
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitro-enabled catalytic enantioselective formal umpolung alkenylation of β-ketoesters†
Abhijnan Ray
Choudhury‡
 ,
Madhu Sudan
Manna‡
and
Santanu
Mukherjee
*
,
Madhu Sudan
Manna‡
and
Santanu
Mukherjee
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: sm@orgchem.iisc.ernet.in; Fax: +91-80-2360-0529; Tel: +91-80-2293-2850
First published on 2nd August 2017
Abstract
A formal umpolung strategy is presented for the enantioselective installation of an alkenyl group with a terminal double bond at a tertiary center. This one-pot two-step sequence relies on the unique features of the nitro group, which after inverting the polarity of the alkenylating agent toward the desired bond formation, itself serves as a leaving group. The application of this protocol to cyclic β-ketoesters results in densely functionalized products, bearing an all-carbon quaternary stereocenter including an alkenyl substituent with a terminal double bond, in high yields with excellent enantioselectivities.
Introduction
An all-carbon quaternary stereogenic center bearing an alkenyl substituent is a structural motif found in various families of natural products and bioactive molecules (Fig. 1).1 While enantioselective construction of all-carbon quaternary stereocenters is itself considered challenging,2 those bearing an alkenyl substituent present an additional level of difficulty due to the lack of available synthetic methods for the installation of alkenyl groups at sterically hindered positions.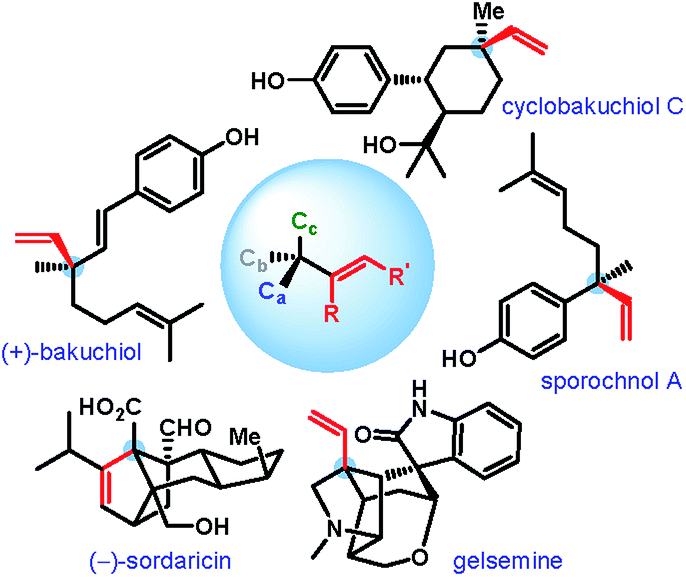 | ||
| Fig. 1 Natural products with alkenyl-bearing all-carbon quaternary stereocenters. | ||
Driven by these challenges, the past decade has witnessed the emergence of a number of strategies for the catalytic enantioselective α-alkenylation of carbonyl and related compounds.3–5 However, the majority of these reactions lead to the products bearing the alkenyl group with an internal double bond (Scheme 1A).3,4 In contrast, enantioselective installation of an alkenyl group with a terminal double bond is far less explored5 and calls for innovative strategies.
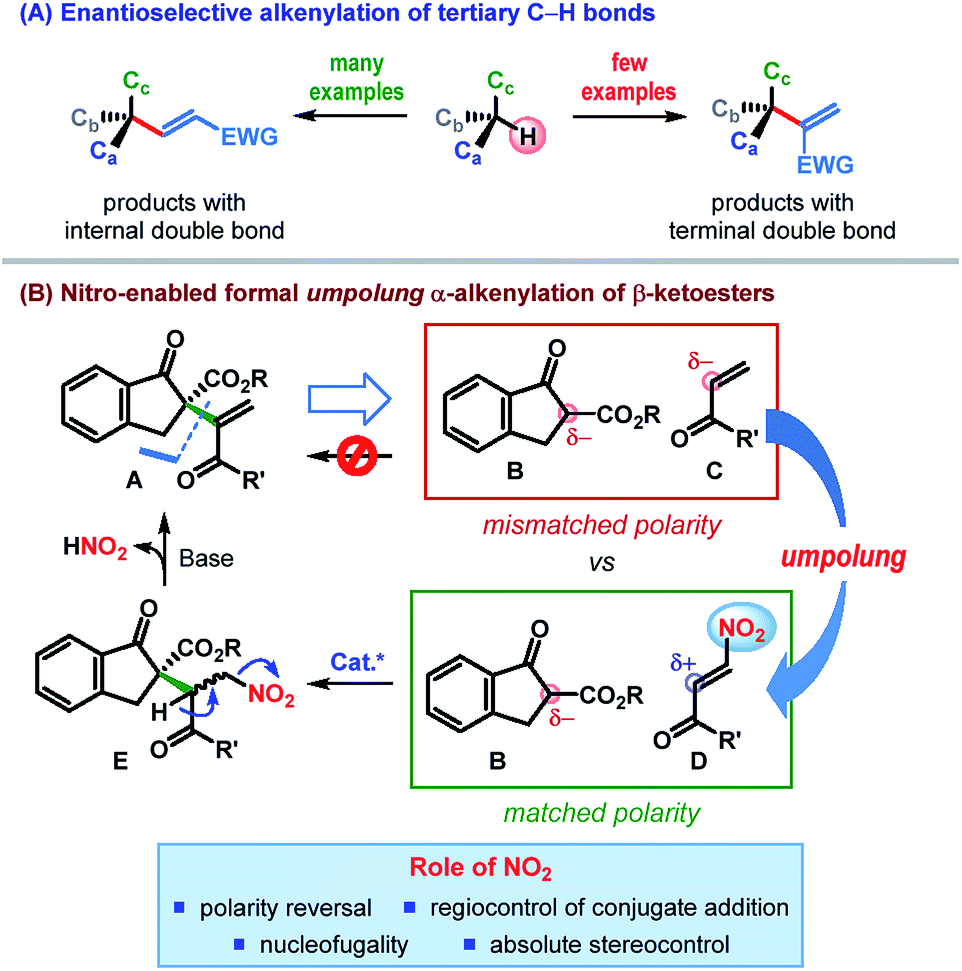 | ||
| Scheme 1 Enantioselective construction of alkenyl-containing all-carbon quaternary stereocenters. | ||
In an effort to address this issue, we contemplated the development of a catalytic enantioselective α-alkenylation of β-ketoesters6 – a reaction which would furnish a product bearing an all-carbon quaternary stereogenic center. The conventional retrosynthetic analysis of a potential product of type A, bearing an alkenyl group with a terminal double bond, leads to a pair of synthons (B & C) having “mismatched polarity”, thereby making such a process electronically improbable (Scheme 1B). Consequently, polarity reversal (umpolung)7 of one of these synthons would be essential to effect this C–C bond formation.
Recent investigations by our laboratory and others have established the nitro functional group as a powerful directing as well as leaving group.8 We reasoned that appending enone C with a nitro group at its β-position would reverse its naturally nucleophilic α-position to an electrophilic center (Scheme 1B). The desired C–C bond formation between the resulting β-nitroenone D and the β-ketoester B would then be electronically feasible (“matched polarity”). In fact, owing to the hydrogen bonding acceptor ability of the nitro group, the conjugate addition could be catalyzed regioselectively with the use of a suitable H-bond donor. Control of the stereochemistry (by the catalyst) at the prochiral center would lead, in the worst case, to an inconsequential mixture of diastereomers (E). Nucleofugality of the nitro group along with the presence of a suitably placed acidic proton in E should eventually facilitate the elimination of nitrous acid, leaving behind an all-carbon quaternary stereogenic center bearing an alkenyl group with a terminal double bond. Apart from the nucleofugality of the nitro group, this strategy is built on the ability of the nitro group to invert the natural polarity of enone (umpolung) as well as to control the regio- and stereoselectivity of the conjugate addition.9
Results and discussion
To ascertain the viability of our postulated formal umpolung alkenylation reaction, we began our study with a β-ketoester derived from 1-indanone (1) as the pronucleophile and phenyl β-nitroenone 2a as the alkenylating agent (Table 1). Based on their proven efficacy in enantioselective transformations of nitro compounds, tertiary amino-(thio)urea derivatives were selected as the potential catalyst candidates.10 Initial screening of ester substituents on 1 revealed the t-Bu group to be optimal, with respect to the diastereoselectivity of the initial conjugate addition step.11 However, irrespective of the ester substituent, Michael addition was found to take place with exquisite regioselectivity to furnish 3 as the sole product. The optimization of the catalyst and reaction conditions was then undertaken for the reaction between tert-butyl β-ketoester 1a and 2a. The undesired background reaction operating at 25 °C (entry 1) was eliminated by conducting the reaction at −60 °C (entry 2). Solvent screening with dihydroquinine-derived thiourea I as the catalyst established 2-MeTHF as the preferred reaction medium.11 Under these conditions, the Michael adduct 3aa was formed in 88% yield with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. (entry 3). As anticipated, treatment of 3aa with DBU in THF at ambient temperature resulted in the desired α-alkenylation product 4aa, bearing an all-carbon quaternary stereocenter, in 85% yield with 94.5:5.5 er. The related (thio)urea derivatives II–V failed to improve the enantioselectivity (entries 4–7). Replacing the 3,5-bis(trifluoromethyl)phenyl group of thiourea in I with 3,5-bis(trifluoromethyl)benzyl resulted in a poorly enantioselective catalyst VI (entry 8). However, catalyst VII containing a cyclohexyl moiety was found not only to be more active, but also furnished the product 4aa with excellent er (entry 9). The elimination step was found to be considerably slower when conducting the two steps in a one-pot manner in 2-MeTHF (entry 10), although it did not influence the enantioselectivity. Finally, a solvent switch (to THF) prior to elimination proved beneficial and provided 4aa with improved yield and identical er (entry 11). Attempts to further improve the enantioselectivity through additives or by tweaking other reaction parameters proved futile.11
:1 dr. (entry 3). As anticipated, treatment of 3aa with DBU in THF at ambient temperature resulted in the desired α-alkenylation product 4aa, bearing an all-carbon quaternary stereocenter, in 85% yield with 94.5:5.5 er. The related (thio)urea derivatives II–V failed to improve the enantioselectivity (entries 4–7). Replacing the 3,5-bis(trifluoromethyl)phenyl group of thiourea in I with 3,5-bis(trifluoromethyl)benzyl resulted in a poorly enantioselective catalyst VI (entry 8). However, catalyst VII containing a cyclohexyl moiety was found not only to be more active, but also furnished the product 4aa with excellent er (entry 9). The elimination step was found to be considerably slower when conducting the two steps in a one-pot manner in 2-MeTHF (entry 10), although it did not influence the enantioselectivity. Finally, a solvent switch (to THF) prior to elimination proved beneficial and provided 4aa with improved yield and identical er (entry 11). Attempts to further improve the enantioselectivity through additives or by tweaking other reaction parameters proved futile.11
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | t [h] | Yield [%] of 3aab | Yield [%] of 4aab | er of 4aac |
| a Reactions were carried out on a 0.1 mmol scale using 1.0 equiv. of 1a and 1.2 equiv. of 2a. b Yields correspond to the isolated product. c The enantiomeric ratio (er) was determined by HPLC analysis. d Reaction at 25 °C. e Using 2.0 equiv. of DBU. f One-pot reaction in 2-MeTHF. Time required for the 2nd step was 40 h. g One-pot reaction with solvent switch. | |||||
| 1d | None | 6 | 90 | 90 | — |
| 2 | None | 48 | <5 | — | — |
| 3 | I | 48 | 88 | 85 | 94.5:5.5 |
| 4 | II | 46 | 95 | 87 | 94:6 |
| 5 | III | 48 | >99 | 86 | 92:8 |
| 6 | IV | 48 | 90 | 86 | 92:8 |
| 7 | V | 48 | 90 | 85 | 93:7 |
| 8 | VI | 48 | >99 | 88 | 70:30 |
| 9 | VII | 24 | >99 | 88 | 97.5:2.5 |
| 10e,f | VII | 24 | — | 48 | 97.5:2.5 |
| 11e,g | VII | 24 | — | 91 | 97.5:2.5 |
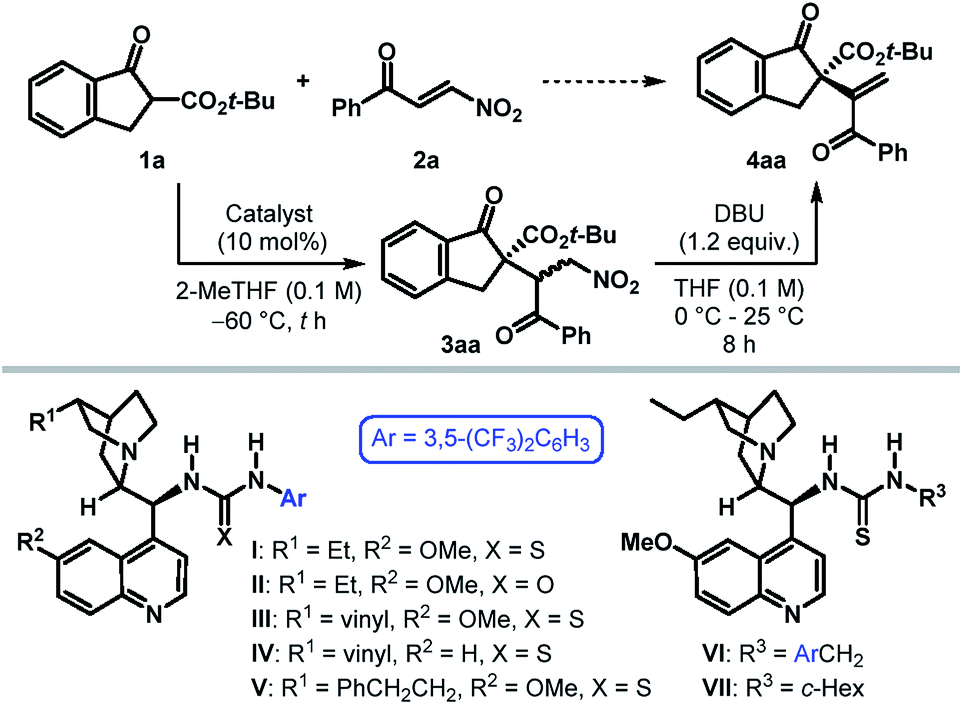
Table 2 summarizes the scope and limitations of this catalytic enantioselective formal umpolung alkenylation protocol under the optimized catalyst and reaction conditions (Table 1, entry 11). Our protocol was found to be operational with a variety of electronically tuned and sterically diverse β-nitroenones 2 (Table 2A). Different para- and meta-substituted aryl β-nitroenones (2b–h) could be employed to generate the desired alkenylated products 4ab–ah in high yields with excellent enantioselectivities (entries 2–8). Controlling the enantioselectivity of the reactions involving ortho-substituted aromatic substrates is generally a cause of concern in asymmetric synthesis. We were delighted to find our protocol to be equally suited for ortho-substituted aryl β-nitroenones (2i–j) as the products (4ai–aj) were formed with excellent enantioselectivities (entries 9–10). The same trend was observed for the products (4ak–ao) derived from β-nitroenones bearing naphthyl (2k–l), 2-thienyl (2m) and highly electron rich aryl substituents (2n–o) (entries 11–15). An excellent er prevailed even in the case of an alkyl-substituted β-nitroenone, although the product 4ap was obtained only with modest yield (entry 16). The formation of an internal olefin (in 22% yield) accounts for the diminished yield in this case.11
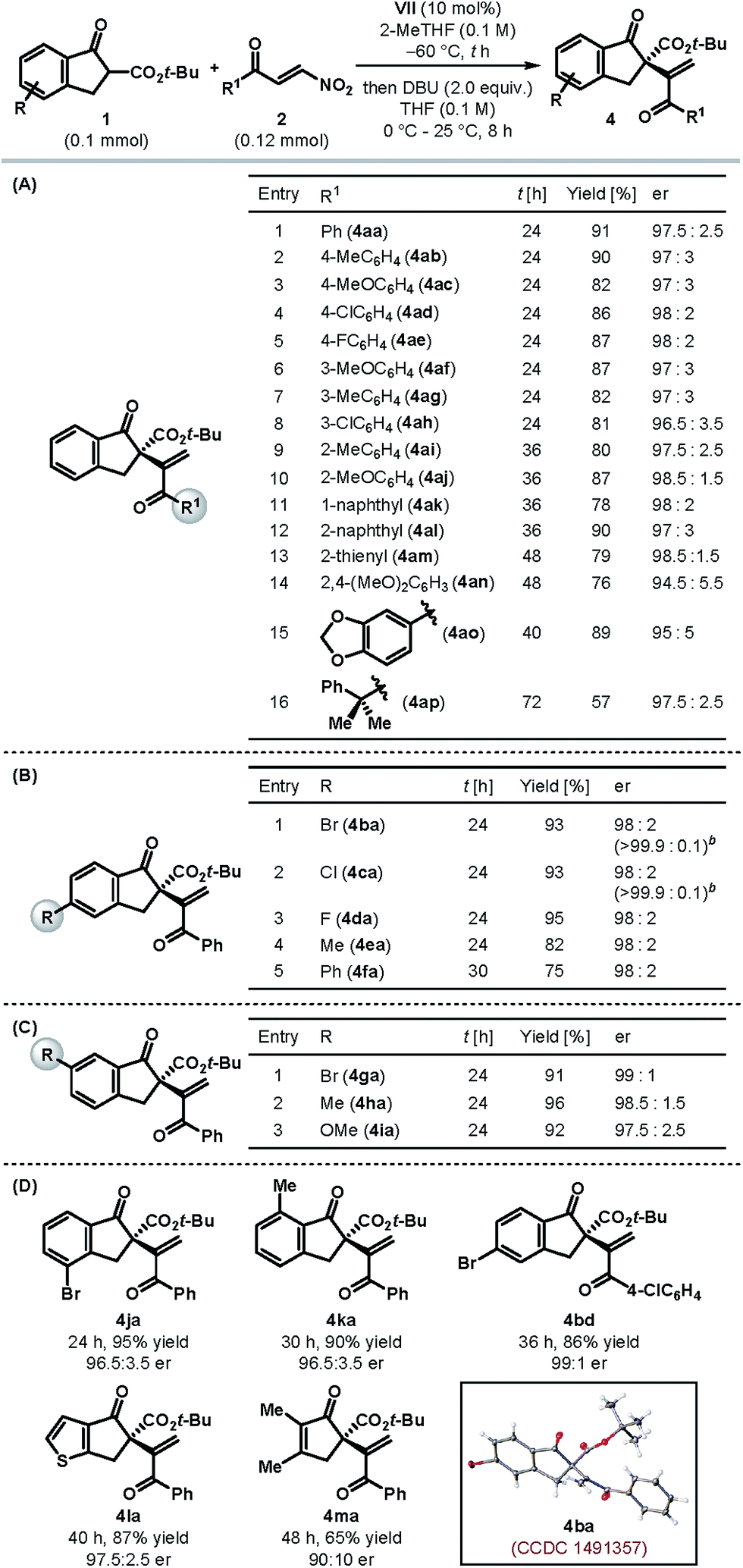
The scope of tert-butyl β-ketoesters with respect to the substituents on the aryl ring was also tested. As illustrated in Table 2B, various 5-substituted 1-indanone derived β-ketoesters (1b–f) were found to be suitable substrates and the products 4ba–fa were obtained generally in high yields with excellent enantioselectivities. Please note that the alkenylated products 4ba and 4ca could be obtained essentially in enantiopure form after a single recrystallization. Similarly, 6-substituted 1-indanone derived β-ketoesters (1g–i) resulted in the products 4ga–ia with superb er (Table 2C). Substituents on the 4- and 7-positions of 1-indanone, too, were tolerated and furnished the products 4ja and 4ka, respectively, in high yields and enantioselectivities (Table 2D). In fact, tert-butyl β-ketoesters derived from cyclopentenones (1l–m) were also found to be competent substrates and the desired products 4la–ma were obtained in high yields with good to excellent enantioselectivities. However, β-ketoesters derived from larger ring ketones (e.g. 1-tetralone and 1-benzosuberone) and acyclic ketones were found to be completely unreactive under our optimized reaction conditions,11 and hence mark the present limitation of our protocol.
To rule out the possibility of enantioenrichment of the products through self-disproportionation of enantiomers (SDE),12 we performed a SDE test through achiral chromatography on silica gel. No noticeable enantioenrichment/depletion was observed in this process, thereby confirming that the enantiomeric ratios reported in Tables 1 and 2 are a direct outcome of the catalytic reactions.
Single crystal X-ray diffraction analysis of the alkenylated product 4ba (Table 2D) enabled us to establish its absolute configuration, which was found to be (S).13 The configurations of the other products, shown in Table 2, were tentatively assigned by analogy as the same, assuming a similar catalytic mechanism was followed.
To demonstrate the scalability of our protocol, a reaction between tert-butyl β-ketoester 1a and phenyl β-nitroenone 2a was carried out on a 1.0 mmol scale with only 5 mol% of catalyst VII (Scheme 2). Albeit with a longer reaction time, 4aa was obtained in nearly quantitative yield with the same level of enantioselectivity as that obtained with the small scale reaction. Moreover, catalyst recovery was possible with 77% yield.
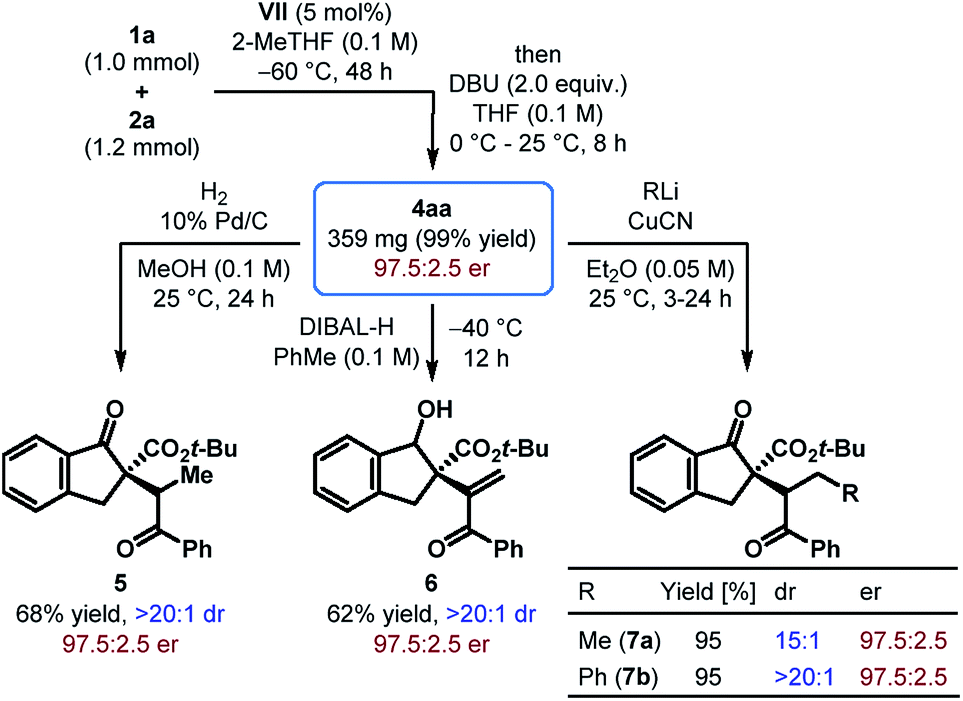 | ||
| Scheme 2 Scalability of the reaction and synthetic elaboration of the alkenylation product. | ||
The utility of the products obtained through the enantioselective formal umpolung alkenylation reaction was displayed through a few synthetic modifications. For example, hydrogenation of 4aa under Pd-catalysis led to the reduction of the alkene and furnished 5 as a single diastereomer in 68% yield (Scheme 2). Chemo- and diastereoselective reduction of the cyclic ketone in 4aa over the enone functionality was achieved with DIBAL-H and the alcohol 6 was formed in 62% yield. The Cu(I)-mediated addition of organolithium reagents to 4aa proceeded regioselectively in a 1,4-fashion and the resulting products 7a–b were generated in excellent yields with good to outstanding dr. In all these reactions, the products were obtained without compromising the stereochemical integrity of the starting material.
Conclusions
In conclusion, the intricate task of installing an alkenyl group with a terminal double bond enantioselectively at a tertiary center has been accomplished through the reaction between two seemingly polarity mismatched coupling partners. The result is a highly efficient and enantioselective formal umpolung α-alkenylation of cyclic β-ketoesters. This reaction proceeds through a one-pot two-step sequence consisting of a catalytic regio-, diastereo- and enantioselective conjugate addition to β-nitroenones followed by a base mediated nitrous acid elimination. Catalyzed by a dihydroquinine-based bifunctional tertiary aminothiourea derivative, this process delivers synthetically versatile products bearing an all-carbon quaternary stereocenter including an alkenyl substituent with a terminal double bond in high yields with excellent enantioselectivities. We expect this alkenylation strategy to be applicable to other classes of substrate as well. These possibilities are currently under investigation in our laboratory.Acknowledgements
Financial support from the Science and Engineering Research Board (SERB) [Grant No. SB/S1/OC-63/2013], Council of Scientific and Industrial Research (CSIR) [Grant No. 02(0207)/14/EMR-II] and DAE-BRNS [Grant No. 2013/37C/56/BRNS/2440] is gratefully acknowledged. A. R. C. (a Shyama Prasad Mukherjee Fellow) and M. S. M. thank the CSIR for the doctoral fellowships. We sincerely thank Prodip Howlader (IPC, IISc, Bangalore) for his help with the X-ray structure analysis.Notes and references
- (a) M. Büschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156–4186 CrossRef PubMed; (b) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed.
- (a) F. Vetica, R. M. de Figueiredo, M. Orsini, D. Tofani and T. Gasperi, Synthesis, 2015, 47, 2139–2184 CrossRef CAS; (b) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed; (c) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC; (d) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS; (e) Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005 Search PubMed.
- For α-alkenylation of unbranched aldehydes, see: (a) Y. Li, L. Ibsen and K. A. Jørgensen, Org. Lett., 2017, 19, 1200–1203 CrossRef CAS PubMed; (b) T. Kano, H. Sugimoto, H. Maruyama and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 8462–8465 CrossRef CAS PubMed; (c) J. M. Stevens and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11756–11759 CrossRef CAS PubMed; (d) E. Skucas and D. W. C. MacMillan, J. Am. Chem. Soc., 2012, 134, 9090–9093 CrossRef CAS PubMed; (e) H. Kim and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 398–399 CrossRef CAS PubMed.
- α-Alkenylation for constructing all-carbon quaternary stereocenters bearing an internal alkene: (a) J. Guo, L. Lin, Y. Liu, X. Li, X. Liu and X. Feng, Org. Lett., 2016, 18, 5540–5543 CrossRef CAS PubMed; (b) Q. Lan, X. Wang, S. Shirakawa and K. Maruoka, Org. Process Res. Dev., 2010, 14, 684–686 CrossRef CAS; (c) A. M. Taylor, R. A. Altman and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 9900–9901 CrossRef CAS PubMed; (d) Z. Chen, M. Furutachi, Y. Kato, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 2218–2220 CrossRef CAS PubMed; (e) P. Elsner, L. Bernardi, G. D. Salla, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2008, 130, 4897–4905 CrossRef CAS PubMed; (f) M. Bell, T. B. Poulsen and K. A. Jørgensen, J. Org. Chem., 2007, 72, 3053–3056 CrossRef CAS PubMed; (g) T. B. Poulsen, L. Bernardi, M. Bell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 6551–6554 CrossRef CAS PubMed; (h) M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 5672–5673 CrossRef CAS PubMed; (i) A. Chieffi, K. Kamikawa, J. Åhman, J. M. Fox and S. L. Buchwald, Org. Lett., 2001, 3, 1897–1900 CrossRef CAS PubMed.
- α-Alkenylation for constructing all-carbon quaternary stereocenters bearing a terminal alkene: (a) J. Alemán, E. Reyes, B. Richter, J. Overgaard and K. A. Jørgensen, Chem. Commun., 2007, 3921–3923 RSC; (b) B. K. Corkey and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17168–17169 CrossRef CAS PubMed. For a chiral auxiliary-based approach, see: (c) T. Fujimoto, K. Endo, H. Tsuji, M. Nakamura and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 4492–4496 CrossRef CAS PubMed . For a few isolated examples, see: ref. 4c and e.
- Selected reactions of β-ketoesters leading to all-carbon quaternary stereocenters: (a) M. Chen, Z.-T. Huang and Q.-Y. Zheng, Org. Biomol. Chem., 2015, 13, 8812–8816 RSC; (b) Q.-H. Deng, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2012, 134, 10769–10772 CrossRef CAS PubMed; (c) D. B. Ramachary, R. Madhavachary and M. S. Prasad, Org. Biomol. Chem., 2012, 10, 5825–5829 RSC; (d) S. Sternativo, A. Calandriello, F. Costantino, L. Testaferri, M. Tiecco and F. Marini, Angew. Chem., Int. Ed., 2011, 50, 9382–9385 CrossRef CAS PubMed; (e) T. A. Moss, D. R. Fenwick and D. J. Dixon, J. Am. Chem. Soc., 2008, 130, 10076–10077 CrossRef CAS PubMed; (f) J. Alemán, B. Richter and K. A. Jørgensen, Angew. Chem., Int. Ed., 2007, 46, 5515–5519 CrossRef PubMed; (g) T. B. Poulsen, L. Bernardi, J. Alemán, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2007, 129, 441–449 CrossRef CAS PubMed; (h) F. Wu, H. Li, R. Hong and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 947–950 CrossRef CAS PubMed. For a review, see: (i) T. Govender, P. I. Arvidsson, G. E. M. Maguire, H. G. Kruger and T. Naicker, Chem. Rev., 2016, 116, 9375–9437 CrossRef CAS PubMed.
- D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239–258 CrossRef.
- (a) B. A. Vara and J. N. Johnston, J. Am. Chem. Soc., 2016, 138, 13794–13797 CrossRef CAS PubMed; (b) R. Sarkar and S. Mukherjee, Org. Lett., 2016, 18, 6160–6163 CrossRef CAS PubMed; (c) Y. Li, F. Tur, R. P. Nielsen, H. Jiang, F. Jensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2016, 55, 1020–1024 CrossRef CAS PubMed; (d) L. Cerisoli, M. Lombardo, C. Trombini and A. Quintavalla, Chem.–Eur. J., 2016, 22, 3865–3872 CrossRef CAS PubMed; (e) M. S. Manna and S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 130–133 CrossRef CAS PubMed; (f) C. Gianelli, R. Lopez, Á. Puente, M. Zalacain and C. Palomo, Eur. J. Org. Chem., 2012, 2774–2779 CrossRef CAS; (g) R. López, M. Zalacain and C. Palomo, Chem.–Eur. J., 2011, 17, 2450–2457 CrossRef PubMed; (h) D. Seebach, R. Henning and T. Mukhopadhyay, Chem. Ber., 1982, 115, 1705–1720 CrossRef CAS . Also see ref. 3a.
- N. Ono, in The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 Search PubMed.
- For pioneering examples, see: (a) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367–6370 CrossRef CAS PubMed; (b) J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481–4483 RSC; (c) B. Vakulya, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS PubMed; (d) T. Okino, S. Nakamura, T. Furukawa and Y. Takemoto, Org. Lett., 2004, 6, 625–627 CrossRef CAS PubMed; (e) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed . For reviews, see: ; (f) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229–1279 CrossRef CAS; (g) Y. Takemoto, Chem. Pharm. Bull., 2010, 58, 593–601 CrossRef CAS PubMed; (h) S. J. Connon, Chem. Commun., 2008, 2499–2510 RSC.
- See the ESI† for details.
- V. A. Soloshonok, C. Roussel, O. Kitagawad and A. E. Sorochinsky, Chem. Soc. Rev., 2012, 41, 4180–4188 RSC.
- CCDC 1491357† contains the crystallographic data for 4ba.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and analytical data. CCDC 1491357 (4ba). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02232h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |