2+.33 In the reduced forms RubNCS and RubCN, the absorptions in red are ascribed to π*(heterocycle) ← π*(radical anion) transitions.‡
Spectroelectrochemistry has been successfully used to assign the patterns observed in the differential spectra of the MLCT excited states of ruthenium polypyridines.24,34–40Fig. 2 shows the differential changes for metal oxidation (upper panel) and ligand reduction (middle panel) and both contributions summed up (bottom panel) for RubNCS+ and RubCN+. The weak intensity of the negative bands, in the regions where the MLCT bands are located, should be noted. This is due to a LMCT feature in the oxidized form, which compensates for the MLCT bleaching.
Transient absorption experiments
Femtosecond transient absorption measurements were performed for RubNCS+ and RubCN+ in argon-deoxygenated DMSO solutions at room temperature. The upper panels of Fig. 3 and S2† show the broadband differential absorption spectral maps and kinetic traces at selected wavelengths upon 505 nm (2.46 eV) illumination. Under these conditions, both complexes exhibit bleaching in the 450–500 nm range at short time delays. Remarkably, within hundreds of ps they transform into photoinduced absorptions (PIAs) with maxima at 475 and ≈508 nm (RubNCS+)§ and 475 nm (RubCN+).Notably, the presence of a NCS− ligand induces an additional LMCT transition in the differential absorption spectra of RubNCS+ with a maximum at 600–610 nm. Its temporal evolution is in the time range of hundreds of ps. In comparison to the spectroelectrochemical features (Fig. 2), the observed LMCT transitions are blue-shifted due to metal orbital destabilization by the imine radical anion of the MLCT excited state. The cyanide complex gives rise to weaker photoinduced absorptions owing to the fact that in this case, only the dπ(Ru) ← π(heterocycle) LMCT and π*(heterocycle) ← π*(radical anion) transitions contribute in this spectral region.
Interestingly, the differential transient absorption signals observed upon 387 nm (3.20 eV) irradiation are strikingly different (Fig. 4 and S3†). RubNCS+ and RubCN+ lack any bleaching. For RubCN+, the only signal below 550 nm arises from PIA at 475 nm, corresponding to the LMCT transitions. RubNCS+ shows similar transitions at 473 and 506 nm, together with a LMCT PIA at 600–610 nm. These bands are remarkably similar to those observed upon irradiation at 505 nm after 1 ns (Fig. S6 and S8†).
Complementary nanosecond measurements with 505 nm (Fig. S6 and S8†) or 387 nm (Fig. S7 and S9†) pumps point to the presence of a single excited state in the long timescale. These nanosecond differential spectra and their corresponding lifetimes (Table S1†) fully match the spectra observed at the end of the picosecond experiments (Fig. S10†).
Kinetic analysis of the transient experiments and decay model
Global fitting of the data after 505 nm excitation led to three different transient species in each case (Fig S4†): a short-lived component with τ < 6 ps, an intermediate-lived component with τ on the order of hundreds of picoseconds, and a long-lived component that decays on the nanosecond timescale. With the aforementioned data in hand, we performed target analyses38,41 based on the model depicted in Fig. 3 (bottom right). The resulting species-associated differential spectra and time constants are presented in Fig. 3 (bottom left) and Table 1, respectively. The validity of the model is discussed below.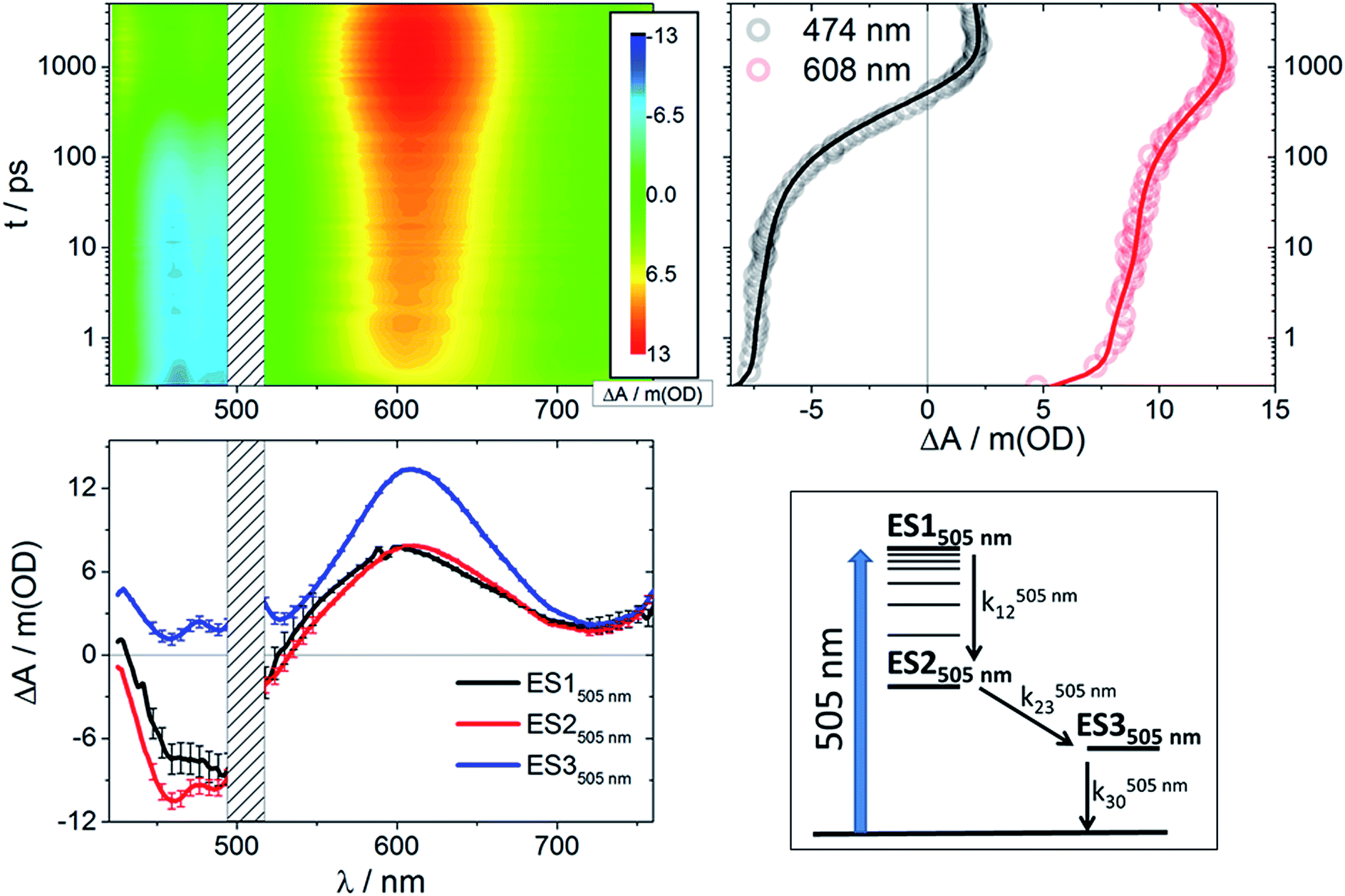 | ||
| Fig. 3 Upper left: Differential absorption 3D map obtained from femtosecond pump-probe experiments (λex = 505 nm) on RubNCS+ in DMSO at room temperature. Upper right: Time absorption profiles (open circles) and corresponding fittings from target analysis (solid lines) for RubNCS+ upon excitation at 505 nm using the model presented in the bottom right side of this figure. Bottom left: Species-associated differential spectra for RubNCS+ upon excitation at 505 nm; ES1505 nm: black, ES2505 nm: red, ES3505 nm: blue. Bottom right: Target model proposed to fit the data. | ||
| X | λ pump = 387 nm | λ pump = 505 nm | |||
|---|---|---|---|---|---|
| k 12/ps−1 (τ12/ps) | k 20/ns−1 (τ20/ns) | k 12/ps−1 (τ12/ps) | k 23 × 103/ps−1 (τ23/ps) | k 30/ns−1 (τ30/ns) | |
| a Values extracted from nanosecond TA experiments. nd: non determined in picosecond experiments. | |||||
| NCS− | 0.262 ± 0.002 (3.8) | 0.028 ± 0.001 (35.9) | 0.330 ± 0.006 (3.0) | 2.67 ± 0.01 (375) | 0.032 ± 0.001 (31.1) |
| CN− | 0.41 ± 0.01 (2.4) | nd (106)a | 0.54 ± 0.02 (1.9) | 3.34 ± 0.05 (300) | nd (112.5)a |
Following excitation at 505 nm, both compounds give rise to an initial excited state spectrum (ES1505 nm) typical of the 3MLCT states seen in ruthenium polypyridine complexes,24,37,40,42–45 with bleaching in the spectral area of the ground state absorption and a weaker positive transient in the spectral range of >550 nm (3D maps and ES1505 nm in Fig. 3 and S2†). For the thiocyanate complex an intense dπ(Ru) ← π(NCS) LMCT transition was found around 600–610 nm.23 The second species (ES2505 nm) has a very similar spectral pattern. Both complexes reveal a third component (ES3505 nm) with a distinct additional photoinduced absorption at <550 nm, but no bleaching was observed in this spectral region. Also, for the thiocyanate complex, an enhancement of the LMCT (600–610 nm) band relative to the short timescale species is clearly discernable.
Global analysis of the results following photoexcitation at 387 nm leads to only two different transient species (Fig. S5†): a short-lived component with τ < 6 ps and a long-lived component with τ on the nanosecond timescale. Target analyses employed with the two-excited-state model depicted in Fig. 4 (bottom right) result in the spectral data presented in Fig. 4 (bottom left) with the time constants listed in Table 1. Immediately upon 387 nm photoexcitation, RubNCS+ and RubCN+ show a lack of ground state bleaching (3D maps and ES1387 nm in Fig. 4 and S3†). The initial species (ES1387 nm) transforms into the final form (ES2387 nm) very rapidly and without significant modifications to the spectrum. RubNCS+ shows the presence of strong LMCT photoinduced absorptions at longer wavelengths. Its spectrum looks strikingly similar to that recorded at long time delays following 505 nm photoexcitation (ES3505 nm).
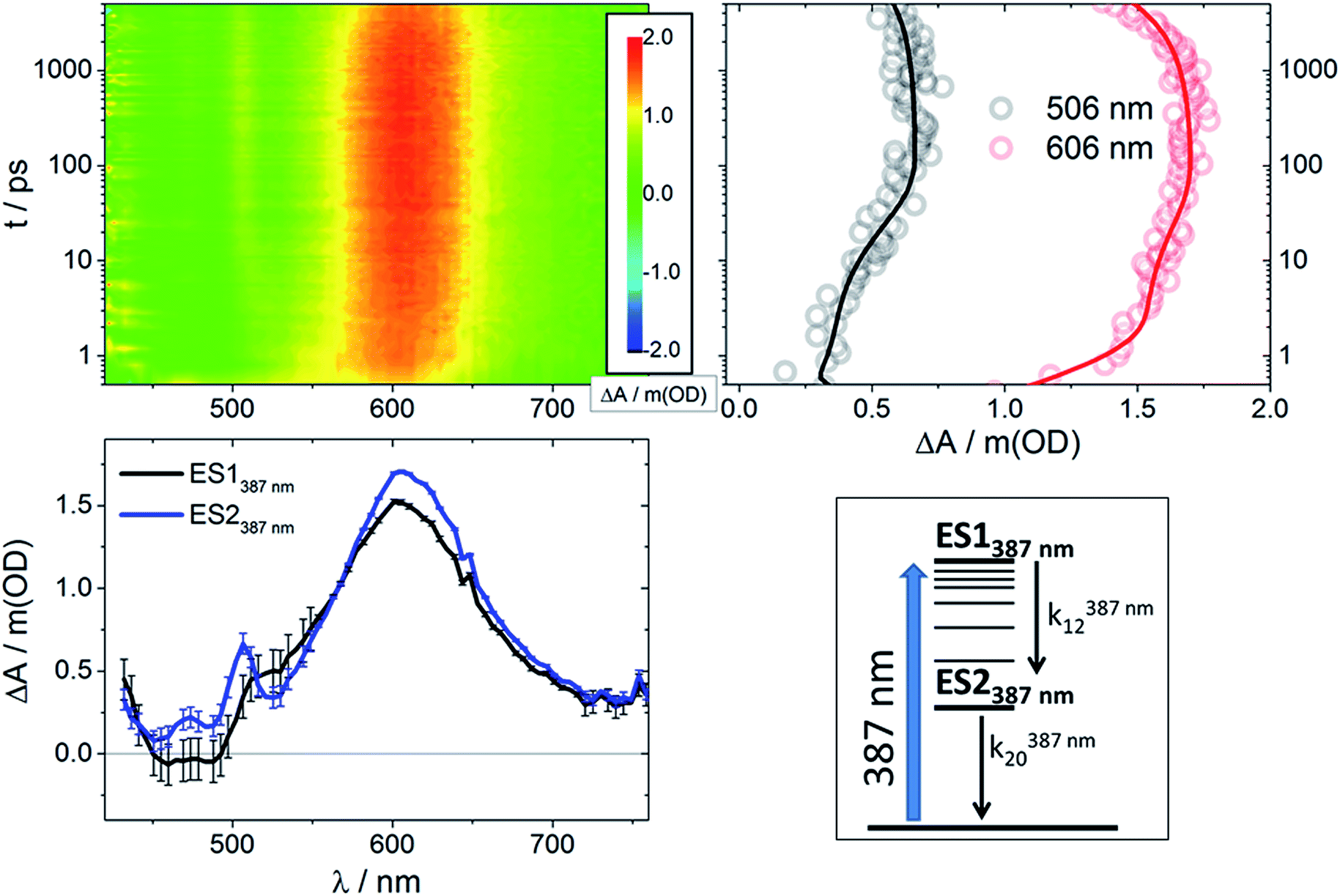 | ||
| Fig. 4 Upper left: Differential absorption 3D map obtained from femtosecond pump-probe experiments (λex = 387 nm) on RubNCS+ in DMSO at room temperature. Upper right: Time absorption profiles (open circles) and corresponding fittings from target analysis (solid lines) for RubNCS+ upon excitation at 387 nm using the model presented at the bottom right side of this figure. Bottom left: Species-associated differential spectra for RubNCS+ upon excitation at 387 nm; ES1387 nm: black, ES2387 nm: blue. Bottom right: Target model proposed to fit the data. | ||
Prior to addressing the nature of the different excited states, a few remarks on the target models are needed. Given the different lifetimes obtained for the two components found following UV (387 nm) photoexcitation, only a sequential model seems applicable (Fig. 5). Upon visible (505 nm) photoexcitation, three components were identified and two potential models were considered. A branched model, in which ES1505 nm feeds ES2505 nm and ES3505 nm, both of which depopulate independently via ground state (GS) recovery, does not give satisfactory results. It results in an ES3505 nm spectrum with very broad negative signals at wavelengths longer than 550 nm. In the experimental spectra neither stimulated emission nor GS absorption is noted at long wavelengths. Consequently, this branched model fails to reflect the fast dynamics accurately. A second model, with sequential steps, was applied, in which ES1505 nm feeds ES2505 nm and subsequently ES3505 nm. This sequential model yields three individual spectra that describe the experimental findings very well (Fig. 3).
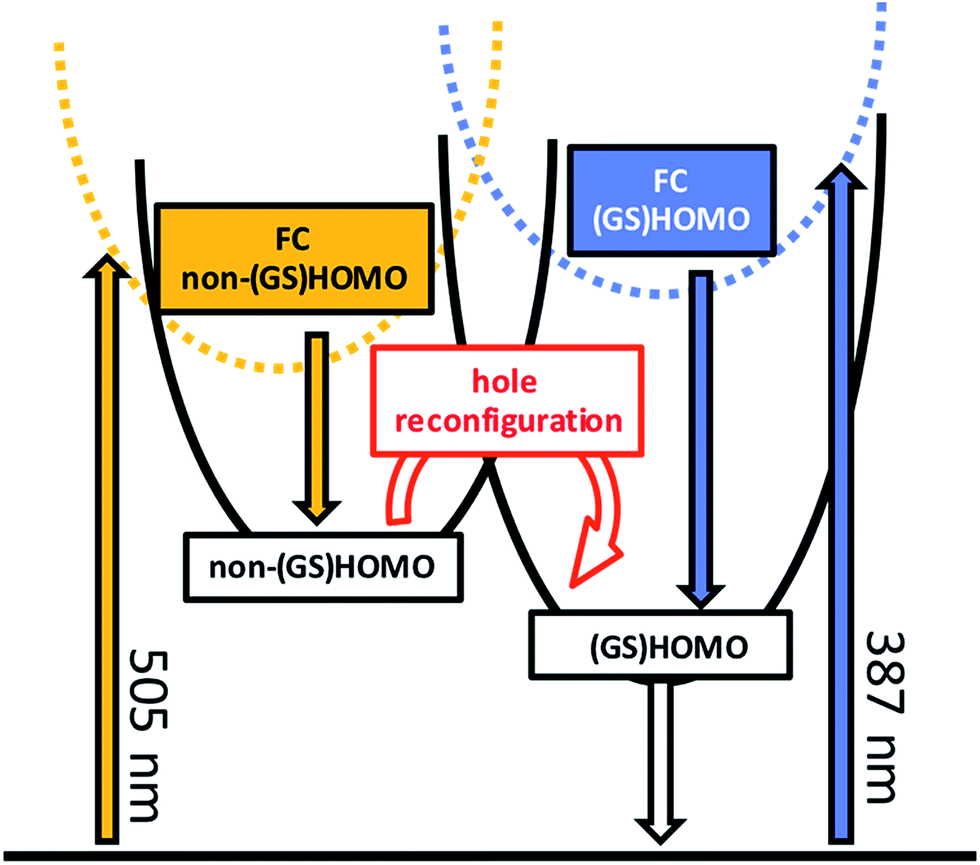 | ||
| Fig. 5 Proposed model describing the photophysical behavior observed for RubNCS+ and RubCN+ upon 505 nm (orange) or 387 nm (blue) illumination. Labels indicate the hole configuration. Highlighted is the proposed hole reconfiguration process characterized by k23. | ||
Regardless of the excitation wavelength, similar long-time dynamics were observed and confirmed by independent picosecond and nanosecond measurements, yielding identical spectra (Fig. S10†) and time constants (Tables 1 and S1†). In line with the previous reports25 the nanosecond measurements show quantitative GS recovery. Upon photoexcitation in different regions of the spectra, both deactivation cascades proceed through different mechanisms, leading to a common excited state with the corresponding lifetimes matching the emission lifetimes.25
Discussion
At the 387 nm and 505 nm excitation wavelengths, which relate to 3.20 eV/25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 cm−1 and 2.46 eV/19800 cm−1, respectively, the GS absorptions of RubNCS+ and RubCN+ are largely dominated by 1MLCT transitions.25 On the femtosecond timescale, intersystem crossing (ISC) between singlet and triplet manifolds leads to the population of the 3MLCT manifold, similar to the results found for other ruthenium polypyridine complexes.46 Following the initial ISC, ground-state absorption bleaching is observed in the differential absorption spectra only upon 505 nm photoexcitation. As such, we propose that different triplet excited states are populated on the early timescale when using different excitation wavelengths.
800 cm−1 and 2.46 eV/19800 cm−1, respectively, the GS absorptions of RubNCS+ and RubCN+ are largely dominated by 1MLCT transitions.25 On the femtosecond timescale, intersystem crossing (ISC) between singlet and triplet manifolds leads to the population of the 3MLCT manifold, similar to the results found for other ruthenium polypyridine complexes.46 Following the initial ISC, ground-state absorption bleaching is observed in the differential absorption spectra only upon 505 nm photoexcitation. As such, we propose that different triplet excited states are populated on the early timescale when using different excitation wavelengths.
From the good agreement between the transient absorption lifetimes and emission lifetimes on the nanosecond timescale, we conclude that ES2387 nm and ES3505 nm are the same states, namely the emissive 3MLCT excited state. The spectral profiles of the emissive states are in good agreement with the spectroelectrochemical assays. Our interpretation of the differential absorption spectra of the long-time emissive states based on the spectroelectrochemical results is therefore appropriate. Thus, we assign a (GS)HOMO(h+)–(GS)LUMO(e−) electronic configuration to these excited states. This is in line with the notion that these excited states are the lowest 3MLCTs.
Considering that the spectral features of ES1387 nm are not markedly different from those of the emissive 3MLCT, we ascribe them to a Franck–Condon state with an electronic configuration similar to that of the emissive states. k12387 nm is assigned to the development of the MLCT manifold47–51 and occurs within a few picoseconds.
When the excitation is shifted to lower energies, namely 505 nm, we observed a different behavior, since in both complexes the initial differential absorption spectra are dominated by a negative signal. This contrasts with the spectra observed upon photoexcitation at 387 nm, which fail to exhibit any negative absorptions. This disparate response suggests that the electronic configuration of ES1505 nm differs from that of ES1387 nm.
We considered different alternatives for the identity of ES1505 nm. In systems featuring π-extended ligands or donor/acceptor groups, 3IL (intra-ligand π–π*)52–57 or 3ILCT (inter-ligand charge transfer)54,58,59 states play the role of energy reservoirs. In our case of {(tpm)(bpy)}, it is unlikely to deal with states at energies low enough to equilibrate with or be thermally populated from 3MLCTs. Firstly, IL states are not observed in [Ru(bpy)3]2+.43,46,47,60 Secondly, tpm GS π–π* absorptions occur around 200 nm, an energy much higher than our excitation. Thirdly, if ES1505 nm was an ILCT state, a hole shift from the Ru(III) present in the initially populated MLCT to the tpm would be required. However, tpm and bpy are harder to oxidize than Ru(II) and, in turn, it is unlikely that holes move in the excited state to any of the iminic ligands, precluding the participation of any ILCT state.
Alternatively, 3MC states could account for ES1505 nm. 3MC states are, however, not observed upon 387 nm photoexcitation, despite the 0.74 eV or 6000 cm−1 of excess electronic and vibrational energy in comparison with the 505 nm excitation. This is inconsistent with a thermally activated process as seen, for example, in the 3MLCT to 3MC state transformation.61,62 Furthermore, in the 3MC states, Ru(II) features unpaired electrons in both parent t2g and eg orbitals, enabling dπ(Ru) ← π(NCS) LMCT absorptions. Importantly, such LMCT absorptions, which originate from the 3MC states, are blue-shifted with respect to those originating from the 3MLCT states, as observed in similar systems.23 For ES1505 nm, a PIA at 600–610 nm is observed at the same energy and with the same shape as that assigned to a LMCT transition in ES1387 nm. Both of these observations point to the MLCT nature of ES1505 nm, and, thus, we assign it as a high energy 3MLCT.¶
As ES1505 nm and the emissive ES3505 nm states are both best described as 3MLCTs with a bpy-localized orbital occupied by an excited electron and a metal-centered hole, they should bear the excited hole or the excited electron in different orbitals. If the difference between these states is based on the location of the excited electron, the prototypical [Ru(bpy)3]2+ should behave in the same way as RubNCS+ and RubCN+, as the bpy-centered orbitals that feature the excited electron are rather independent of the ancillary ligands and the symmetry around the metal center. However, none of the transient absorption experiments performed by different groups in the references gave rise to the dynamics presented in this contribution. Particularly, Hauser and co-workers explored the excited state dynamics of [Ru(bpy)3]2+ with femtosecond transient absorption spectroscopy in the visible63 and near-infrared64 regions upon photoexcitation at 400 nm (3.1 eV). No dynamics other than the GS recovery in the hundreds of nanoseconds timescale are, however, noted after 20 ps. Additionally, Shaw and Papanikolas reported the monoexponential behavior of [Ru(bpy)3]2+ with a 10–12 ps lifetime upon 475 nm (2.6 eV) photoexcitation,65 consistent with the observations by Hammarström and co-workers upon 480 nm excitation47 on the sub-nanosecond timescale.
We strongly believe that the difference in configuration arises from the hole occupation, which could involve one of the several accessible Ru-centered orbitals of similar energy.66 In the following section, we use the electronic structural model for the emitting localized MLCT excited states of ruthenium and osmium polypyridines developed by Kober and Meyer.22 This model is based upon the results of, for example, Crosby et al.67,68 In the tris-, bis- and mono-bpy complexes of Ru(II), four closely-spaced MLCT states exist at very low energies. Three of them are positioned within 200 cm−1 of each other, while the fourth one is at least 800 cm−1 higher in energy. A pseudo C2v symmetry is considered, in which the z axis is the C2 axis of the bpy radical anion in the excited state (Fig. 1). For the tris-, bis- and mono-bpy complexes, in each case the four MLCT states originate from the same spatial electronic configuration. This configuration evolves, by means of spin symmetry, in four states that transform into four different symmetries. The highest lying state, namely “the fourth MLCT”, features more singlet character than any of the other states. Importantly, several temperature-dependent experiments support this notion and show that the fourth MLCT is thermally populated from the equilibrated MLCT.69,70 For example, its enhanced singlet spin character accelerates its decay to the ground state and, in turn, depopulates the MLCT manifold.71,72 We believe that the aforementioned behaviour is not responsible for our observation, as the emitting MLCT lifetimes lack any appreciable changes when changing the excitation energy. Additionally, common to all four states is their similar configuration. As such, it would be difficult to explain the very different spectroscopy. The exact nature of the high-energy MLCT state is, nevertheless, unknown to us. In fact, as this state has a strongly mixed spin and orbital character, we cannot rule out that our high energy state is the fourth MLCT.
We hypothesize that the discrepancies between the emissive states and ES1505 nm might be due to the different spatial orientation of the orbital containing the hole. For example, a consequence of a different hole configuration in the intermediate and in the emissive MLCT might be related to the interactions with the X ligand. In one of these MLCT states, the hole might sit in a d orbital with the symmetry required to interact with the X ligand. As a result, in that state the hole might be extended over the {Ru–X} moiety. This could give rise to different spectroscopy for this state, in comparison with a conventional MLCT state, where the hole sits in an orbital of a different symmetry and is therefore closer to a pure metal-based description. This would allow for photoinduced absorptions to mask the bleaching in the case of the emissive states, but not in the case of ES1505 nm, and would also account for the observed enhancement of the PIA for RubNCS+ at 600–610 nm. The same argument would also explain the difference between the electrochemical (GS)HOMO and the spectroscopic non-(GS)HOMO orbitals. We postulate that ES1505 nm is a Franck–Condon 3MLCT excited state with the hole sitting in a metal-centered orbital different to the (GS)HOMO. ES1505 nm features a sub-10 ps lifetime and a spectral profile that is not markedly different from that of ES2505 nm. The interconversion between ES1505 nm and ES2505 nm is associated with the development of the MLCT manifold on a timescale of a few picoseconds as observed in related molecules.47–51
In our interpretation, k23505 nm (Fig. 5) relates to an internal conversion between two 3MLCT excited states. It can also be described as a hole reconfiguration. In RubNCS+, the remarkable enhancement of the dπ(Ru) ← π(NCS) LMCT transition (Fig. 3) would be the direct consequence of the hole moving to the (GS)HOMO. In short, better orbital overlap and an intensified dπ(Ru) ← π(NCS) LMCT transition in the emissive state would evolve. In contrast, the symmetry of the non-(GS)HOMO metal-centered orbital would provide poorer overlap with NCS− in ES2505 nm, which renders the corresponding LMCT transition less intense.
k
23
505 nm is associated with an activation barrier, whose origin is intriguing. We consider two alternative explanations, which are both based on our interpretation that the internal conversion is a non-(GS)HOMO → (GS)HOMO hole reconfiguration. On one hand, it is well-known that the one-electron oxidation of ruthenium cyanides and thiocyanates has a strong impact on metal–ligand bond distances as well as intra-ligand bond distances. For example, the average Ru–C distance in K4[Ru(CN)6] increases from 1.912 Å upon oxidation to 2.050 Å in K3[Ru(CN)6] due to reduced backbonding effects.73 Additionally, it has been shown that the N-bound thiocyanate is best described as {N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–S} for Ru(II), but {N
C–S} for Ru(II), but {N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS} for Ru(III).30 It is known from spectroelectrochemical near-infrared measurements and theoretical calculations23,30 that in ruthenium thiocyanate and ruthenium cyanide complexes the (GS)HOMOs are spread well over such ligand-centered orbitals. Thus, the (GS)HOMO(h+)–(GS)LUMO(e−) MLCT electronic configurations for RubNCS+ and RubCN+ mimic those of the Ru(III) species, as shown by spectroelectrochemistry. We hypothesize that, given the different overlap with the X ligand, a non-(GS)HOMO → (GS)HOMO hole reconfiguration is likely to affect the metal-X ligand and intra-X ligand bond distances. An immediate consequence would be the considerable internal reorganization energies and activation barriers. Likewise, Ru–N(imine) distances could also be subject to reorganization, from which activation barriers would evolve. We find it difficult, however, to believe that such a barrier originates from a transition between states of the same spatial electronic symmetry, regardless of their total symmetry. In any case, femtosecond mid-IR experiments would be very valuable in determining the origin of this barrier. Of great importance would be the C–N stretchings of CN− and NCS−, as well as the C–C vibrations of the bpy ligand.74,75
CS} for Ru(III).30 It is known from spectroelectrochemical near-infrared measurements and theoretical calculations23,30 that in ruthenium thiocyanate and ruthenium cyanide complexes the (GS)HOMOs are spread well over such ligand-centered orbitals. Thus, the (GS)HOMO(h+)–(GS)LUMO(e−) MLCT electronic configurations for RubNCS+ and RubCN+ mimic those of the Ru(III) species, as shown by spectroelectrochemistry. We hypothesize that, given the different overlap with the X ligand, a non-(GS)HOMO → (GS)HOMO hole reconfiguration is likely to affect the metal-X ligand and intra-X ligand bond distances. An immediate consequence would be the considerable internal reorganization energies and activation barriers. Likewise, Ru–N(imine) distances could also be subject to reorganization, from which activation barriers would evolve. We find it difficult, however, to believe that such a barrier originates from a transition between states of the same spatial electronic symmetry, regardless of their total symmetry. In any case, femtosecond mid-IR experiments would be very valuable in determining the origin of this barrier. Of great importance would be the C–N stretchings of CN− and NCS−, as well as the C–C vibrations of the bpy ligand.74,75
Our rationale also explains why the hole reconfiguration phenomenon within MLCT manifolds of ruthenium polypyridines has not yet been reported. It is primarily a consequence of the very low symmetry of {Ru(tpm)(bpy)} complexes, which results in a splitting of the dπ orbitals and in 3MLCTs with spectral features, depending on the particular hole configuration. The minor density of MLCT states, stemming from the presence of only one bpy in our complexes, enables the rather unusual phenomena. We believe that a similar behavior could be observed in different low-symmetry polypyridinic Ru complexes, such as {Ru(tpy)(bpy)}, etc. Notably, femtosecond transient absorption measurements have already been carried out on some of them,40 but their lifetimes are usually compromised by MC states precluding observations analogous to those presented here.
Experimental
(RubNCS)PF6 and (RubCN)PF6 were synthesized as previously reported.25 Acetonitrile for spectroelectrochemical measurements was distilled and dried over CaH2. UV-visible spectra were recorded with a Hewlett-Packard 8453 diode array spectrometer (range 190–1100 nm). All the spectroelectrochemical (SEC) experiments were performed using a three-electrode OTTLE cell,76 with millimolar solutions of the samples using [TBA]PF6 0.1 M as the supporting electrolyte. Ultrafast transient absorption (TA) experiments were conducted using an amplified Ti/sapphire laser system (Clark MXR CPA2101, FWHM = 150 fs, λexc = 387 nm and 505 nm, 200 nJ per pulse) with TA pump/probe EOS and Helios detection systems from Ultrafast Systems. White light was generated using a sapphire crystal. The optical densities (ODs) of the samples were around 0.5 at the excitation wavelengths. Argon-degassed anhydrous DMSO (99.9% from Aldrich) was employed to eliminate oxygen. A magic angle configuration was employed to avoid rotational dynamics and the chirp generated in the broadband probe was corrected with a polynomial fit before data analysis. Global and target analyses were performed using the GloTarAn software and the R package TIMP.77 Further details are given in the ESI.†Conclusions
The picosecond excited state dynamics of RubNCS+ and RubCN+ have been characterized through the powerful combination of transient absorption measurements and spectroelectrochemistry. Their lowest triplet excited state is the emissive 3MLCT excited state, which is reached as a final reservoir following a cascade of excited state deactivations that start either upon 387 nm or 505 nm illumination. The differential absorption spectroscopy of these states can be accurately reproduced by superimposing those of the one-electron oxidized and one-electron reduced forms. As such, their electronic configuration corresponds to a (GS)HOMO(h+)–LUMO(e−) charge transfer state.While photoexcitation at 387 nm results in, after a few picoseconds, an excited state with a (GS)HOMO(h+)–(GS)LUMO(e−) configuration, 505 nm photoexcitation allows for the observation of an intermediate 3MLCT state, in which the hole sits in a metal-centered orbital of different symmetry. The disparity between the electronic configuration of the hole in the intermediate and the emissive 3MLCTs has two important consequences. On one hand, both states feature very different fingerprint absorptions in transient absorption measurements. On the other hand, the reconfiguration is impeded by a kinetic barrier. As such, the conversion is followed spectroscopically and kinetically on the 300 ps timescale.
Our rare findings suggest that it is possible to take advantage of higher energy 3MLCT states prior to their conversion to low-energy triplets. The corresponding intermediate is a stronger oxidant than the emissive 3MLCT and thus is promising for oxidative catalysis. As such, it is intriguing to explore systems with similar low symmetry to determine the exact parameters that allow the trapping of the higher energy 3MLCT states. In this direction, we are currently exploring the design of coordination compounds, which would enable the utilization of the energy of the intermediate 3MLCT states prior to their dissipation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Universidad de Buenos Aires (UBACyT q534), CONICET (PIP 0659) and ANPCyT (PICT 2013 0069), the National Science Foundation Grant (CHE-1058638), the DFG grant (GU517/23-1) and the “Solar Technologies Go Hybrid” (SolTech) initiative of the State of Bavaria. L. M. B. is a member of the scientific staff of CONICET. A. C. gratefully acknowledges a postdoctoral fellowship from DAAD and Ministerio de Educación, Ciencia y Tecnología (Argentina), and P. S. O. a doctoral fellowship from CONICET. G. E. P. thanks Prof. Adrián Roitberg for selflessly sharing his knowledge and DAAD for personal funding. A. C. thanks ALN for a fruitful collaborative environment.Notes and references
- A. Breivogel, M. Meister, C. Förster, F. Laquai and K. Heinze, Chemistry, 2013, 19(41), 13745–13760 CrossRef CAS PubMed.
- D. L. Ashford, C. R. K. Glasson, M. R. Norris, J. J. Concepcion, S. Keinan, M. K. Brennaman, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2014, 53(11), 5637–5646 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigeletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- J. Romanova, Y. Sadik, M. R. Ranga Prabhath, J. D. Carey and P. D. Jarowski, J. Phys. Chem. C, 2017, 121(4), 2333–2343 CAS.
- Q. Zeng, F. W. Lewis, L. M. Harwood and F. Hartl, Coord. Chem. Rev., 2015, 304–305, 88–101 CrossRef CAS.
- M. Yamamoto and K. Tanaka, Chempluschem, 2016, 81(10), 1028–1044 CrossRef CAS.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138(42), 14152–14158 CrossRef CAS PubMed.
- Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6(5), 3063–3074 RSC.
- A. Nakada, T. Nakashima, K. Sekizawa, K. Maeda and O. Ishitani, Chem. Sci., 2016, 7, 4634–4731 RSC.
- M. Falkenström, O. Johansson and L. Hammarström, Inorg. Chim. Acta, 2007, 360, 741–750 CrossRef.
- D. F. Zigler, Z. A. Morseth, L. Wang, D. L. Ashford, M. K. Brennaman, E. M. Grumstrup, E. C. Brigham, M. K. Gish, R. J. Dillon, L. Alibabaei, G. J. Meyer, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2016, 138(13), 4426–4438 CrossRef CAS PubMed.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38(1), 115–164 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- C. W. Stark, W. J. Schreier, J. Lucon, E. Edwards, T. Douglas and B. Kohler, J. Phys. Chem. A, 2015, 119, 4813–4824 CrossRef CAS PubMed.
- L. Flamigni, S. Encinas, F. Barigelletti, F. M. MacDonnell, K.-J. Kim, F. Puntoriero and S. Campagna, Chem. Commun., 2000,(13), 1185–1186 RSC.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- Y. Sun and C. Turro, Inorg. Chem., 2010, 49(11), 5025–5032 CrossRef CAS PubMed.
- E. E. Beauvilliers and G. J. Meyer, Inorg. Chem., 2016, 55(15), 7517–7526 CrossRef CAS PubMed.
- G. E. Shillito, C. B. Larsen, J. R. W. McLay, N. T. Lucas and K. C. Gordon, Inorg. Chem., 2016, 55(21), 11170–11184 CrossRef CAS PubMed.
- Q. Pan, F. Mecozzi, J. P. Korterik, D. Sharma, J. L. Herek, J. G. Vos, W. R. Browne and A. Huijser, J. Phys. Chem. C, 2014, 118(36), 20799–20806 CAS.
- D. M. Dattelbaum, E. M. Kober, J. M. Papanikolas and T. J. Meyer, Chem. Phys., 2006, 326(1), 71–78 CrossRef CAS.
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1984, 23, 3877–3886 CrossRef CAS.
- A. Cadranel, G. E. Pieslinger, P. Tongying, M. K. Kuno, L. M. Baraldo and J. H. Hodak, Dalton Trans., 2016, 45(13), 5464–5475 RSC.
- K. Barthelmes, M. Jäger, J. Kübel, C. Friebe, A. Winter, M. Wächtler, B. Dietzek and U. S. Schubert, Inorg. Chem., 2016, 55(11), 5152–5167 CrossRef CAS PubMed.
- A. Cadranel, P. Alborés, S. Yamazaki, V. D. Kleiman and L. M. Baraldo, Dalton Trans., 2012, 41(17), 5343–5350 RSC.
- G. M. Bryant and J. E. Fergusson, Aust. J. Chem., 1971, 24, 275–286 CrossRef CAS.
- J. N. Braddock and T. J. Meyer, J. Am. Chem. Soc., 1973, 95(10), 3158 CrossRef CAS.
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1983, 22(11), 1614–1616 CrossRef CAS.
- M. K. Nazeeruddin, S. M. Zakeeruddin and K. Kalyanasundaram, J. Chem. Phys., 1993, 97, 9607–9612 CrossRef CAS.
- S. Kämper, A. Paretzki, J. Fiedler, S. Zalis, W. Kaim, S. Záliš, W. Kaim, S. Zalis and W. Kaim, Inorg. Chem., 2012, 51(4), 2097–2104 CrossRef PubMed.
- X. Li, M. K. Nazeeruddin, M. Thelakkat, P. R. F. Barnes, R. Vilar and J. R. Durrant, Phys. Chem. Chem. Phys., 2011, 13, 1575–1584 RSC.
- Y. Tachibana, J.-E. Moser, M. Gratzel, D. R. Klug, J. R. Durrant, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem., 1996, 100(96), 20056–20062 CrossRef CAS.
- P. S. Braterman, J.-I. Song and R. D. Peacock, Spectrochim. Acta, Part A, 1992, 48(6), 899–903 CrossRef.
- A. E. Curtright and J. K. McCusker, J. Phys. Chem. A, 1999, 103, 7032–7041 CrossRef CAS.
- A. M. Brown, C. E. McCusker and J. K. McCusker, Dalton Trans., 2014, 43, 17635–17646 RSC.
- N. H. Damrauer, T. R. Boussie, M. Devenney, J. K. Mccusker and R. V. April, J. Am. Chem. Soc., 1997, 119(35), 8253–8268 CrossRef CAS.
- J. K. McCusker, Acc. Chem. Res., 2003, 36(12), 876–887 CrossRef CAS PubMed.
- J. D. Henrich, H. Zhang, P. K. Dutta and B. Kohler, J. Phys. Chem. B, 2010, 114(45), 14679–14688 CrossRef CAS PubMed.
- C. Herrero, A. Quaranta, R.-A. Fallahpour, W. Leibl and A. Aukauloo, J. Phys. Chem., 2013, 117(19), 9605–9612 CAS.
- J. T. Hewitt, J. J. Concepcion and N. H. Damrauer, J. Am. Chem. Soc., 2013, 135(34), 12500–12503 CrossRef CAS PubMed.
- M. Bräutigam, M. Wächtler, S. Rau, J. Popp and B. Dietzek, J. Phys. Chem. C, 2012, 116(1), 1274–1281 Search PubMed.
- C. E. McCusker and J. K. McCusker, Inorg. Chem., 2011, 50(5), 1656–1669 CrossRef CAS PubMed.
- N. H. Damrauer and J. K. McCusker, J. Phys. Chem. A, 1999, 103, 8440–8446 CrossRef CAS.
- J. T. Hewitt, P. J. Vallett and N. H. Damrauer, J. Phys. Chem. A, 2012, 116(47), 11536–11547 CrossRef CAS PubMed.
- Q. Sun, S. Mosquera-Vazquez, L. M. Lawson Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, J. Am. Chem. Soc., 2013, 135(37), 13660–13663 CrossRef CAS PubMed.
- N. Damrauer, G. Cerullo, A. Yeh, T. Boussie, C. Shank and J. McCusker, Science, 1997, 275(5296), 54–57 CrossRef CAS PubMed.
- S. Wallin, J. Davidsson, J. Modin and L. Hammarström, J. Phys. Chem. A, 2005, 109, 4697–4704 CrossRef CAS PubMed.
- J. T. Hewitt, P. J. Vallett and N. H. Damrauer, J. Phys. Chem. A, 2012, 116(47), 11536–11547 CrossRef CAS PubMed.
- G. B. Shaw, D. J. Styers-Barnett, E. Z. Gannon, J. C. Granger and J. M. Papanikolas, J. Phys. Chem. A, 2004, 108(23), 4998–5006 CrossRef CAS.
- S. a. McFarland, F. S. Lee, K. a W. Y. Cheng, F. L. Cozens and N. P. Schepp, J. Am. Chem. Soc., 2005, 127, 7065–7070 CrossRef CAS PubMed.
- A. A. Rachford and J. J. Rack, J. Am. Chem. Soc., 2006, 128, 14318–14324 CrossRef CAS PubMed.
- P. Manca, M. I. Pilo, G. Sanna, A. Zucca, G. Bergamini and P. Ceroni, Chem. Commun., 2011, 47, 3413–3415 RSC.
- A. I. Baba, J. R. Shaw, J. A. Simon, R. P. Thummel and R. H. Schmehl, Coord. Chem. Rev., 1998, 171, 43–59 CrossRef CAS.
- M. B. Majewski, N. R. D. Tacconi, F. M. MacDonnell and M. O. Wolf, Chem.–Eur. J., 2013, 19(25), 8331–8341 CrossRef CAS PubMed.
- Y. Sun, M. E. Ojaimi, R. Hammitt, R. P. Thummel and C. Turro, J. Phys. Chem. B, 2010, 114(45), 14664–14670 CrossRef CAS PubMed.
- A. F. Morales, G. Accorsi, N. Armaroli, F. Barigelletti, S. J. A. Pope and M. D. Ward, Inorg. Chem., 2002, 41(25), 6711–6719 CrossRef CAS PubMed.
- X. Y. Wang, A. Del Guerzo and R. H. Schmehl, J. Photochem. Photobiol., C, 2004, 5(1), 55–77 CrossRef CAS.
- M. B. Majewski, N. R. D. Tacconi, F. M. MacDonnell and M. O. Wolf, Inorg. Chem., 2011, 50(20), 9939–9941 CrossRef CAS PubMed.
- M. B. Majewski, J. G. Smith, M. O. Wolf and B. O. Patrick, Eur. J. Inorg. Chem., 2016, 1470–1479 CrossRef CAS.
- A. T. Yeh, C. V. Shank and J. K. McCusker, Science, 2000, 289(5481), 935–938 CrossRef CAS PubMed.
- J. Van Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98(16), 4853–4858 CrossRef CAS.
- Q. Sun, S. Mosquera-vazquez, Y. Suffren, J. Hankache, N. Amstutz, L. Max, L. Daku, E. Vauthey and A. Hauser, Coord. Chem. Rev., 2015, 283, 87–99 CrossRef.
- Q. Sun, S. Mosquera-Vazquez, L. M. Lawson Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, J. Am. Chem. Soc., 2013, 135(37), 13660–13663 CrossRef CAS PubMed.
- Q. Sun, B. Dereka, E. Vauthey, L. M. Lawson Daku and A. Hauser, Chem. Sci., 2017, 8, 223–230 RSC.
- G. B. Shaw and J. M. Papanikolas, J. Phys. Chem. B, 2002, 106, 6156–6162 CrossRef CAS.
- D. W. Thompson, A. Ito and T. J. Meyer, Pure Appl. Chem., 2013, 85(7), 1257–1305 CrossRef CAS.
- G. D. Hager and G. A. Crosby, J. Am. Chem. Soc., 1975, 97(24), 7031–7037 CrossRef CAS.
- D. Hager, R. J. Watts and G. A. Crosby, J. Am. Chem. Soc., 1975, 97(24), 7037–7042 CrossRef.
- M. Sykora and J. R. Kincaid, Inorg. Chem., 1995, 34(7), 5852–5856 CrossRef CAS.
- R. M. O’Donnell, P. G. Johansson, M. Abrahamsson and G. J. Meyer, Inorg. Chem., 2013, 52(12), 6839–6848 CrossRef PubMed.
- R. S. Lumpkin, E. M. Kober, L. A. Worl, Z. Murtaza and T. J. Meyer, J. Phys. Chem., 1990, 94, 239–243 CrossRef CAS.
- A. Harriman and G. Izzet, Phys. Chem. Chem. Phys., 2007, 9(8), 944–948 RSC.
- J. Bendix, P. Steenberg and I. Søtofte, Inorg. Chem., 2003, 42(15), 4510–4512 CrossRef CAS PubMed.
- T. Mukuta, A. Inagaki, S. Koshihara and K. Onda, ChemistrySelect, 2016, 1, 2802–2807 CrossRef CAS.
- Q. Sun, B. Dereka, E. Vauthey, L. M. Lawson Daku and A. Hauser, Chem. Sci., 2017, 8, 223–230 RSC.
- W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38(12), 3373–3382 RSC.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49(3), 1–22 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02670f |
| ‡ The cyclic voltammetric measurements of saturated solutions of tpm in acetonitrile failed to disclose any appreciable cathodic processes all the way up to −2.5 V vs. Ag/AgCl (Fig. S1†), hampering the recording of spectral fingerprints associated with the tpm reduction. |
| § The last maximum is obscured by pump scattering. |
| ¶ In the complexes reported here, depopulation of the long-lived emissive 3MLCT state is linked to GS recovery. 3MC excited states are, however, likely to be thermally populated from the emissive 3MLCT excited state and reduce the overall lifetimes relative to the case of [Ru(bpy)3]2+. Therefore, depopulation of the emissive states includes direct conversion to the GS and also an intermediate internal conversion step to the 3MC excited states, whose lifetimes are too short to be detected.62 |
| This journal is © The Royal Society of Chemistry 2017 |