
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A complementary pair of enantioselective switchable organocatalysts†
Guillaume
De Bo
 ,
David A.
Leigh
*,
Charlie T.
McTernan
and
Shoufeng
Wang
,
David A.
Leigh
*,
Charlie T.
McTernan
and
Shoufeng
Wang
School of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.leigh@manchester.ac.uk
First published on 23rd August 2017
Abstract
A pair of enantioselective switchable bifunctional catalysts are shown to promote a range of conjugate addition reactions in up to 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 e.r. and 95% conversion. Each catalyst can be switched OFF using conditions that switch the other catalyst ON. Catalyst ON:OFF ratios of up to 98:2 and 1:99 were achieved, with a ratio of reaction rates of up to 16:1 between the ON and OFF states, maintained over complete ON–OFF–ON and OFF–ON–OFF cycles. However, simultaneous operation of the catalyst pair in the same reaction vessel, which in principle could allow product handedness to be switched by simple E–Z isomerisation of the catalyst pair, was unsuccessful. In this first generation complementary pair of enantioselective switchable organocatalysts, the OFF state of one catalyst inhibits the ON state of the other.
:5 e.r. and 95% conversion. Each catalyst can be switched OFF using conditions that switch the other catalyst ON. Catalyst ON:OFF ratios of up to 98:2 and 1:99 were achieved, with a ratio of reaction rates of up to 16:1 between the ON and OFF states, maintained over complete ON–OFF–ON and OFF–ON–OFF cycles. However, simultaneous operation of the catalyst pair in the same reaction vessel, which in principle could allow product handedness to be switched by simple E–Z isomerisation of the catalyst pair, was unsuccessful. In this first generation complementary pair of enantioselective switchable organocatalysts, the OFF state of one catalyst inhibits the ON state of the other.
Introduction
Trigger effects and feedback loops are key processes used to control enzyme activity in cells.1 They help to maintain homeostasis and enable lengthy reaction pathways to be carried out in complex chemical environments. Such switchable catalytic behaviour is mimicked in an array of artificial systems2 in order to modulate rate,3 reaction pathway4 and, somewhat less frequently, stereoselectivity.5 However, state-of-the-art catalytic systems are often sensitive to structural modification and their incorporation into molecular switches and other devices often leads to significant erosion of selectivity or activity.4c,5d This is especially true for bifunctional organocatalysts,6a which typically require the cooperation of a (chiral) base and a hydrogen-bond donor.6b Here we demonstrate a modular approach to enantioselective switchable bifunctional organocatalysts that provides a high degree of control over catalyst activity and stereoselectivity with little loss of efficacy of organocatalytic function. By judicious choice of the regiochemistry of attachment of the switching unit, a complementary pair of enantioselective catalysts was prepared using this approach, in which each catalyst could be switched ON under conditions that switched the other catalyst OFF.The catalyst pair is based on a bifunctional cinchona alkaloid-squaramide motif as the active catalyst,7 with a hydrogen bond acceptor, nitrobenzene, as an additional potentially inhibiting unit (Scheme 1). The E/Z isomerisation of a pyridyl-acyl hydrazone switch8,9 is used to control the activity of the catalyst. In 1, the catalytically inactive OFF state is generated by operating the switch with UV light, bringing the nitro group into close proximity with the squaramide (Scheme 1a). By changing the regiochemistry of the pyridine ring (2), the same stimulus can be used to turn catalyst 2 ON through the equivalent isomerisation process forcing the nitro group away from the squaramide (Scheme 1b). The resulting pair of enantioselective switchable bifunctional organocatalysts can be switched between states with various stimuli (light, heat or pH), giving ON:OFF ratios of up to 98:2 and 1:99. In their ON state each catalyst promotes Michael additions with an enantiomeric ratio (e.r.) of up to 95:5.
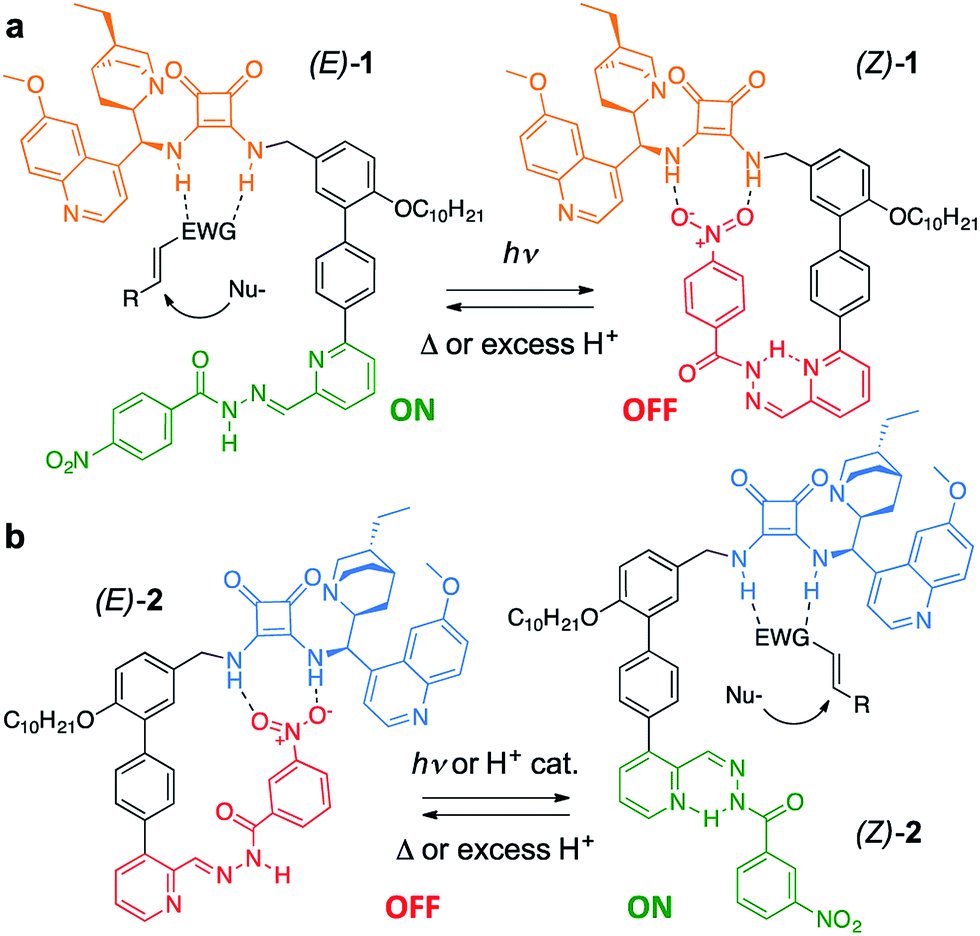 | ||
| Scheme 1 A pair of enantioselective switchable bifunctional organocatalysts. | ||
Results and discussion
Switchable catalysts 1 and 2 were assembled (Scheme 2) around a biphenyl unit 3 featuring (i) an n-decyl chain to ensure sufficient organic solvent solubility of the final catalyst, (ii) a protected amine to anchor the catalytic units 4 or 5, and (iii) a boronic ester through which to attach the switching unit. Complementary switching behaviour was achieved by coupling 3 with 2,3-substituted bromopyridine 6 or its 2,6-isomer 7. The squaramide inhibiting unit was installed by condensing a nitrobenzohydrazide with aldehydes 8 or 9 to form hydrazones 10 and 11. Finally, amine deprotection followed by addition of hydroquinine-derived mono-squaramide 4 or hydroquinidine-derived mono-squaramide 5 furnished catalysts 1 and 2, respectively. Catalysts 1 and 2 were synthesised in 14 steps in overall yields of 18% and 12%, respectively (see ESI† for full synthetic procedures and characterisation data).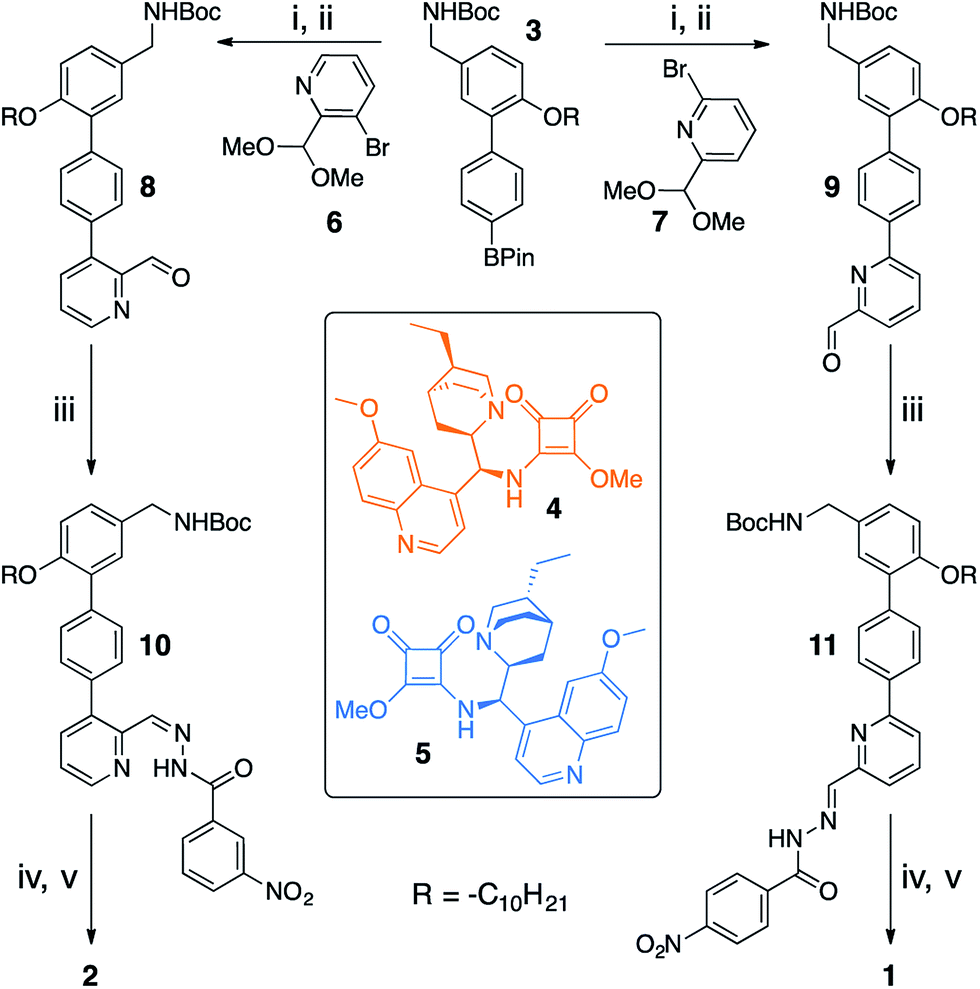 | ||
| Scheme 2 Modular assembly of enantioselective switchable catalysts 1 and 2. Reagents and conditions: (i) 6 or 7, Pd(PPh3)4, THF, 2 M Na2CO3, 60 °C, 4 days. (ii) p-Toluenesulfonic acid, THF, H2O, 50 °C, 3 h. (iii) 3- or 4-Nitrobenzohydrazide, aniline, CH2Cl2, r.t., 16 h. (iv) CF3CO2H, CH2Cl2, r.t., 1 h. (v) 4 or 5, DMF, Et3N, MeOH, r.t., 16 h. | ||
With 1 and 2 in hand our attention turned to switching between the E- and Z-states of the catalyst pair. Catalyst 1 was typically formed in a 80:20 E:Z ratio from Scheme 2, and optimised light-switching conditions provided a 21:79 E:Z ratio at 395 nm (Table 1, entry 1 and Table S1†). The most effective conditions for Z-to-E isomerisation proved to be using CF3CO2H in ethyl acetate at 50 °C, which afforded 98:2 ratio of E:Z-1 (Table 1, entry 3 and Table S2†).
| Entry | Catalyst | Conditions |
E:Z |
|---|---|---|---|
| 1 | 1 | CH2Cl2:EtOAc 1:4, 395 nm, 700 mW, 20 min, 0.3 mg ml−1 |
21:79 |
| 2 | 1 | DMF, 90 °C, 5 h, 0.2 mg ml−1 | 90:10 |
| 3 | 1 | EtOAc, 0.1% CF3CO2H, 60 min, 50 °C, 0.02 mg ml−1 | 98:2 |
| 4 | 2 | CH2Cl2:EtOAc 1:4, 395 nm, 700 mW, 20 min, 0.3 mg ml−1 |
21:79 |
| 5 | 2 | EtOAc, 0.1% CF3CO2H, 60 min, 50 °C, 0.02 mg ml−1 | 99:1 |
| 6 | 2 | CHCl3, 0.25 eq. CF3CO2H, 24 h, 40 °C, 0.1 mg ml−1 | 5:95 |
As with catalyst 1, catalyst 2 formed a 21:79 E:Z mixture under irradiation at 395 nm (entry 4, Table 1). A 99:1 E:Z ratio was reached upon heating 2 with 0.1% CF3CO2H in EtOAc (entry 5). Somewhat surprisingly, 0.25 equiv. CF3CO2H in CHCl3 promoted E-to-Z isomerisation to give a 5:95 E:Z ratio of isomers (entry 6), although these conditions proved ineffective with catalyst 1. The ability to use either light or one of two different acid-based conditions to promote switching augurs well for operating the catalytic groups in more complex settings.
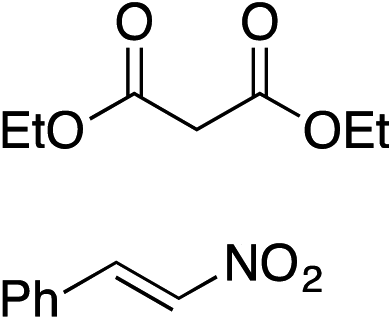
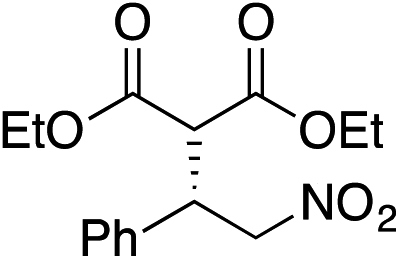
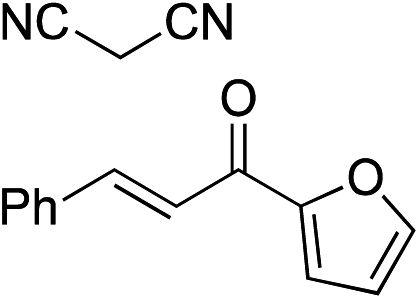
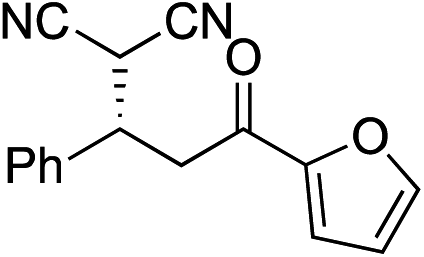
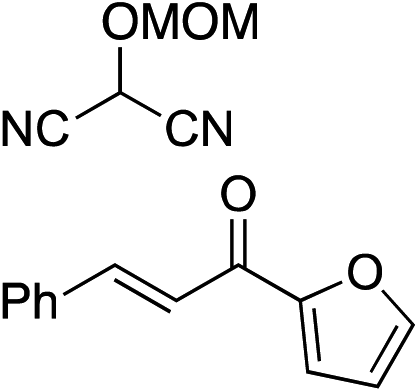
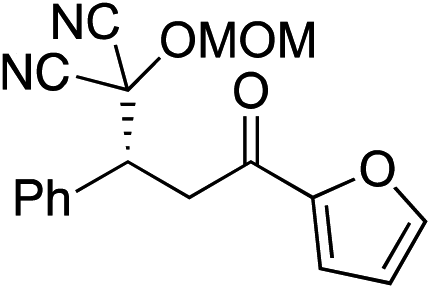
Bifunctional cinchona-squaramide catalysts are known to promote a range of conjugate addition reactions,10 so we focused on examining this reaction class with 1 and 2 (Table 2). The addition of malonitrile or masked thiol to chalcone derivatives was catalysed at low catalyst loadings with good conversions and e.r. (Table 2, entries 1, 2 and 5), as was the addition of 1,3-diphenyl-1,3-propandione or diethyl malonate to β-nitrostyrene (Table 2, entries 3 and 4). Masked acyl cyanides could be generated with excellent conversions and very good e.r. (Table 2, entry 6).11 Overall, the conversions and e.r. are similar to previously reported bifunctional organocatalysts (Table 2, entries 1–5), despite the switching process leaving some catalyst in the OFF state (catalyst 1 used as 98:2 E:Z; catalyst 2 used as 5:95 E:Z). The results show that catalytic motifs can be incorporated into more complex switching structures without significantly impairing catalytic performance.
| Entry | Reagents | Producta | Conv. (S:R), 1 |
Conv. (S:R), 2 |
Prev. reportb (S:R) |
|---|---|---|---|---|---|
|
a Products of catalyst 1 depicted.
b Catalyst structures and reaction conditions are shown in Table S3, ESI. MOM = methoxymethyl. In all cases catalyst 1 was used as 98:2 E:Z and catalyst 2 as 1:99 E:Z.
|
|||||
| 1 |
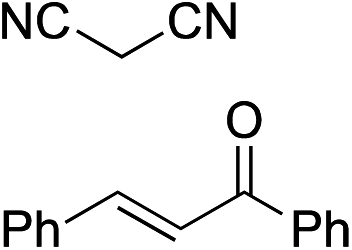
|
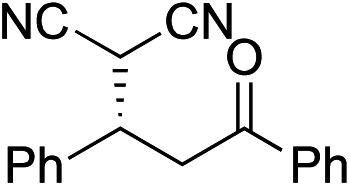
|
91% (83:17) |
90% (23:77) |
82% (95:5)10c |
| 2 |
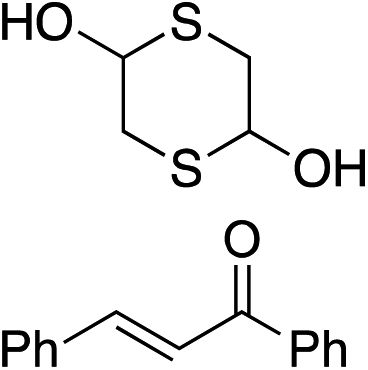
|
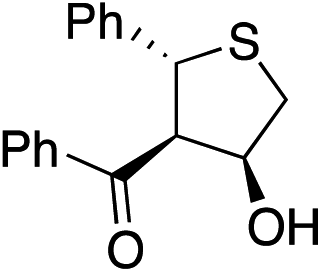
|
93% (25:75) |
94% (70:30) |
81% (94:6)10d |
| 3 |
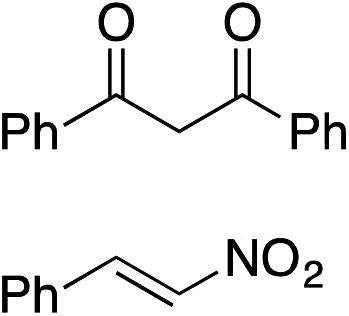
|
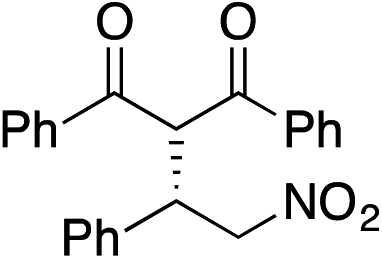
|
81% (91:9) |
90% (10:90) |
89% (95:5)10f,h |
| 4 |
|
|
88% (89:11) |
70% (20:80) |
83% (95:5)10h |
| 5 |
|
|
95% (95:5) |
93% (7:93) |
96% (93:7)10e |
| 6 |
|
|
95% (87:13) |
95% (16:84) |
— |
The limiting factor that determines the efficacy of turning catalysis ON and OFF with a switchable catalyst is generally not how effective the ON state is as a catalyst, but rather the lack of catalysis by the OFF state.2a Often the OFF state still catalyses the reaction to some extent, which is compounded if the switching leaves even a small amount of the ON state of the catalyst present.2a We used the formation of 14 by addition of malonitrile (12) to chalcone 13 (Fig. 1a) to investigate the relative reactivity of the ON and OFF states of each member of the catalyst pair (Fig. 1b and c). The reactions were monitored by 1H NMR and, as expected from the imperfect photostationary state (E:Z = 21:79 for the OFF state of 1), a 6:1 ON:OFF ratio of initial rates was observed when using 5 mol% of catalyst 1 (Fig. 1b). With a 3.5 mol% loading of catalyst 2 (E:Z = 99:1 for the OFF state), an ON:OFF ratio of 16:1, excellent for the current generation of artificial switchable catalysts, was achieved (Fig. 1c). Finally we investigated the switching of catalysis ON and OFF during the chemical reaction of 12 and 13 (see ESI†). A full switching cycle was carried out starting from either the ON or OFF state of catalyst 2 (3.5 mol%, initial E:Z ratio 99:1 (ON) or 2:98 (OFF)). After 6 h the E-to-Z (or Z-to-E) stimulus was applied (0.1% CF3CO2H, 60 min, 50 °C or 395 nm, 700 mW, 20 min) and after 24 h (for initially OFF 2) or 21 h (for initially ON 2) the opposing stimulus was applied reverting catalyst 2 to its initial state. In both cases good control of reaction rate over the entire cycle was achieved, without compromising the stereoselectivity of product formation (see ESI†).
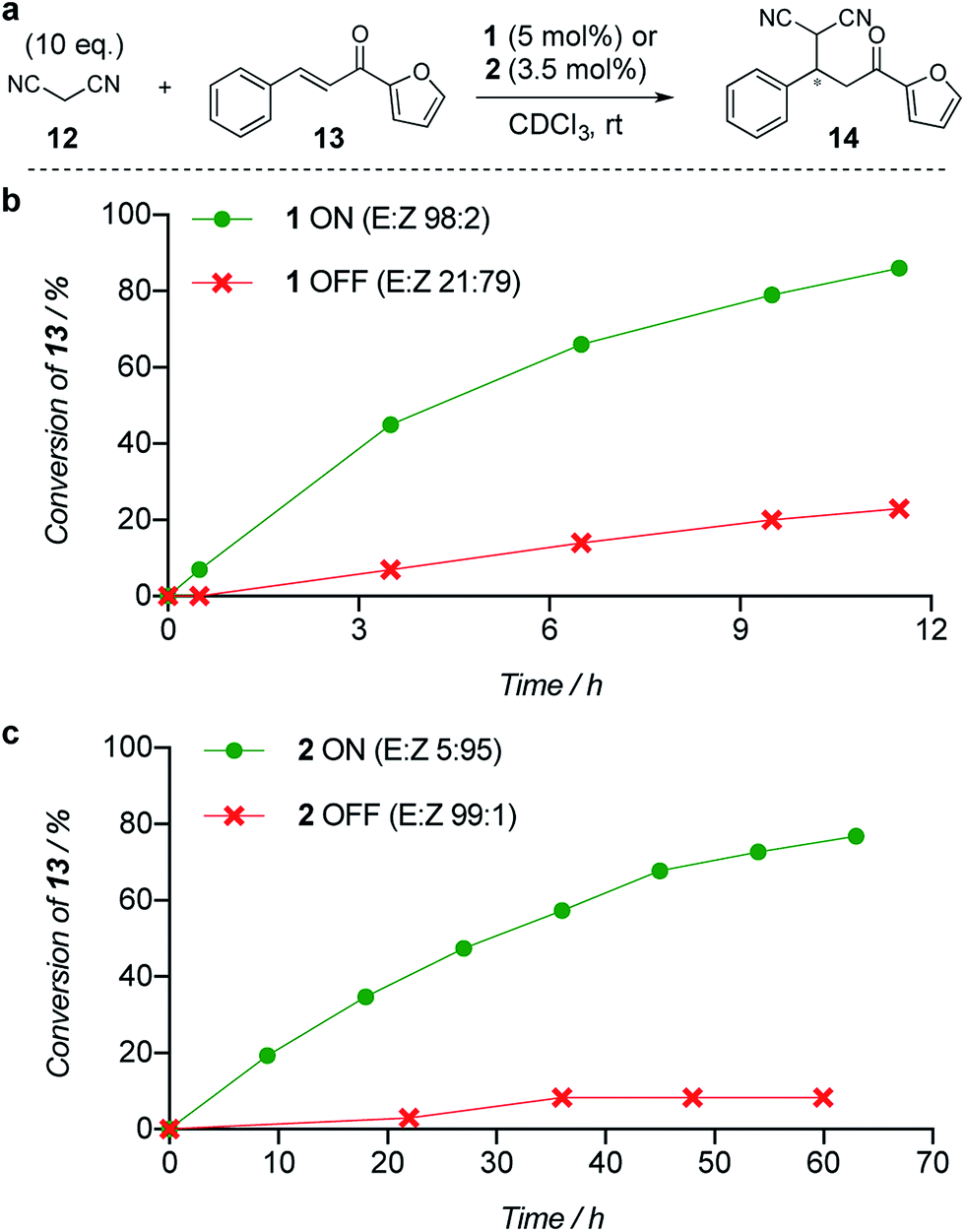 | ||
| Fig. 1 Relative reactivity of ON and OFF states of (b) catalyst 1 and (c) catalyst 2 towards the addition of malonitrile (12) to chalcone (13). Solid lines are a guide to the eye. | ||
The ultimate goal of a complementary pair of switchable enantioselective catalysts is the enticing prospect of being able to operate them simultaneously in the same reaction vessel so that the handedness of the product produced by the catalysis can be controlled by switching OFF one catalyst while the same stimulus switches ON the other. Our attempts to achieve this with 1 and 2 proved unsuccessful, with the organocatalysis remaining OFF when either stimulus was applied. Bifunctional cinchona-squaramide catalysts have a tendency to aggregate12 and for 1 and 2 it appears that the OFF state of one catalyst binds to the ON state of the other catalyst, preventing catalysis by either state of either member of the catalyst pair. Inhibiting association between the catalysts must be an important consideration in future designs of complementary pairs of switchable enantioselective catalysts.
Conclusions
A complementary pair of switchable enantioselective catalysts, 1 and 2, were prepared, each capable of catalyzing Michael additions of carbon and sulfur nucleophiles with chalcones in 70–95% yield with good stereoselectivity (up to 95:5 e.r.). The switchable catalysts have similar activities to previously reported non-switchable catalysts and so incorporating the bifunctional organocatalytic groups into a more complex, switchable, molecular structure does not significantly impact on the intrinsic efficacy of the catalytic groups. The two switchable catalysts generate opposite enantiomers, so both hands of products can be accessed, and a single stimulus switches one member of the catalyst pair OFF and the other ON. Photochemical or acid-catalysed isomerisation provides excellent E:Z ratios in each switching direction, and significant differences in ON versus OFF rates of catalysis were obtained, which were maintained throughout full ON–OFF–ON and OFF–ON–OFF cycles. Future work will focus on the design of systems where the complementary pair of catalysts can be operated simultaneously in the same reaction vessel.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Engineering and Physical Sciences Research Council (UK). We thank the Royal Society for a University Research Fellowship (to G. D. B.) and Research Professorship (to D. A. L.).Notes and references
- (a) T. W. Traut, Allosteric Regulatory Enzymes, Springer, New York, 2008 Search PubMed; for recent examples of feedback loops in synthetic systems, see:; (b) D. Ray, J. T. Foy, R. P. Hughes and I. Aprahamian, Nat. Chem., 2012, 4, 757 CrossRef CAS PubMed; (c) S. Pramanik and I. Aprahamian, J. Am. Chem. Soc.2016, 138, 15142 Search PubMed.
- (a) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed.
- (a) A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1981, 94 RSC; (b) F. Würthner and J. Rebek, Angew. Chem., Int. Ed., 1995, 34, 446 CrossRef; (c) H. Sugimoto, T. Kimura and S. Inoue, J. Am. Chem. Soc., 1999, 121, 2325 CrossRef CAS; (d) R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Am. Chem. Soc., 2003, 125, 2224 CrossRef CAS PubMed; (e) M. V. Peters, R. S. Stoll, A. Kühn and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 5968 CrossRef CAS PubMed; (f) R. S. Stoll, V. M. Peters, A. Kühn, S. Heiles, R. Goddard, M. Bühl, C. M. Thiele and S. Hecht, J. Am. Chem. Soc., 2009, 131, 357 CrossRef CAS PubMed; (g) H. J. Yoon, J. Kuwabara, J. H. Kim and C. A. Mirkin, Science, 2010, 330, 66 CrossRef CAS PubMed; (h) O. B. Berryman, A. C. Sather, A. Lledó and J. Rebek, Angew. Chem., Int. Ed., 2011, 50, 9400 CrossRef CAS PubMed; (i) T. Imahori, R. Yamaguchi and S. Kurihara, Chem.–Eur. J., 2012, 18, 10802 CrossRef CAS PubMed; (j) V. Blanco, A. Carlone, K. D. Hanni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166 CrossRef CAS PubMed; (k) B. M. Neilson and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 12693 CrossRef CAS PubMed; (l) D. Wilson, N. Branda and R. Turning, Angew. Chem., Int. Ed., 2012, 51, 5431 CrossRef CAS PubMed; (m) M. Schmittel, S. De and S. Pramanik, Angew. Chem., Int. Ed., 2012, 51, 3832 CrossRef CAS PubMed; (n) M. Schmittel, S. Pramanik and S. De, Chem. Commun., 2012, 48, 11730 RSC; (o) B. M. Neilson and C. W. Bielawski, Organometallics, 2013, 32, 3121 CrossRef CAS; (p) S. De, S. Pramanik and M. Schmittel, Dalton Trans., 2014, 43, 10977 RSC; (q) V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, J. Am. Chem. Soc., 2014, 136, 15775 CrossRef CAS PubMed; (r) V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, J. Am. Chem. Soc., 2014, 136, 4905 CrossRef CAS PubMed; (s) C. M. McGuirk, J. Mendez-Arroyo, A. M. Lifschitz and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 16594 CrossRef CAS PubMed; (t) A. M. Lifschitz, R. M. Young, J. Mendez-Arroyo, C. L. Stern, C. M. McGuirk, M. R. Wasielewski and C. A. Mirkin, Nat. Commun., 2015, 6, 6541 CrossRef CAS PubMed; (u) A. J. Teator, Y. Tian, M. Chen, J. K. Lee and C. W. Bielawski, Angew. Chem., Int. Ed., 2015, 54, 11559 CrossRef CAS PubMed; (v) A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969 CrossRef CAS PubMed; (w) S. Semwal and J. Choudhury, ACS Catal., 2016, 6, 2424 CrossRef CAS; (x) C. M. McGuirk, J. Mendez-Arroyo, A. I. d'Aquino, C. L. Stern, Y. Liu and C. A. Mirkin, Chem. Sci., 2016, 7, 6674 RSC; (y) A. C. H. Jans, A. Gómez-Suárez, S. P. Nolan and J. N. H. Reek, Chem.–Eur. J., 2016, 22, 14836 CrossRef CAS PubMed; (z) N. Mittal, S. Pramanik, I. Paul, S. De and M. Schmittel, J. Am. Chem. Soc., 2017, 139, 4270 CrossRef CAS PubMed.
- (a) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264 CrossRef CAS PubMed; (b) S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255 CrossRef CAS PubMed; (c) J. Beswick, V. Blanco, G. De Bo, D. A. Leigh, U. Lewandowska, B. Lewandowski and K. Mishiro, Chem. Sci., 2015, 6, 140 RSC; (d) S. Gaikwad, A. Goswami, S. De and M. Schmittel, Angew. Chem., Int. Ed., 2016, 55, 10512 CrossRef CAS PubMed; (e) C.-S. Kwan, A. S. C. Chan and K. C.-F. Leung, Org. Lett., 2016, 18, 976 CrossRef CAS PubMed.
- (a) D. Sud, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2005, 44, 2019 CrossRef CAS PubMed; (b) Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 9254 CrossRef CAS PubMed; (c) J. Wang and B. L. Feringa, Science, 2011, 331, 1429 CrossRef CAS PubMed; (d) S. Mortezaei, N. R. Catarineu and J. W. Canary, J. Am. Chem. Soc., 2012, 134, 8054 CrossRef CAS PubMed; (e) M. Vlatković, L. Bernardi, E. Otten and B. L. Feringa, Chem. Commun., 2014, 50, 7773 RSC; (f) S. Mortezaei, N. R. Catarineu, X. Duan, C. Hu and J. W. Canary, Chem. Sci., 2015, 6, 5904 RSC; (g) D. Zhao, T. M. Neubauer and B. L. Feringa, Nat. Commun., 2015, 6, 6652 CrossRef CAS PubMed; (h) C.-T. Chen, C.-C. Tsai, P.-K. Tsou, G.-T. Huang and C.-H. Yu, Chem. Sci., 2017, 8, 524 RSC; (i) A. Martinez Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R. A. Orenes, M. Alajarin and J. Berna, Chem. Sci., 2017, 8, 3775 RSC; (j) G. Romanazzi, L. Degennaro, P. Mastrorilli and R. Luisi, ACS Catal., 2017, 7, 4100 CrossRef CAS.
- (a) O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051 RSC; for a recent review on bifunctional amine-squaramide organocatalysts see: (b) P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253 CrossRef CAS.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS PubMed.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem.–Eur. J., 2011, 17, 248 CrossRef CAS PubMed.
- For other examples of acyl hydrazone switches, see: (a) X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963 RSC; (b) L. A. Tatum, X. Su and I. Aprahamian, Acc. Chem. Res., 2014, 47, 2141 CrossRef CAS PubMed; (c) D. J. van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982 CrossRef CAS PubMed.
- (a) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed; (b) R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC; (c) W. Yang, Y. Jiu and D.-M. Du, Org. Biomol. Chem., 2012, 10, 332 RSC; (d) J.-B. Ling, Y. Su, H.-L. Zhu, G.-Y. Wang and P.-F. Xu, Org. Lett., 2012, 14, 1090 CrossRef CAS PubMed; (e) N. Molleti, K. N. Rana and V. K. Singh, Org. Lett., 2012, 14, 4322 CrossRef CAS PubMed; (f) B. Liu, X. Han, Z. Dong, H. Lv, H.-B. Zhou and C. Dong, Tetrahedron: Asymmetry, 2013, 24, 1276 CrossRef CAS; (g) T. J. Auvil, A. G. Schafer and A. E. Mattson, Eur. J. Org. Chem., 2014, 13, 2633 CrossRef; (h) K. S. Rao, R. Trivedi and M. L. Kantam, Synlett, 2015, 26, 221 CrossRef CAS; (i) H. Y. Bae and C. E. Song, ACS Catal., 2015, 5, 3613 CrossRef CAS; (j) E. Veverková, S. Bilka, R. Baran and R. Šebesta, Synthesis, 2016, 48, 1474 CrossRef.
- K. S. Yang, A. E. Nibbs, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050 CrossRef CAS PubMed.
- H. B. Jang, H. S. Rho, J. S. Oh, E. H. Nam, S. E. Park, H. Y. Bae and C. E. Song, Org. Biomol. Chem., 2010, 8, 3918 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02462b |
| This journal is © The Royal Society of Chemistry 2017 |