
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Completing the series of boron-nucleophilic cyanoborates: boryl anions of type NHC–B(CN)2−†‡
Richard
Böser
 a,
Lisa C.
Haufe
a,
Matthias
Freytag
a,
Peter G.
Jones
a,
Gerald
Hörner
*b and
René
Frank
*a
a,
Lisa C.
Haufe
a,
Matthias
Freytag
a,
Peter G.
Jones
a,
Gerald
Hörner
*b and
René
Frank
*a
aTechnical University of Braunschweig, Department of Life Sciences, Institute of Analytical and Inorganic Chemistry, Hagenring 30, 38106, Braunschweig, Germany. E-mail: r.frank@tu-braunschweig.de
bTechnical University of Berlin, Department of Chemistry, Institute of Bioinorganic Chemistry, Strasse des 17. Juni 135, 10623 Berlin, Germany. E-mail: gerald.hoerner@tu-berlin.de
First published on 3rd July 2017
Abstract
Since the first seminal report of boron-centred nucleophiles, the area of boryl anions has developed only sporadically and requires further systematisation. The boryl anions of type NHC–B(CN)2− (NHC = N-heterocyclic carbene) described herein complete a consistent series with the known anions cAAC–B(CN)2− [cAAC = cyclic(alkyl)(amino)carbene] and B(CN)32−. A novel approach towards NHC-stabilised cyanoboranes based on alkylthio-cyano exchange at boron is presented, and in contrast to other methods affords the products in better purity and yield. Reduction of suitable NHC–dicyanoboranes gave two unprecedented examples of NHC–B(CN)2− boryl anions. The latter were shown to react as boron-centred nucleophiles with facile formation of B–E bonds, where E = C, Si, Sn, P, Au. Bonding analysis by DFT calculations suggests a systematic variation of the energy of the boron-centred HOMO depending on the carbene, which in turn can control the nucleophilic character.
Introduction
The electron-deficient nature of boron has limited the reactivity of mononuclear boron centres to Lewis-acidity. This is obvious for compounds of type BR3 with a vacant p-orbital but also holds for four-coordinate borates BR4−. Although the latter serve as transfer reagents of the nucleophile R−, the boron centre displays Lewis-acidic properties to stabilise the substituent R−, with the most prominent example being BH4− as a common reducing agent.1 Boron-centred nucleophiles were long ago considered as attractive alternatives to classical boron reagents; attempts to target them date back 50 years, but include erroneous reports.2 For example, the preparation of nucleophilic boryl anions of type BR2−, R = n-Bu, Ph, was claimed in at least two cases but was unambiguously refuted later.3 The discovery of the first (structurally confirmed) anions with boron-centred nucleophilicity (I, X = CH, R = H, Scheme 1) in 2006 therefore represented a significant breakthrough.4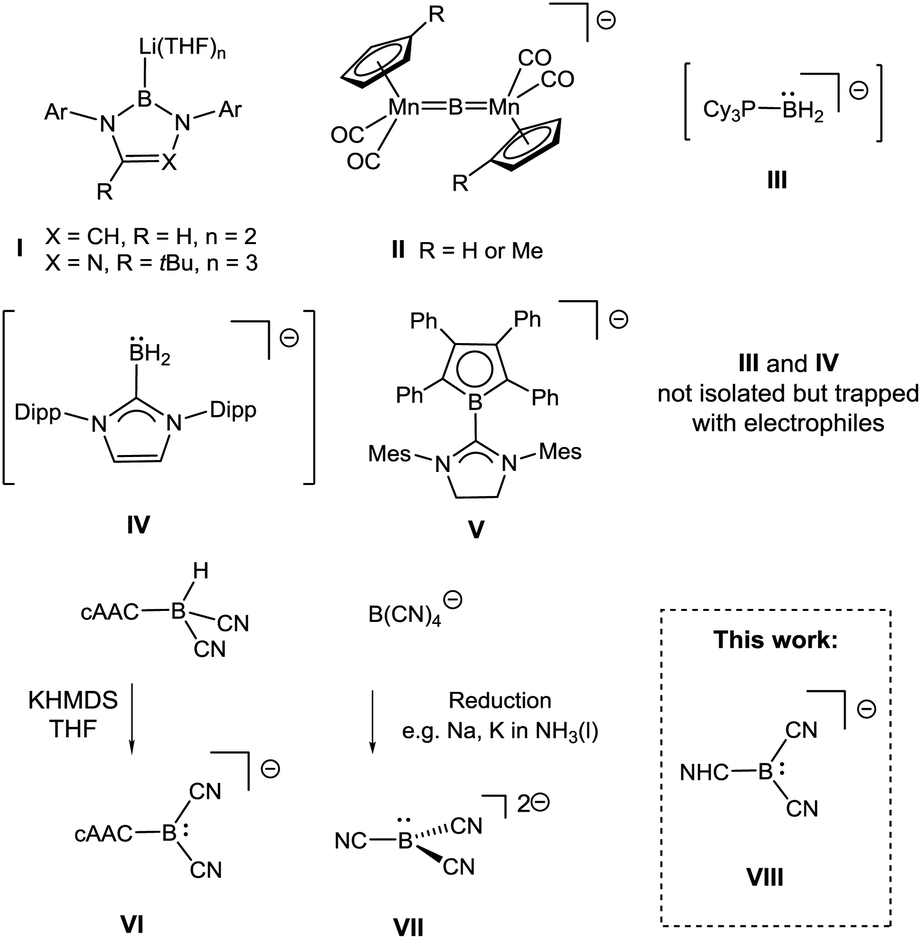 | ||
| Scheme 1 Anionic species with boron-centred nucleophilicity. Cations omitted. For rare examples of neutral boron nucleophiles see ref. 5, Ar = aryl, Mes = 2,4,6-Me3C6H2, Dipp = 2,6-(iPr)2C6H3. | ||
These boron nucleophiles opened up routes to species such as boryl complexes of electropositive metals or metalloids,6 which are difficult to obtain with traditional electrophilic boron reagents. Most remarkably, the stronger electron-releasing character of boryl anions I compared to carbanions has led to the isolation of species for which no organometallic precedents are known.7 Recent examples documenting this behaviour include stable radicals ˙MR2, M = Ga, In, Tl8 or a germanium analogue of vinylidene Ge![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) GeR2,9 and these results have stimulated the quest for further anionic boryl species. Subsequently, a triazaborol-3-yl anion (I, X = N, R = tBu, Scheme 1) was prepared,10 and nucleophilic behaviour at the bridging boron atom was found in an anionic dimanganese borylene complex II.11 Attempts to stabilise the proposed six-electron species of type BR2− involved the use of strong σ-donor ligands L; the parent anionic species L–BH2−, III (L = PCy3)12 and IV (L = IDipp)13 were obtained, although isolation or crystallisation of pure material was impossible because of instability at ambient temperature.
GeR2,9 and these results have stimulated the quest for further anionic boryl species. Subsequently, a triazaborol-3-yl anion (I, X = N, R = tBu, Scheme 1) was prepared,10 and nucleophilic behaviour at the bridging boron atom was found in an anionic dimanganese borylene complex II.11 Attempts to stabilise the proposed six-electron species of type BR2− involved the use of strong σ-donor ligands L; the parent anionic species L–BH2−, III (L = PCy3)12 and IV (L = IDipp)13 were obtained, although isolation or crystallisation of pure material was impossible because of instability at ambient temperature.
In contrast, the NHC-substituted π-borolyl anion V displayed better stability, but its behaviour as a true nucleophile was called into question because of strong evidence of radical pathways.14 Cyano moieties facilitate boron-centred nucleophilicity since the π-acidic character stabilises pz-located lone pairs. Thus, the boryl anion VI cAAC–B(CN)2− [cAAC = cyclic (alkyl)(amino)carbene] was obtained in a remarkable deprotonation reaction from the parent hydroborane cAAC–BH(CN)2.15 Tricyanoborate anion VII B(CN)32− was obtained by the reduction of B(CN)4− or BF(CN)3− or by deprotonation of BH(CN)3−.16,17 Both VI and VII react as boron-centred nucleophiles, although steric congestion caused by the cAAC-moiety strongly means that VI only reacts with small electrophiles. N-Heterocyclic carbenes (NHCs) behave as strong σ-donor (but also as weak acceptor) ligands18 and, considering their ability to stabilise main group elements, it is surprising that boryl anions of type NHC–B(CN)2−VIII are unknown. We therefore set out to develop routes to anions VIII with various the NHC moieties, with the intention of studying the nucleophilic behaviour of such species.
Results and discussion
The preparation of anions VI and VII by deprotonation of the parent hydroboranes by strong bases prompted us to study analogous reactions with IMes–BH(CN)2 [IMes = cyclo-C{N(Mes)CH}2, Mes = 2,4,6-Me3C6H2]. However, the use of various bases of hydride, hydrocarbyl or amide character in THF or 1,4-dioxane proved unsuccessful in our hands; no deprotonation was observed and the starting material was recovered unchanged. While the stronger π-acidity of cAAC-type compared to NHC-type carbenes19 better stabilises the anion VI, the deprotonation of BH(CN)3− is driven by the poor solubility of the alkali metal salts of VII in organic solvents.17 We interpret the ineffective deprotonation of IMes–BH(CN)2 as an absence of such conditions. An alternative approach was then chosen, which involves borane precursors of type NHC–BX(CN)2 where X is a reducible leaving group such as halide. The introduction of cyano groups into carbene–borane adducts has already been accomplished by nucleophilic replacement of boron-bound triflate moieties by cyanide, i.e. B–OTf → B–CN.20 However, the respective mono- or dicyanoborane adducts are obtained as impure samples containing ca. 20% of inseparable isonitrile boranes with B–NC moieties. For an improved synthetic protocol towards mono- or dicyanoboranes, we first established the NHC–borane adducts L–B(SEt)3 (1A–C, Scheme 2), in which electronic and steric properties of the NHC–moiety L (A–C) vary widely. For isolable N-heterocyclic carbenes A and B the adduct formation proceeds in reactions with B(SEt)3 to afford 1A or 1B, respectively. Carbene C is prone to rapid dimerization but can be made in situ by deprotonation of the imidazolium iodide [LH]I, L = C, and is then trapped by B(SEt)3 to give 1C.21 Evidence for the formation of 1A–C is provided by the high-field shift by ca. 60 ppm and the significant signal narrowing in the 11B{1H}-NMR spectra, e.g. δ(11B) = 60.2 ppm, ω1/2 = 72 Hz for B(SEt)3vs. δ(11B) = −2.0 ppm, ω1/2 = 3 Hz for 1B, which is expected for a shift from three- to four-coordinated boron centres. Structural authentication by X-ray crystallography is provided for compounds 1B and 1C (section ESI‡) and shows the expected bond geometries for this class of compounds.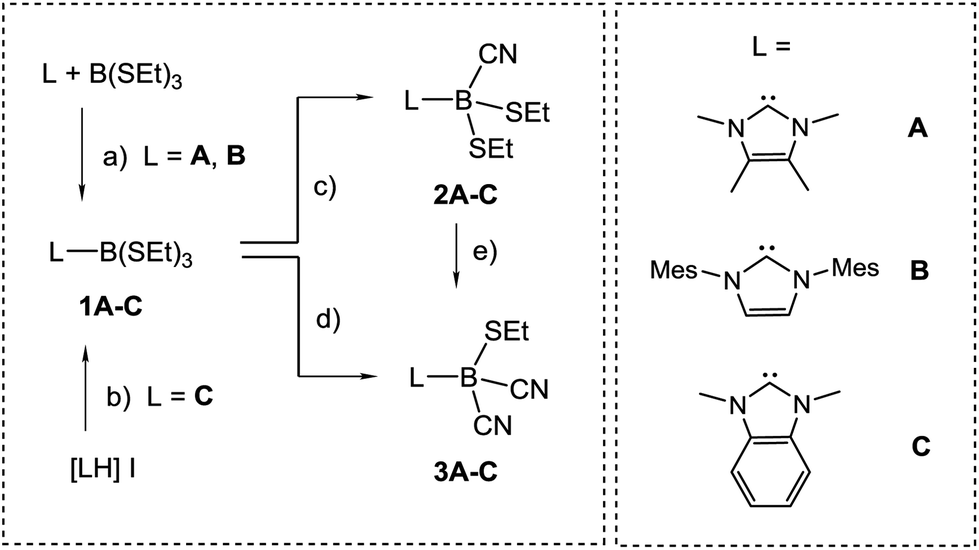 | ||
| Scheme 2 Reagents and conditions. (a) Toluene, rt, 5 min; (b) Na[N(SiMe3)2], THF, −78 °C to rt, 72 h; (c) 1 eq. Me3SiCN, toluene, 45 °C, 24 h; (d) 2.3 eq. Me3SiCN, cat. B(SEt)3, toluene, 95 °C, 24 h; (e) 1.3 eq. Me3SiCN, toluene, 95 °C, 24 h. | ||
For the introduction of nitrile groups, we found Me3SiCN to be an excellent cyanation agent. Thus, the reaction of 1A–C with Me3SiCN (1 eq.) in toluene neatly afforded the monocyanoboranes 2A–C. Although carbon bound alkylthio moieties are commonly poor leaving groups, their nucleophilic replacement has been observed in boron chemistry before.22 In the 11B{1H}-NMR spectra the monocyanoboranes 2A–C are high-field shifted in comparison to the starting material, e.g. δ(11B) = −13.7 ppm, ω1/2 = 3 Hz for 2B. The IR-spectra display typical bands for the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch vibration, e.g. at 2194 cm−1 for 2B. X-ray crystallography confirmed the constitution of compounds 2B and 2C and again showed bond lengths and angles in the expected range (section ESI‡). Attempts to obtain dicyanoboranes 3A–C were performed with analytically pure samples of 1A–C or 2A–C with an excess of Me3SiCN at various temperatures, but decomposition was invariably observed. In contrast, impure crude products 1A–C neatly afforded the desired compounds 3A–C upon treatment with a slight excess (0.3 eq.) of Me3SiCN at elevated temperature. The compounds 3A–C show 11B{1H}-NMR-signals shifted further upfield, e.g. δ(11B) = −24.8 ppm, ω1/2 = 3 Hz for 3B, and display two bands in the IR-spectra, as is expected for both the symmetric and antisymmetric CN stretch vibration, e.g. 2124 cm−1 and 2200 cm−1 in 3B. Structural elucidation by X-ray crystallography confirmed the identity of 3A with bonding parameters in the expected range (section ESI‡). The fact that the dicyanoboranes 3A–C were only accessible from crude products 1A–C or 2A–C was confusing, and we hypothesised that trace amounts of B(SEt)3 in crude samples could be responsible for this unusual result. This assumption is corroborated by the observation that analytically pure samples of 1A–C or 2A–C readily afforded dicyanoboranes 3A–C when they were doped with catalytic amounts of B(SEt)3. For further insight into this system we reacted B(SEt)3 with Me3SiCN in toluene at ambient temperature. Solution NMR-spectra of the reaction mixture revealed only one new boron species δ(11B) = −28.1 ppm, and we obtained compound 4, which rapidly crystallised from the solution (Scheme 3). The formulation of compound 4 as a formal isonitrile-borane adduct Me3Si–NC–B(CN)2SEt was confirmed by X-ray crystallography, and in particular the assignment of the carbon and nitrogen atoms within the linear isonitrile moiety of 4 is unambiguous. This result is in sharp contrast to a previous report in which the analogous reaction of B(SMe)3 with Me3SiCN under comparable conditions gave the nitrile–borane adduct 5.23 Although X-ray crystallographic data had not been reported by the authors the nature of 5 as a nitrile–borane adduct was clarified based on 11B{1H}-NMR chemical shifts, i.e. δ(11B) = 0.3 ppm for 5, whereas we found δ(11B) = −28.1 ppm for the dissolved crystals of 4. The reason for the formation of different products from very similar reactions is, however, currently unclear.
N stretch vibration, e.g. at 2194 cm−1 for 2B. X-ray crystallography confirmed the constitution of compounds 2B and 2C and again showed bond lengths and angles in the expected range (section ESI‡). Attempts to obtain dicyanoboranes 3A–C were performed with analytically pure samples of 1A–C or 2A–C with an excess of Me3SiCN at various temperatures, but decomposition was invariably observed. In contrast, impure crude products 1A–C neatly afforded the desired compounds 3A–C upon treatment with a slight excess (0.3 eq.) of Me3SiCN at elevated temperature. The compounds 3A–C show 11B{1H}-NMR-signals shifted further upfield, e.g. δ(11B) = −24.8 ppm, ω1/2 = 3 Hz for 3B, and display two bands in the IR-spectra, as is expected for both the symmetric and antisymmetric CN stretch vibration, e.g. 2124 cm−1 and 2200 cm−1 in 3B. Structural elucidation by X-ray crystallography confirmed the identity of 3A with bonding parameters in the expected range (section ESI‡). The fact that the dicyanoboranes 3A–C were only accessible from crude products 1A–C or 2A–C was confusing, and we hypothesised that trace amounts of B(SEt)3 in crude samples could be responsible for this unusual result. This assumption is corroborated by the observation that analytically pure samples of 1A–C or 2A–C readily afforded dicyanoboranes 3A–C when they were doped with catalytic amounts of B(SEt)3. For further insight into this system we reacted B(SEt)3 with Me3SiCN in toluene at ambient temperature. Solution NMR-spectra of the reaction mixture revealed only one new boron species δ(11B) = −28.1 ppm, and we obtained compound 4, which rapidly crystallised from the solution (Scheme 3). The formulation of compound 4 as a formal isonitrile-borane adduct Me3Si–NC–B(CN)2SEt was confirmed by X-ray crystallography, and in particular the assignment of the carbon and nitrogen atoms within the linear isonitrile moiety of 4 is unambiguous. This result is in sharp contrast to a previous report in which the analogous reaction of B(SMe)3 with Me3SiCN under comparable conditions gave the nitrile–borane adduct 5.23 Although X-ray crystallographic data had not been reported by the authors the nature of 5 as a nitrile–borane adduct was clarified based on 11B{1H}-NMR chemical shifts, i.e. δ(11B) = 0.3 ppm for 5, whereas we found δ(11B) = −28.1 ppm for the dissolved crystals of 4. The reason for the formation of different products from very similar reactions is, however, currently unclear.
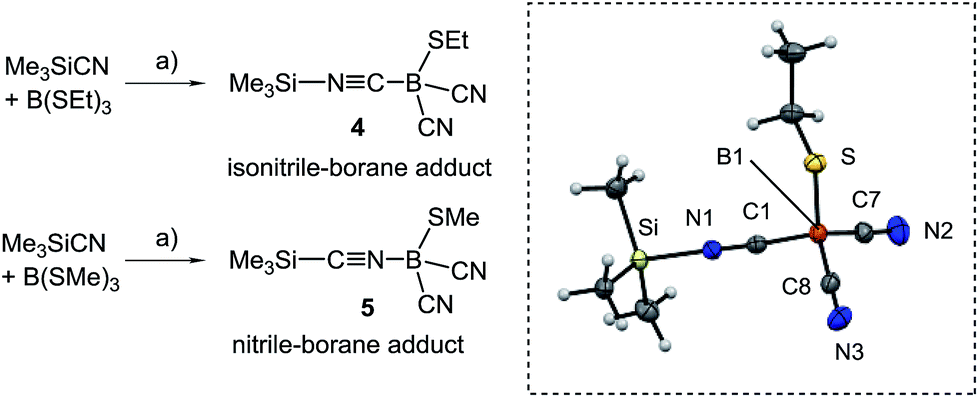 | ||
| Scheme 3 Reagents and conditions. (a) Toluene, rt, 30 min. Dashed box: molecular structure of isonitrile–borane adduct 4. Thermal ellipsoids are presented at 50% probability levels. Selected bond lengths (Å) and angles (°). Si–N1 1.836(1), N1–C1 1.141(2), S–B1 1.898(2), B1–C8 1.588(2), B1–C7 1.588(2), B1–C1 1.608(2), N2–C7 1.144(2), N3–C8 1.146(2), C1–N1–Si 177.54(10), N1–C1–B1 176.10(12). | ||
We further investigated the role of 4 in the dicyanation step and found that analytically pure samples of 1A–C or 2A–C doped with catalytic amounts of isolated 4 also were efficiently dicyanated. Compound 4 is proposed in this system as an active source of silyl cations SiMe3+ (silylium ions) originating from heterolytic dissociation (Scheme 4). Silylium ions are widely known to be efficient Lewis acidic catalysts.24 The alkylthio groups in 2A–C could be activated by the formation of sulfonium salts, from which thioether Me3SiSEt can readily be eliminated with concomitant formation of boryl cations L–B(CN)(SEt)+. The latter could react with Me3SiCN to form dicyanoboranes 3A–C and re-form compound 4.
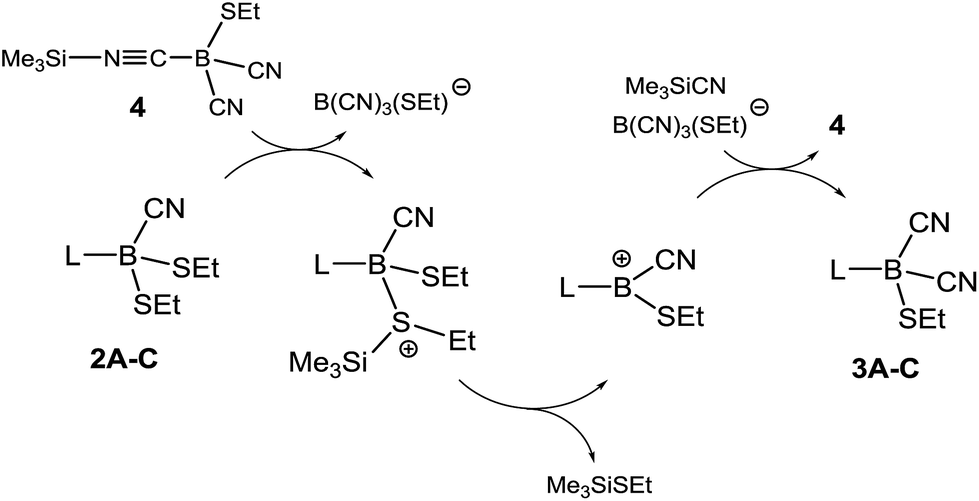 | ||
| Scheme 4 Proposed mechanism for the formation of dicyanoboranes 3A–C. L = carbene moiety A, B or C. | ||
The involvement of silylium ions was further investigated with trimethylsilyl perchlorate, Me3SiOClO3,25 which is a confirmed silylium transfer reagent. Indeed, analytically pure samples of 1A–C or 2A–C reacted with Me3SiCN (2.3 eq. or 1.3 eq., respectively) in the presence of Me3SiOClO3 (catalytic amounts) to afford dicyanoboranes 3A–C, which provides strong support for the involvement of silylium cations in the dicyanation step. We observed no indication of the introduction of a third cyano group to give the percyanated species L–B(CN)3.
In approaches towards boryl anions of type VIII, we considered the ethylthio moieties in 3A–C as conceivable reducible leaving groups (Scheme 5).
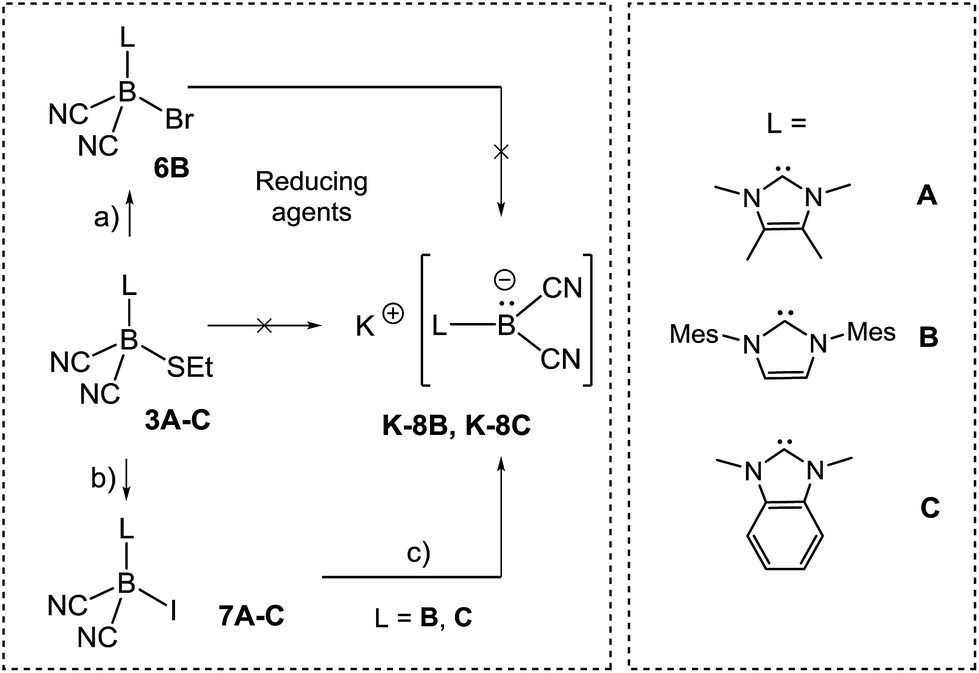 | ||
| Scheme 5 Reagents and conditions. (a) Br2, CH2Cl2, rt, 1 min; (b) MeI, adapted conditions 7A–C; (c) for K-8B: 6 eq. KC8, THF, rt, 20 min; for K-8C: 6 eq. KC8, DME, rt, 20 min; for K-8B or K-8C: 6 eq. K in NH3, −60 °C, 20 min. | ||
However, attempts to reduce 3A–C, e.g. with KC8 or NaC10H8, gave a mixture of several species as assessed by 11B{1H}-NMR monitoring. A more successful approach involved the introduction of halogen atoms and their subsequent removal by reducing agents. Compound 3B reacted with elemental bromine to give bromoborane 6B with an 11B{1H} chemical shift of δ(11B) = −29.3 ppm, ω1/2 = 38 Hz. The same compound 6B was formed from the parent hydroborane IMes–BH(CN)2 and bromine. Although B–Br bonds are expected to be labile, the reduction of bromoborane 6B did not afford the boryl anion of type VIII. With the intention of introducing better leaving groups, we found that dicyanoboranes 3A–C react with methyl iodide to give iodoboranes 7A–C. In contrast, the parent hydroborane IMes–BH(CN)2 did not react with elemental iodine to yield the iodinated compound 7B. The 11B{1H}-NMR spectra indicate a significant upfield shift of the signals with concomitant line broadening, e.g. δ(11B) = −24.8 ppm, ω1/2 = 3 Hz for 3Bvs. δ(11B) = −41.7 ppm, ω1/2 = 46 Hz for 7B, which is consistent with spin–orbit coupling effects of iodine showing normal halogen dependence (NHD-effect).26 Structural authentication of compounds 7A–C is provided by X-ray crystallographic analysis (Fig. 1). The iodoboranes 7A–C display the expected tetrahedral geometry at boron. The bond lengths B1–I decrease in the order 7A → 7B → 7C and correlate with the σ-donating properties of the carbene, which fall from 7A to 7C. Similarly, the bond angles I–B1–C1 increase systematically from 7A to 7C. The reduction of iodoboranes 7A–C was attempted with KC8 (in THF or DME) and gave compounds K–8B and K–8C in 80–85% yield. The reactions required an excess of KC8 (6 eq.), lower quantities led to unidentified side products as indicated by 11B{1H}-NMR spectroscopy. The alternative reduction of 7B or 7C with K/NH3 afforded samples of comparable yield and purity.
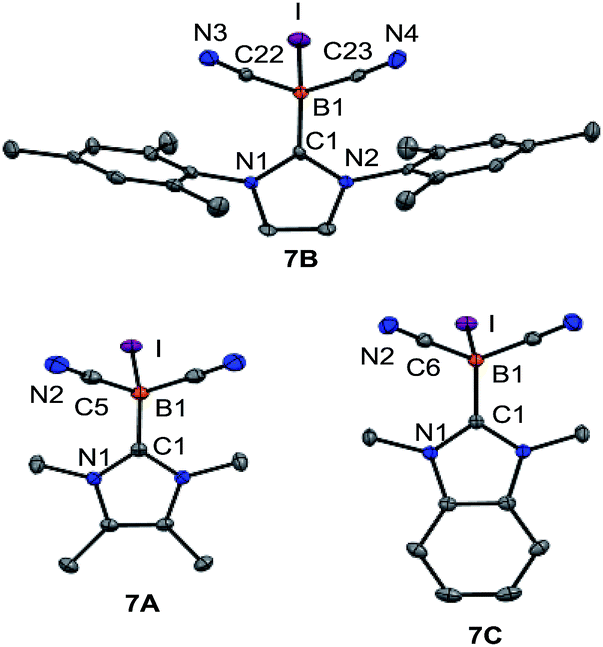 | ||
| Fig. 1 Molecular structures of iodoboranes 7A–C. Thermal ellipsoids are presented at 50% probability levels. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and bond angles (°). For 7A (CH2Cl2 omitted for clarity): I–B1 2.291(3), B1–C1 1.596(4), B1–C5 1.584(2), N2–C5 1.146(3), C1–B1–I 106.14(16), for 7B, with three molecules in the asymmetric unit, average values are: I–B1 2.281(3), B1–C1 1.607(3), B1–C22 1.586(3), B1–C23 1.589(3), N3–C22 1.143(3), N4–C23 1.144(3), C1–B1–I 106.81(14), for 7C: I–B1 2.271(2), B1–C1 1.605(3), B1–C6 1.584(2), N2–C6 1.146(2), C6–B1–I 108.35(10). | ||
In sharp contrast, the reduction of iodoborane 7A under the same conditions did not yield the expected compound K-8A. Samples recorded after the reduction were 11B-NMR silent, whereas the anions 8B and 8C give rise to signals at δ(11B) = −28.3 ppm and δ(11B) = −24.1 ppm, respectively. Crystals suitable for X-ray crystallography were obtained from a solution of K-8B in THF by slow diffusion of pentane (Fig. 2 and section ESI‡). Compound K-8B crystallised as a nonamer of bridged K[IMes–B(CN)2] units. Compound K-8C crystallised in the presence of 18-crown-6 to afford [K(18-cr-6)][BAC–B(CN)2], [K(18-cr-6)]-8C, (Fig. 2).
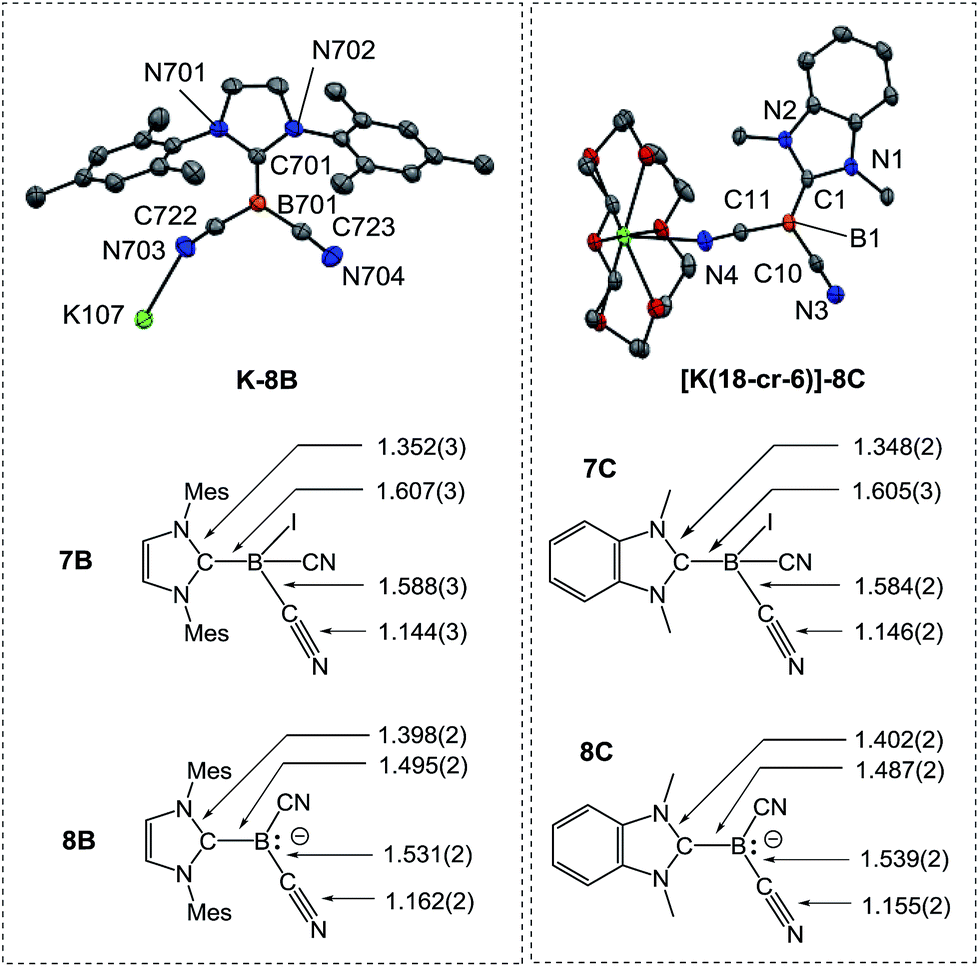 | ||
| Fig. 2 Molecular structures of compounds K-8B and [K(18-cr-6)]-8C. For K-8B a representative K[IMes–B(CN)2] unit of the nonamer is illustrated. [K(18-cr-6)]-8C crystallises as a bridged dimer located on a centre of inversion and only the symmetry independent unit is presented (for the dimer see section ESI‡). Thermal ellipsoids are presented at 50% probability levels. Hydrogen atoms omitted for clarity. The bond lengths in iodoboranes 7B, 7C and boryl anions 8B, 8C (reported in Å) represent average values calculated from each crystallographically independent molecule in the asymmetric unit. | ||
The boron centres in the anions feature trigonal planar geometry. All B–C bonds of the boryl anions are shortened compared to the iodoboranes, whereby the bonds to the carbene are most affected. A lengthening of the C–N bonds is observed for both the nitrile groups and carbenes. These observations indicate strong resonance stabilisation of the boron-centred lone pair with the pz-orbital at the carbene carbon atom and the π*-orbital of the nitrile groups. These resonance effects are also obvious in the IR-spectra, in which the CN stretch vibrations are red-shifted, e.g. 2207 cm−1 in 7Bvs. 2090 cm−1 and 2123 cm−1 in 8B. Theoretical calculations performed at the B3LYP-D3/TZVP level reliably reproduced both the bond lengths of the boryl anions 8B, 8C and the IR-spectra with respect to position and relative intensity of the CN stretch vibrations (section ESI‡).
For a systematic investigation we calculated frontier orbitals of the reported boryl anions VI and VII and compared them with our novel anions of type VIII. As expected, the HOMOs of the boryl anions are essentially boron-centred and display significant delocalisation into nitrile groups or carbene moieties (where applicable, section ESI‡). The energy levels of the HOMOs show a systematic increase in correlation with the falling π-acceptor properties of the substituents L in the anions L–B(CN)2n−, L = carbene or CN− (Fig. 3). In particular, the anions 8B and 8C occupy a central position between the reported anions VI and VII. Interestingly, the attempted anion 8A shows no peculiarities of its orbitals, and the failure to synthesize it must be of kinetic rather than thermodynamic origin. The decreasing π-acceptor character of the substituent L in the anions gives rise to a concomitant decrease of the bond order B–C between the boron centre and the substituent L, which is also consistent with increasingly negative Mulliken charges at boron. The nucleophilic activity of a Lewis base can be correlated with the energy level of its lone pair. In view of the Klopman–Salem-concept27 and in consideration of the structural similarity of the anions L–B(CN)2− (boron as the nucleophilic centre, similar orbital shapes) the nucleophilicity can be expected to rise with increasing HOMO-energy. The distinct colours [K-8C – bright orange (λmax < 400 nm) vs.K-8B – deep red (shoulder, λmax = 480 nm)] are readily apparent from the lower HOMO–LUMO gap in 8B. Detailed excited-state calculations reveal a HOMO → LUMO+1 transition (fosc = 0.028, λmax = 480 nm) in 8B, which represents a charge-transfer from the essentially boron-centred lone-pair towards the asymmetric π*-orbitals of the mesityl substituent; the further red-shifted HOMO → LUMO transition is substantially less intense (fosc = 0.005, λmax = 518 nm). In contrast, the HOMO → LUMO transition in 8C occurs from the lone pair into π*-orbitals of the phenylene moieties and is calculated to be below 400 nm (section ESI‡).
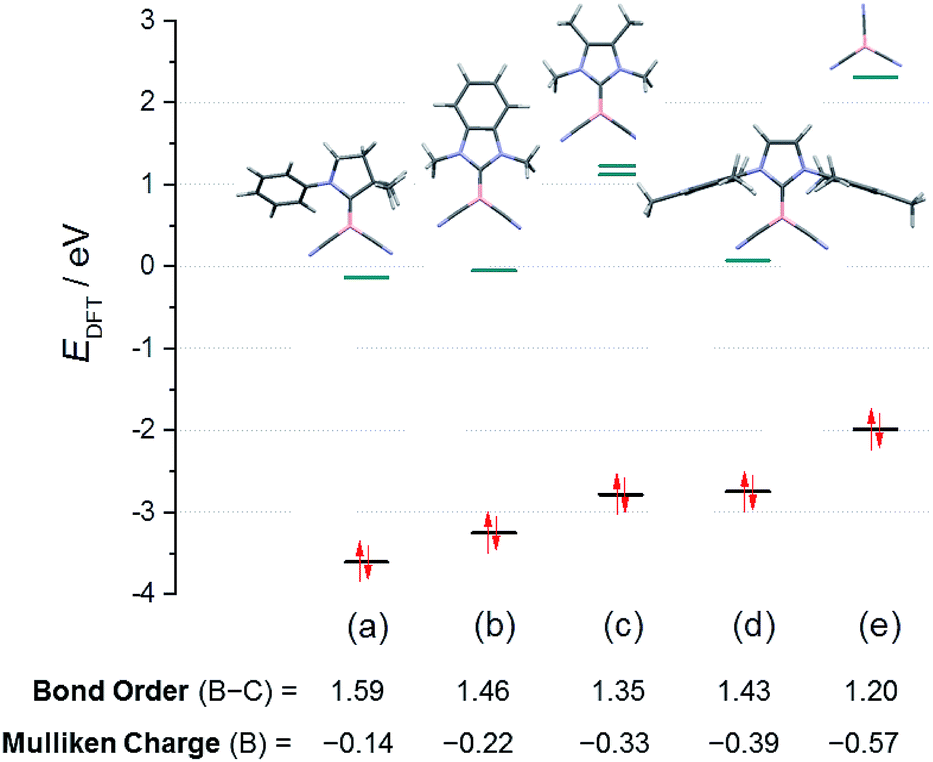 | ||
| Fig. 3 Energy levels of the frontier orbitals in the boryl anion series L–B(CN)2−, L = carbene or CN−: (a) truncated anion VI; (b) 8C; (c) hypothetical 8A; (d) 8B; (e) anion VII. | ||
The reactivity of the boryl anions 8B and 8C was probed with electrophiles, revealing a boron-centred nucleophilicity in both cases (Scheme 6). Thus, the reaction of K-8B with methyl iodide or gold electrophiles afforded the methylated species 9 or the gold boryl complex 10, giving rise to signals at δ(11B) = −27.6 or −29.7 ppm, respectively. Structural characterisation by X-ray crystallography was performed for 9 (section ESI‡), but no suitable crystals of 10 could be obtained. The identity of 10 is, among other data, unambiguously confirmed by the coupling of the 11B with the 31P nucleus, i.e.2J(31P–11B) = 65 Hz observed in both 11B{1H} and 31P{1H}-NMR spectra. Only three examples of gold boryl complexes are currently known28 but due to the lack of X-ray crystallographic analysis structural comparison cannot be drawn. The steric congestion by the carbene IMes in 8B prevented simple reactions with bulkier electrophiles including Me3ECl (E = Si, Sn). Reactions of the sterically less crowded 8C with main group electrophiles (including bulkier representatives) cleanly afforded boron-substituted products 11–14, which were characterised by X-ray crystallography except for 12 (section ESI‡). Compound 13 shows a characteristic signal of δ(11B) = −35.9 ppm in the 11B{1H}-NMR spectrum accompanied by well resolved tin satellites 1J(117Sn–11B) = 325 Hz, 1J(119Sn–11B) = 338 Hz, while 14 resonates at δ(11B) = −29.6 ppm as a doublet, 1J(31P–11B) = 24 Hz.
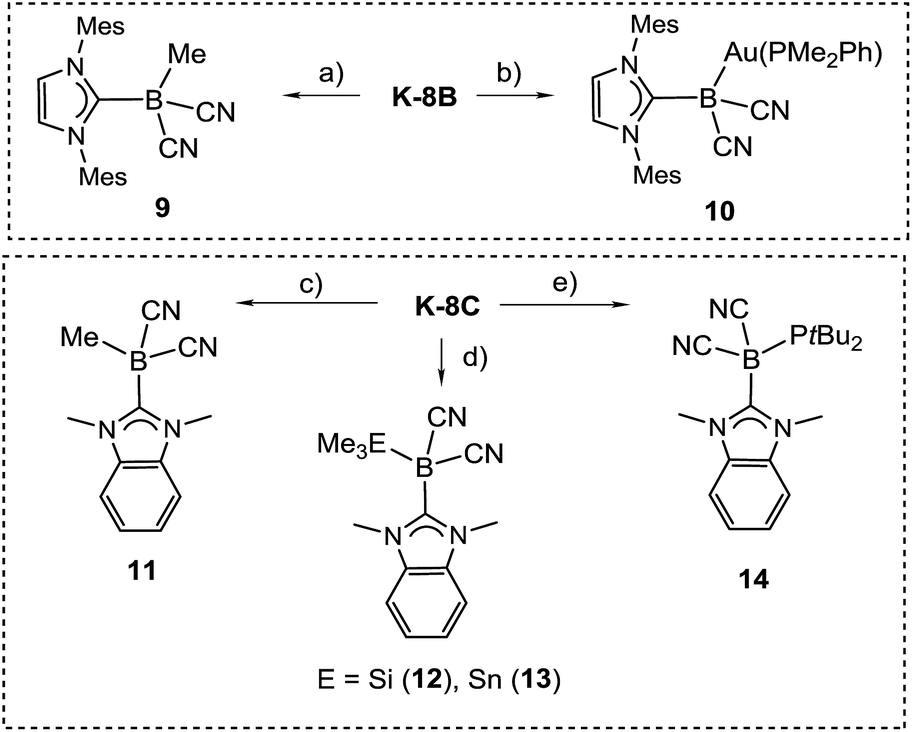 | ||
| Scheme 6 Reactions indicating the nucleophilic behaviour of boryl anions 8B and 8C. Reagents and conditions. (a) MeI, THF, rt, 10 min; (b) AuCl(PMe2Ph), THF, rt, 2 h; (c) MeI, THF, rt, 10 min; (d) Me3ECl, THF, 15 min, rt; (e) ClP(tBu)2, DME, 1 h, rt. | ||
Conclusions
The boryl anions of type NHC–B(CN)2− described herein complete a consistent series with the known anions cAAC–B(CN)2− (VI) and B(CN)32− (VII). Since N-heterocyclic carbenes are a thoroughly studied ligand class, their incorporation into NHC–B(CN)2−-systems essentially gives rise to the full scope of usual advantages, including a systematic variation of steric and electronic parameters, and in particular careful control of the nucleophilic properties at the boron centre. The novel approach towards NHC-stabilised cyanoboranes employs alkylthio-cyano exchange at boron and cleanly affords the mono- or dicyanated products [NHC–B(CN)(SEt)2 or NHC–B(CN)2SEt] while avoiding the isomeric isonitriles. The dicyanation step was shown to be silylium-catalysed. Facile iodination of dicyanated boranes to give NHC–B(CN)2I was shown to occur with methyl iodide. Only the iodoboranes were able to afford two novel boryl anions of type NHC–B(CN)2− upon reduction, while other conceivable leaving groups – e.g. Br, SEt – were ineffective. Crystal structures and DFT calculations suggest a boron-centred lone pair, which is resonance-stabilised by π-acidic NHC and CN substituents. The energy level of the chiefly boron-centred HOMO, and thus the nucleophilicity, can be controlled by the π-acidity of the carbene. The species NHC–B(CN)2− showed distinct boron-centred nucleophilicity with facile formation of B–E bonds, where E = C, Si, Sn, P, Au. Future investigations will concentrate on the preparation of further examples and the exploitation in salt metathesis reactions to form M–B bonds, M = metal or metalloid.Acknowledgements
This work was generously supported by the Fonds der Che-mischen Industrie (FCI). R. B. and R. F. are kindly indebted for their scholarships and financial support. G. H. acknowledges support by the Deutsche Forschungsgemeinschaft (SFB 658, Elementary processes in molecular switches at surfaces).Notes and references
- J. Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, Wiley-VCH, New York, 2nd edn, 1997 Search PubMed; H. C. Brown and P. V. Ramachandran, in Reductions in Organic Synthesis. Recent Advances and Practical Applications, ed. A. F. Abdel-Magid, American Chemical Society, Washington DC, 1996, ACS Symposium Series, vol. 641, pp. 1−30 Search PubMed.
- D. Stephan, Angew. Chem., Int. Ed., 2017, 56, 5984–5992 ( Angew. Chem. , 2017 , 129 , 6078–6086 ) CrossRef CAS PubMed.
- (n-Bu)2B−: claimed by R. W. Auten and C. A. Kraus, J. Am. Chem. Soc., 1952, 74, 3398–3401 CrossRef CAS , refuted by K. Smith and K. Swaminathan, J. Chem. Soc., Dalton Trans., 1976, 2297–2300; Ph2B−: claimed by J. L. R. Williams, J. C. Doty, P. J. Grisdale, R. Searle, T. H. Regan, G. P. Happ and D. P. Maier, J. Am. Chem. Soc., 1967, 89, 5153–5157 CrossRef , refuted by J. D. Wilkey and G. B. Schuster, J. Org. Chem., 1987, 52, 2117–2122.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed; Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef PubMed.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed; D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837–7839 RSC; L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280–9283 ( Angew. Chem. , 2014 , 126 , 9434–9437 ) CrossRef PubMed; D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef PubMed; L. Kong, R. Ganguly, Y. Li and R. Kinjo, Chem. Sci., 2015, 6, 2893–2902 RSC; B. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef PubMed; H. Braunschweig, R. D. Dewhurst, L. Pentecost, K. Radacki, A. Vargas and Q. Ye, Angew. Chem., Int. Ed., 2016, 55, 436–440 ( Angew. Chem. , 2016 , 128 , 447–451 ) CrossRef PubMed.
- Key metals or metalloids include Mg: M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570–9571 CrossRef CAS PubMed ; Mg, Cu, Zn: M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377–1392 CrossRef ; Al: N. Dettenrieder, H. M. Dietrich, C. Schädle, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2012, 51, 4461–4465 ( Angew. Chem. , 2012 , 124 , 4537–4541 ) CrossRef PubMed ; Si, Ge, Sn: A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef PubMed ; Pb, Cd, Hg: A. V. Protchenko, D. Dange, A. D. Schwarz, C. Y. Tang, N. Phillips, P. Mountford, C. Jones and S. Aldridge, Chem. Commun., 2014, 50, 3841–3844 RSC ; Ti, Hf: T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162–14163 CrossRef PubMed ; Mn, Fe, Co, Re: R. Frank, J. Howell, R. Tirfoin, D. Dange, C. Jones, D. M. P. Mingos and S. Aldridge, J. Am. Chem. Soc., 2014, 136, 15730–15741 CrossRef PubMed ; Co: R. Frank, J. Howell, J. Campos, R. Tirfoin, N. Phillips, S. Zahn, D. M. P. Mingos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 9586–9590 ( Angew. Chem. , 2015 , 127 , 9722–9726 ) CrossRef PubMed.
- The strong σ-donating effect of boryl ligands has been recognised recently: J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed.
- A. V. Protchenko, D. Dange, J. R. Harmer, C. Y. Tang, A. D. Schwarz, M. J. Kelly, N. Phillips, R. Tirfoin, K. H. Birjkumar, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Nat. Chem., 2014, 6, 315–319 CrossRef CAS PubMed.
- A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022–1026 CrossRef CAS PubMed.
- W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS PubMed.
- H. Braunschweig, M. Burzler, R. D. Dewhurst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5650–5653 ( Angew. Chem. , 2008 , 120 , 5732–5735 ) CrossRef CAS PubMed.
- T. Imamoto and T. Hikosaka, J. Org. Chem., 1994, 59, 6753–6759 CrossRef CAS.
- J. Monot, A. Solovyev, H. Bonin-Dubarle, E. Derat, D. P. Curran, M. Robert, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2010, 49, 9166–9169 ( Angew. Chem. , 2010 , 122 , 9352–9355 ) CrossRef CAS PubMed.
- H. Braunschweig, C.-W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041–2044 ( Angew. Chem. , 2010 , 122 , 2085–2088 ) CrossRef CAS PubMed; R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 ( Angew. Chem. , 2014 , 126 , 5557–5561 ) CrossRef PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590–7592 ( Angew. Chem. , 2013 , 125 , 7739–7742 ) CrossRef CAS PubMed.
- E. Bernhardt, V. Bernhardt-Pitchougina, H. Willner and N. V. Ignatiev, Angew. Chem., Int. Ed., 2011, 50, 12085–12088 ( Angew. Chem. , 2011 , 123 , 12291–12294 ) CrossRef CAS PubMed; J. Landmann, J. A. P. Sprenger, R. Bertermann, N. Ignat'ev, V. Bernhardt-Pitchougina, E. Bernhardt, H. Willner and M. Finze, Chem. Commun., 2015, 51, 4989–4992 RSC.
- J. Landmann, F. Keppner, D. B. Hofmann, J. A. P. Sprenger, M. Häring, S. H. Zottnick, K. Müller-Buschbaum, N. V. Ignat'ev and M. Finze, Angew. Chem., Int. Ed., 2017, 56, 2795–2799 CrossRef CAS PubMed.
- S. Díez-Gonzales, N-Heterocyclic carbenes, from laboratory curiosities to efficient synthetic tools, RSC Publishing, 2011 Search PubMed; S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, Wiley VCH, 2014 Search PubMed.
- M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 ( Angew. Chem. , 2010 , 122 , 8992–9032 ) CrossRef CAS PubMed.
- A. Solovyev, Q. Chu, S. J. Geib, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 15072–15080 CrossRef CAS PubMed.
- Y. Liu, P. E. Lindner and D. M. Lemal, J. Am. Chem. Soc., 1999, 121, 10626–10627 CrossRef CAS.
- H. Nöth and U. Schuchardt, Chem. Ber., 1974, 107, 3104–3112 CrossRef.
- D. Williams, B. Pleune, J. Kouvetakis, M. D. Williams and R. A. Andersen, J. Am. Chem. Soc., 2000, 122, 7735–7741 CrossRef CAS . A further example of an isonitrile–borane adduct of type Me3SiNC–B(CN)3 is reported: E. Bernhardt, M. Berkei, H. Willner and M. Schürmann, Z. Anorg. Allg. Chem., 2003, 629, 677–685 CrossRef.
- T. Müller, Struct. Bonding, 2014, 155, 107–162 CrossRef.
- U. Wannagat, F. Brandmair, W. Liehr and H. Niederprüm, Z. Anorg. Allg. Chem., 1950, 302, 185–198 CrossRef CAS; U. Wannagat and W. Liehr, Angew. Chem., 1957, 69, 783–784 Search PubMed.
- M. Kaupp, O. L. Malkina, V. G. Malkin and P. Pyykkö, Chem.–Eur. J., 1998, 4, 118–126 CrossRef CAS.
- G. Klopmann, J. Am. Chem. Soc., 1968, 90, 223–225 CrossRef CAS; L. Salem, J. Am. Chem. Soc., 1968, 90, 543–552 CrossRef; L. Salem, J. Am. Chem. Soc., 1968, 90, 553–556 CrossRef.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 ( Angew. Chem. , 2007 , 119 , 6830–6833 ) CrossRef CAS PubMed ; ref. 15; W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef PubMed.
Footnotes |
| † Dedicated to Prof. Dr Evamarie Hey-Hawkins on the occasion of her 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, full characterisation, crystallographic data and computational details. CCDC 1550754 (1B), 1550755 (2B), 1550760 (7B), 1550767 (K-8B), 1550769 (9), 1550740 (1C), 1550756 (2C), 1550774 (7C), 1550768 {[K(18-cr-6)]-8C}, 1550770 (11), 1550771 (13), 1550772 (14), 1550757 (3A), 1550759 (7A), 1550758 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02238g |
| This journal is © The Royal Society of Chemistry 2017 |