
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid reversible borane to boryl hydride exchange by metal shuttling on the carborane cluster surface†
Bennett J.
Eleazer
a,
Mark D.
Smith
a,
Alexey A.
Popov
 *b and
Dmitry V.
Peryshkov
*a
*b and
Dmitry V.
Peryshkov
*a
aDepartment of Chemistry and Biochemistry, University of South Carolina, 631 Sumter St., Columbia, South Carolina 29208, USA. E-mail: peryshkov@sc.edu
bLeibniz Institute for Solid State and Materials Research, Helmholtzstrasse 20, 01069 Dresden, Germany. E-mail: A.Popov@ifw-dresden.de
First published on 25th May 2017
Abstract
In this work, we introduce a novel concept of a borane group vicinal to a metal boryl bond acting as a supporting hemilabile ligand in exohedrally metalated three-dimensional carborane clusters. The (POBOP)Ru(Cl)(PPh3) pincer complex (POBOP = 1,7-OP(i-Pr)2-m-2-carboranyl) features extreme distortion of the two-center-two-electron Ru–B bond due to the presence of a strong three-center-two-electron B–H⋯Ru vicinal interaction. Replacement of the chloride ligand with a hydride afforded the (POBOP)Ru(H)(PPh3) pincer complex, which possesses B–Ru, B–H⋯Ru, and Ru–H bonds. This complex was found to exhibit a rapid exchange between hydrogen atoms of the borane and the terminal hydride through metal center shuttling between two boron atoms of the carborane cage. This exchange process, which involves sequential cleavage and formation of strong covalent metal–boron and metal–hydrogen bonds, is unexpectedly facile at temperatures above −50 °C corresponding to an activation barrier of 12.2 kcal mol−1. Theoretical calculations suggested two equally probable pathways for the exchange process through formally Ru(0) or Ru(IV) intermediates, respectively. The presence of this hemilabile vicinal B–H⋯Ru interaction in (POBOP)Ru(H)(PPh3) was found to stabilize a latent coordination site at the metal center promoting efficient catalytic transfer dehydrogenation of cyclooctane under nitrogen and air at 170 °C.
Introduction
Multidentate ligands containing a heteroatom backbone have attracted significant recent attention in ligand design and catalysis. Boron-based ligand systems demonstrate versatility in the possible bonding interactions to a transition metal center as a neutral borane as a Z- or an L-type ligand, an anionic borate as an L-type ligand, or a central anionic boryl moiety as an X-type ligand.1–4 Interconversion between these coordination modes has been demonstrated to provide an additional reactivity manifold through metal–ligand cooperation. Pincer boryl ligands with diazaborole, diarylborane, and carborane fragments have been recently introduced incorporating strongly electron-donating boryl donors in tridentate meridional coordination frameworks (Chart 1).5–19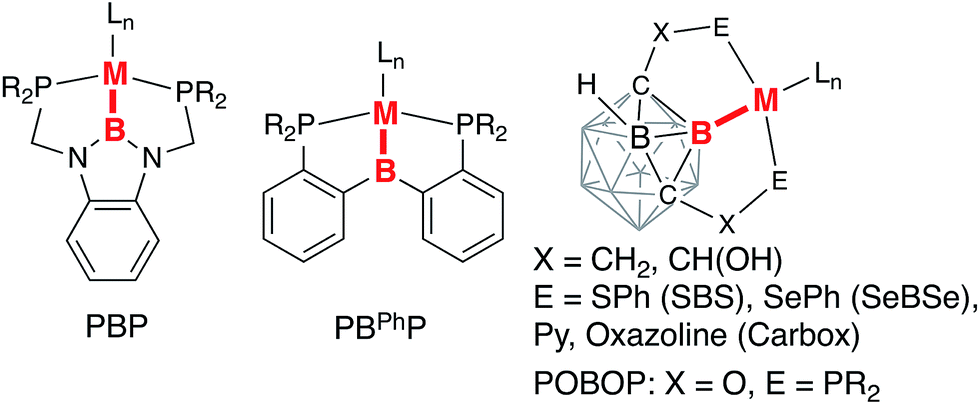 | ||
| Chart 1 Examples of boryl-based pincer complexes. | ||
Polyhedral boron clusters, such as icosahedral C2B10H12 carborane cages, are chemically robust molecular species possessing unique electronic properties and increased steric bulk and often are considered as inorganic three-dimensional “pseudoaromatic” analogs of arenes.20–23 Carboranes have been shown to be promising molecular building blocks for potential application in metal–organic frameworks, organomimetic architectures, luminescent materials, batteries, liquid crystals, coordination chemistry and catalysis.24–36
Icosahedral carborane clusters represent an unusual 3-D ligand framework where one metalated boron vertex of the cage is surrounded by multiple vicinal B–H bonds. These B–H bonds can serve as hemilabile neutral ligands to the metal center and, in principle, can themselves be activated. Recently, we demonstrated that utilization of the carborane cage backbone in the pincer-type chelating ligand POBOP (POBOP = 1,7-OP(i-Pr)2-m-2-carboranyl) led to the close simultaneous contact of the coordinated metal center and two cluster vertices. The double B–H bond activation at adjacent boron vertices by a single ruthenium center resulted in the formation of the first example of a (BB)–carboryne complex with a highly strained three-membered BB > Ru metallacycle.37
The functionalization of boron clusters by B–H bond activation represents an attractive synthetic strategy.38–41 The use of directing groups on a boron cage has been demonstrated to lead to metal-promoted derivatization of neighboring boron vertices.42–44 Regioselectivity of B–H bond activation is highly desired given the high number of potential isomers for an exo-substituted icosahedral cage. In this work, we probed the B–H bond activation process in detail and experimentally assessed the possibility of intramolecular interconversion between the coordinated borane B–H⋯Ru and the metal boryl hydride B–Ru–H interactions or, in other words, reversibility of B–H bond activation and the possibility of migration of the metal center on the carborane cluster surface (Chart 2). The use of the strained three-dimensional POBOP carboranyl pincer framework allowed us to enforce an unprecedented simultaneous coordination of the ruthenium metal center with a terminal hydride ligand to the boron cluster through the metal–boryl bond and vicinal metal–borane bond. Furthermore, the reactivity of this unusual (POBOP)Ru(H)(PPh3) boryl hydride complex featuring a latent open coordination site was probed in reactions with H2, D2, and in catalytic dehydrogenation of cyclooctane.
 | ||
| Chart 2 The pendulum clock-type fluxional behavior of the (POBOP)Ru(H)(PPh3) complex resulting from the rapid interchange between the B–Ru–H boryl hydride and the B–H⋯Ru coordinated borane. | ||
Results and discussion
Synthesis and structure of (POBOP)Ru(Cl)(PPh3)
The reaction of the ligand precursor (POBOP)H (1) and Ru(PPh3)3Cl2 in THF at reflux temperature led to the B–H bond activation of the ligand and the formation of the B-carboranyl pincer complex (POBOP)Ru(Cl)(PPh3) (2, Scheme 1). The product was isolated by recrystallization from an ether/hexanes mixture at −30 °C in high yield (86%) as an orange powder.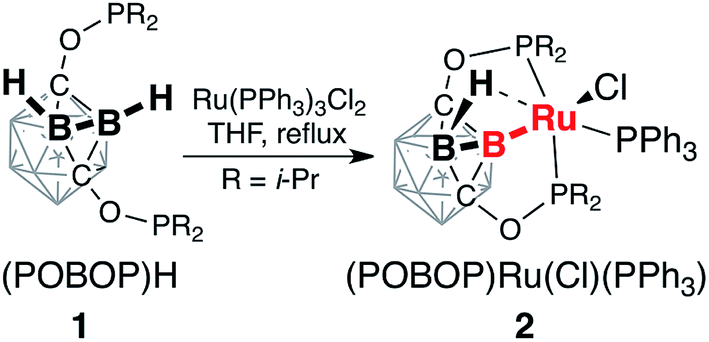 | ||
| Scheme 1 Synthesis of (POBOP)Ru(Cl)(PPh3) (2). | ||
The single crystal X-ray structure of 2 revealed a surprisingly high degree of bond strain in the boron–ruthenium bond imposed by the stabilizing effect of the vicinal B–H⋯Ru interaction and ligand geometry (Fig. 1a and b). The Ru1–B1 bond length is 2.086(2) Å, which is comparable to a two-center-two-electron (2c-2e) Ru–B bond length in previously reported ruthenium diazaborolyl PBP pincer complexes (2.022(4)–2.080(14) Å).2,7–9 The short Ru1⋯B2 (2.417(2) Å) and Ru⋯H2 (1.96(2) Å) distances are indicative of a strong bridging B–H⋯Ru interaction. This apparently strong 3c-2e bond attracts the metal center to the B2 vertex, and, as a consequence, results in a significant distortion of the vicinal 2c-2e Ru1–B1 bond. The B2–B1–Ru1 angle is 77.4(1)°, which deviates drastically from an unstrained exohedral bond angle of 120° for an idealized icosahedral cluster. For comparison, the corresponding exohedral B2–B1–H1 angle in the ligand precursor (POBOP)H is 116.1(9)°. Previously reported 2-B-metalated m-carborane complexes exhibit significantly larger values of the analogous B2–B1–M angle in the range from 104.5(1)° to 120(1)°, with the only exception being the ruthenium BB–carboryne complex previously synthesized in our group featuring two adjacent 2c-2e metal–boron bonds.13–16,37,45 Thus, the presence of the strong B–H⋯Ru interaction in 2 led to the unprecedented distortion of the vicinal metal boryl bond. The coordination geometry of the ruthenium center is, therefore, octahedral, with the borane B–H bond serving as one of the ligands. Triphenylphosphine is located trans- to the coordinated B–H bond with the H2⋯Ru1–P3 angle of 176.5(5)°.
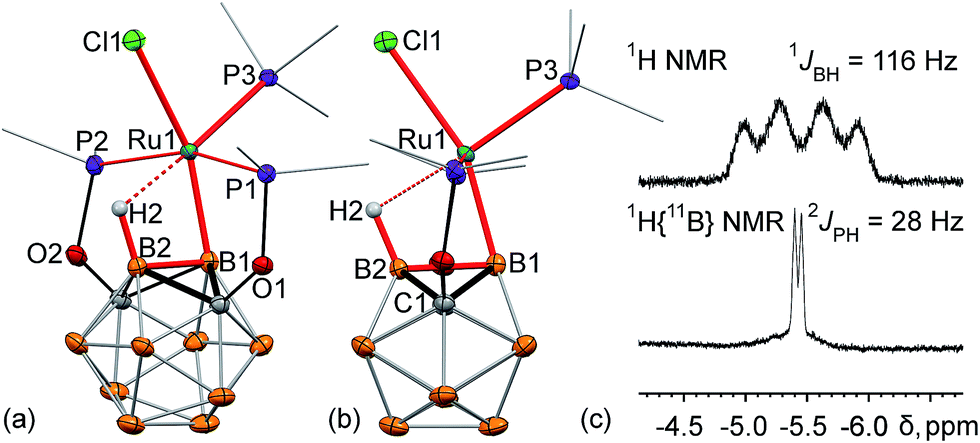 | ||
| Fig. 1 (a, b) Displacement ellipsoid plots (50% probability) of (POBOP)Ru(Cl)(PPh3) (POBOP = 1,7-OP(i-Pr)2-2-dehydro-m-carboranyl) (2). (a): a general view (b): a view perpendicular to the (B2–B1–Ru1) plane. Atoms belonging to isopropyl groups of the ligand arms and phenyl rings of triphenylphosphine have been omitted for clarity. Hydrogen atoms of the boron cluster, except for H2 are not shown. Selected bond distances (Å) and angles (°): Ru1–B1 = 2.086(2), Ru1⋯B2 = 2.417(2), Ru⋯H2 = 1.96(2), Ru1–Cl1 = 2.501(1), B2–B1–Ru1 = 77.4(1), B1–Ru1–Cl1 = 159.6(1), and H2⋯Ru1–P3 = 176.5(5). (c) Fragments of 1H and 1H{11B} NMR spectra of 2 featuring a signal from the bridging hydride B2–H1⋯Ru1 and its coupling to B2 and P3 atom nuclei, respectively. | ||
The strong bridging B–H⋯Ru interaction is persistent in solution as manifested by the characteristic broadened 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 quartet at −5.4 ppm in the 1H NMR spectrum of 2 in C6D6 (Fig. 1c). The 1:1:1:1 splitting pattern is indicative of the coupling of a proton to a 11B nucleus. This signal is shifted considerably upfield in comparison to the remaining hydrogen atoms of the carborane cluster which are represented by a set of overlapping signals in the range from +4 to +1 ppm. In the boron-decoupled 1H{11B} NMR spectrum, this quartet signal (1JBH = 116 Hz) converted into a doublet due to coupling to the 31P nucleus of the triphenylphosphine ligand (2JPH = 28 Hz) coordinated to the metal center, which is another indication of the strong bridging B–H⋯Ru interaction.
:1:1:1 quartet at −5.4 ppm in the 1H NMR spectrum of 2 in C6D6 (Fig. 1c). The 1:1:1:1 splitting pattern is indicative of the coupling of a proton to a 11B nucleus. This signal is shifted considerably upfield in comparison to the remaining hydrogen atoms of the carborane cluster which are represented by a set of overlapping signals in the range from +4 to +1 ppm. In the boron-decoupled 1H{11B} NMR spectrum, this quartet signal (1JBH = 116 Hz) converted into a doublet due to coupling to the 31P nucleus of the triphenylphosphine ligand (2JPH = 28 Hz) coordinated to the metal center, which is another indication of the strong bridging B–H⋯Ru interaction.
Persistent bridging 3c-2e B–H⋯M interactions have been often observed between cationic metal complexes and anionic heteroboranes that can be considered as cation–anion pairs.46–49 For example, the nido-carborane anions with pendant donor groups have been shown to exhibit strong binding to a ruthenium center with signals corresponding to B–H⋯Ru coordination in 1H NMR spectra ranging from −2 ppm (a larger contribution from a B–H extreme form) to −18 ppm (a larger contribution from a Ru–H extreme form).50–52 The intramolecular combination of interactions observed in the complex 2 where the metal center is simultaneously bound to one boron atom of a neutral carborane cluster with a 2c-2e B–Ru bond and to the adjacent boron–hydrogen bond of the same cluster with a 3c-2e B–H⋯Ru bond has not been observed prior to this work. Notably, formation of intermediates of this type may account for rare cases of isomerization of B-metalated boron clusters at high temperatures, where a metal center changes its position on the heteroborane cage.53 Furthermore, the H2-mediated Ru–C to Ru–B bond conversion has been reported for cyclopentadienylcarboranyl complexes, which likely proceeds through intermediates similar to 2.54 To probe the possibility of such “cage walking” by the metal center, we synthesized the ruthenium carboranyl hydride complex (POBOP)Ru(H)(PPh3) in the reaction of the carboranyl chloride complex 2 and a hydride source.
Synthesis and structure of (POBOP)Ru(H)(PPh3)
Reaction of (POBOP)Ru(Cl)(PPh3) and NaH in THF at reflux temperature for 36 h resulted in the clean formation of a single product according to 31P NMR spectral data (Scheme 2). The product was crystallized from diethyl ether as a pale yellow solid in 98% yield. The crystal structure determination revealed an unusual coordination geometry of the ruthenium hydride complex (POBOP)Ru(H)(PPh3) (3) (Fig. 2). Two crystallographically independent but chemically identical molecules were found. The most striking feature of the molecular structure of 3 is the extreme ligand-induced strain of the B–Ru bond as demonstrated by the acute exohedral B2–B1–Ru1 angle of 69.4(2)°. This extreme bond strain results in the remarkable closeness of the covalent 2c-2e boryl B1–Ru1 bond length (2.208(3) Å) and the vicinal 3c-2e coordinated borane (H2)B2⋯Ru1 distance (2.276(3) Å). Importantly, the value of the exohedral B2–B1–Ru1 angle in 3 is the smallest for 2c-2e M–B bonds in any icosahedral carborane complex reported to date with the only exception the ruthenium BB–carboryne complex (B2–B1–Ru angle values 65.5(1)° and 68.4(1)°).37 The PPh3 ligand is located trans- to B–Ru bond with the B1–Ru1–P3 angle of 172.5(1)°. The hydride ligand H1 and the bridging borohydride H2 are located trans- to each other with a H1–Ru1⋯H2 angle of 177(1)°. The Ru1–H1 bond length in the crystal structure of 3 is 1.70(3) Å and the Ru1⋯H2 distance is 1.77(3) Å. These hydrogen atoms were clearly located using the electron density difference map. The metal center is, therefore, in the distorted octahedral environment.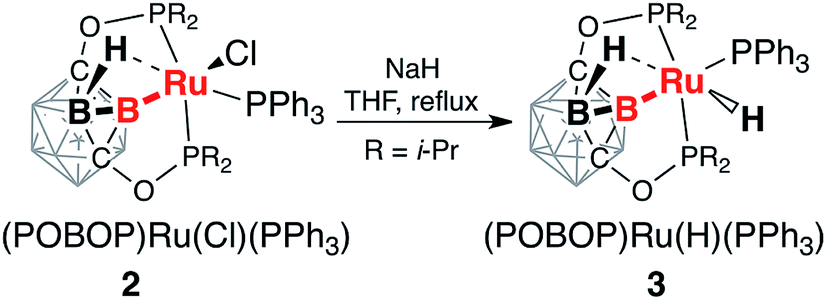 | ||
| Scheme 2 Synthesis of (POBOP)Ru(H)(PPh3) (3). | ||
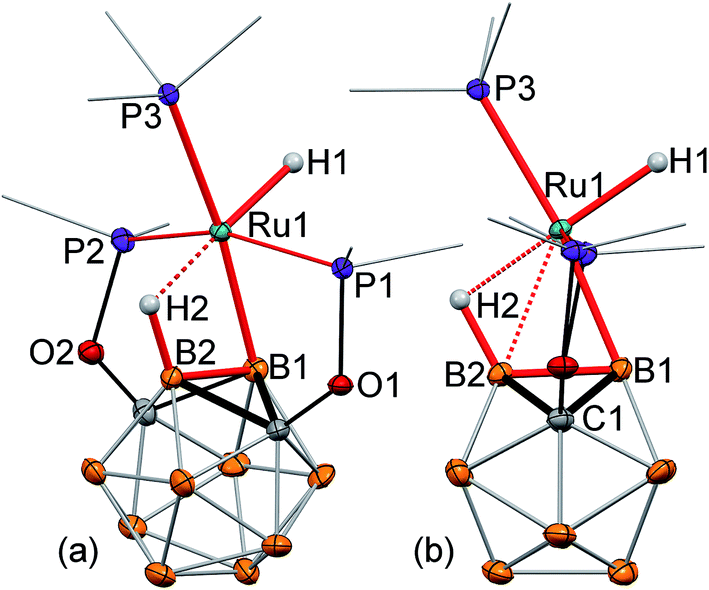 | ||
| Fig. 2 Displacement ellipsoid plots (50% probability) of (POBOP)Ru(H)(PPh3) (3). (a): a general view (b): a view perpendicular to the (B2–B1–Ru1) plane. Atoms belonging to isopropyl groups of the ligand arms and phenyl rings of triphenylphosphine have been omitted for clarity. Hydrogen atoms of the boron cluster, except for H2 are not shown. Selected bond distances (Å) and angles (°): Ru1–B1 = 2.208(3), Ru1⋯B2 = 2.276(3), Ru1–H1 = 1.70(3) Ru1⋯H2 = 1.77(3), Ru1–P3 = 2.343(1), B2–B1–Ru1 = 69.4(2), B1–B2–Ru1 = 65.2(2), B1–Ru1–P3 = 172.5(1), and H1–Ru1⋯H2 = 177(1). | ||
Dynamic behavior of (POBOP)Ru(H)(PPh3) and its variable temperature NMR spectra
The most prominent feature of the 1H NMR spectrum of 3 at room temperature was the presence of a broad signal at −8.8 ppm with an integral intensity corresponding to two hydrogen atoms in the complex (Fig. 3). At the same time, no other signals were found in the range from 0 ppm to −15 ppm. These observations suggested that the Ru–H hydride and the Ru⋯H–B bridging borohydride group in (POBOP)Ru(H)(PPh3) undergo a rapid exchange at room temperature. This exchange process is likely intramolecular due to significant steric hindrance of the POBOP pincer ligand with diisopropylphosphinite arms that preclude bimolecular interactions between complexes. To probe whether dissociation of the PPh3 ligand is responsible for the observed dynamic behavior, an excess of a smaller cone angle, more electron-rich phosphine, PEt3, was added to the solution of (POBOP)Ru(H)(PPh3) in C6D6. No replacement of the coordinated PPh3 in the complex by PEt3 was observed at room temperature for 16 h, suggesting that the dissociation of PPh3 is slow and unlikely to be responsible for the observed rapid borane/boryl hydride exchange (see ESI† for details).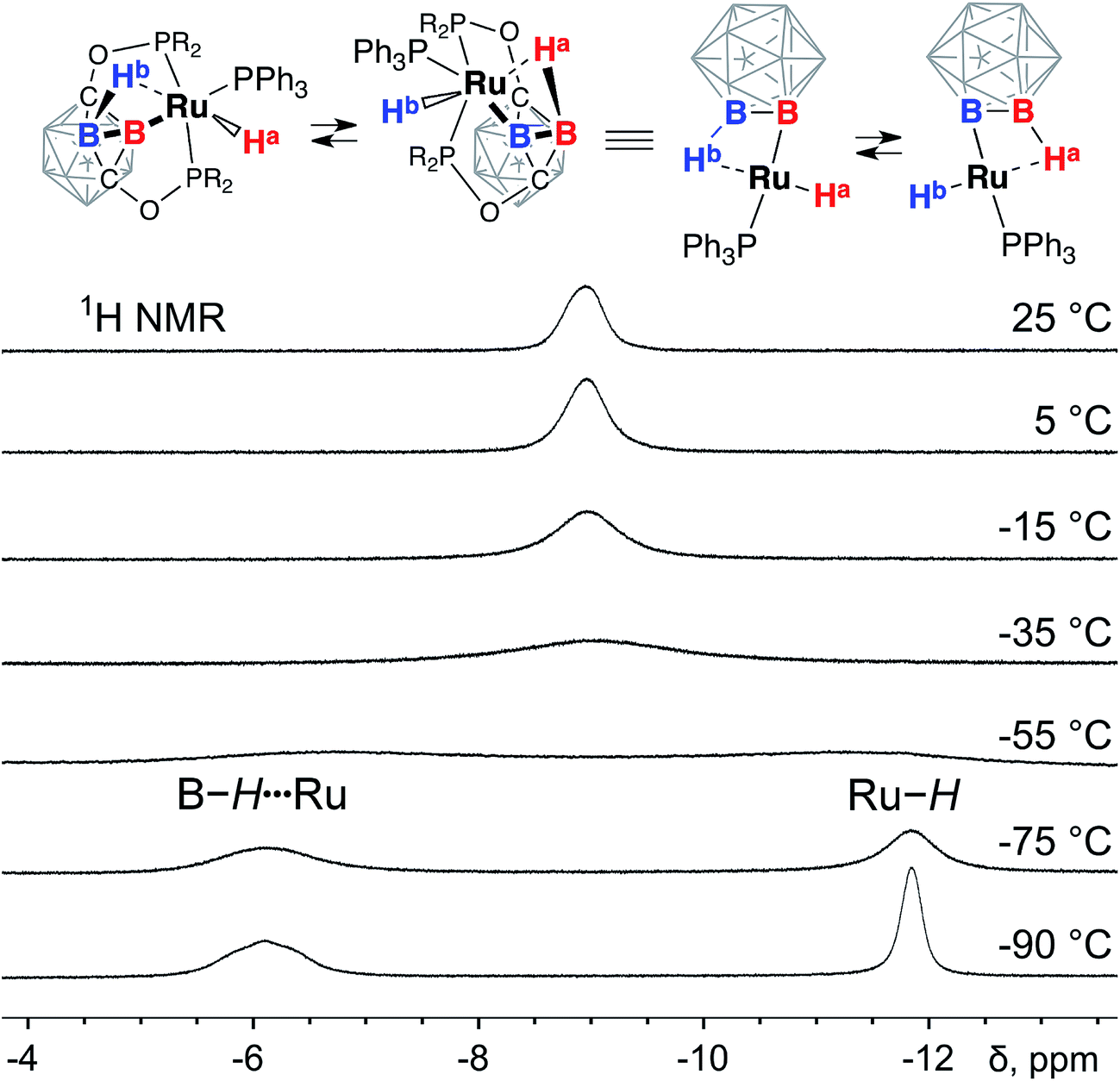 | ||
| Fig. 3 The fragments of 1H NMR spectra of (POBOP)Ru(H)(PPh3) in CD2Cl2 recorded in the temperature range from 25 °C to −90 °C. | ||
The series of the 1H NMR spectra of 3 recorded at the temperature range from +22 °C to −90 °C shed light on the dynamics of the exchange process. Instead of one broad signal at −8.8 ppm at room temperature, two signals at −6.1 ppm and −11.8 ppm were observed at −90 °C. The signal at −6.1 ppm sharpened in the 1H{11B} NMR spectrum indicating its correspondence to the coordinated B–H⋯Ru borane while the signal at −11.8 ppm corresponded to the Ru–H hydride. The coalescence temperature for the exchange process was estimated to be close to −50 °C. Based on this value of the coalescence temperature (223 K), the activation energy ΔG‡ for the exchange transformation was determined to be 12.2 kcal mol−1.
The (POBOP)Ru(H)(PPh3) complex reacted with 1 atm D2 gas at room temperature in benzene solution leading to the formation of 3-d2 featuring selective deuteration and the formation of the B–D⋯Ru borane group and the Ru–D hydride. The 2H NMR spectrum of 3-d2 exhibited one signal at −8.9 ppm at room temperature. Analogously to 3, cooling the solution of 3-d2 in dichloromethane to −90 °C led to the appearance of two distinct signals in the 2H NMR spectrum: one at −6.3 ppm corresponding to the coordinated borane moiety and another at −12.1 ppm corresponding to the metal hydride (see ESI† for details). The activation energy ΔG‡ of the exchange process was determined to be 13.3 kcal mol−1 for 3-d2 based on the observed coalescence temperature of 223 K. The value of kinetic isotope effect for the B–H⋯Ru/Ru–H exchange was estimated to be 6.53 at 223 K.
The rapid intramolecular hydrogen atom exchange along with observation of the H/D exchange upon conversion of 3 to 3-d2 prompted us to explore the reaction of 3 with dihydrogen. Exposure of a degassed solution sample of 3 in C6D6 to 1 atm of H2 led to a partial (ca. 30%) conversion of 3 to new species 5 according to 31P NMR spectral data with the new set of signals at 241.5 ppm (2P, pincer ligand arms) and 42.8 ppm (1P, coordinated PPh3 ligand). Replacement of 1 atm of H2 with 1 atm of N2 led to the complete conversion of 5 back to 3. The 1H NMR spectrum of the mixture of 3 and 5 under dihydrogen atmosphere exhibited a new signal at −10.5 ppm (a sharp doublet of triplets, 1H, Ru–H), and a broadened signal at −4.8 ppm with an integral intensity corresponding to two hydrogen atoms that was assigned to the coordinated dihydrogen (Ru–H2) (Fig. 4). No correlations between these signals were observed in the 1H–1H NOESY NMR spectrum of the mixture of 3 and 5 suggesting the probable trans-mutual orientation of the hydride and dihydrogen ligands. Furthermore, no signals corresponding to bridging B–H⋯Ru interactions were observed, suggesting the replacement of the coordinated borane in 3 by the dihydrogen molecule in 5.
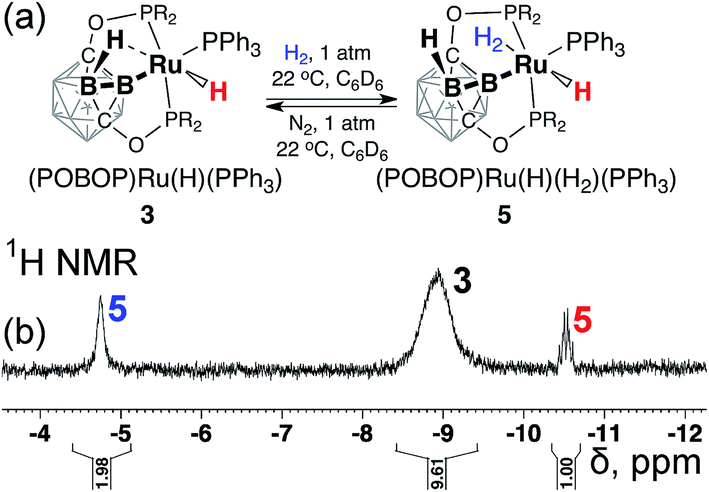 | ||
| Fig. 4 The fragment of the 1H NMR spectrum of the sample of (POBOP)Ru(H)(PPh3) (3) under H2 (1 atm) at room temperature. Signals from the proposed dihydrogen complex 5 (2H, br, −4.8 ppm and 1H, dt, −10.5 ppm) are highlighted. | ||
Proposed exchange mechanisms. Metal center “shuttling” on the cluster surface
One plausible sequence of steps responsible for the exchange between the hydride and borohydride groups in 3 (Scheme 3) may be the reductive elimination of the hydride ligand from the metal center with the formation of the B1–H1 bond and a formally Ru(0) diborane complex 4-1 followed by the oxidative addition of the B2–H2 bond with the formation of Ru1–H2 hydride and Ru1–B2 boryl, and the “pendulum”-like swing of the triphenylphosphine ligand. Another possible mechanism involves the oxidative addition of the B2–H2 bond with the formation of a formally Ru(IV) diboryl dihydride intermediate 4-2 followed by the reductive elimination of one hydride ligand from the metal center and the formation of the B–H bond coordinated to ruthenium ending with the “pendulum”-like swing of the triphenylphosphine ligand. Both these processes produce the same starting complex with the metal center moving from one boron atom to another on the carborane cage. The 11B and 11B{1H} NMR spectra at room temperature exhibited a characteristic singlet at −2 ppm corresponding to an integral intensity of two boron atoms while all other signals appeared as doublets in the 11B spectrum corresponding to the presence of B–H bonds thus suggesting the chemical exchange of the metalated boron atom and the coordinated vicinal borane B–Ru/B–H⋯Ru.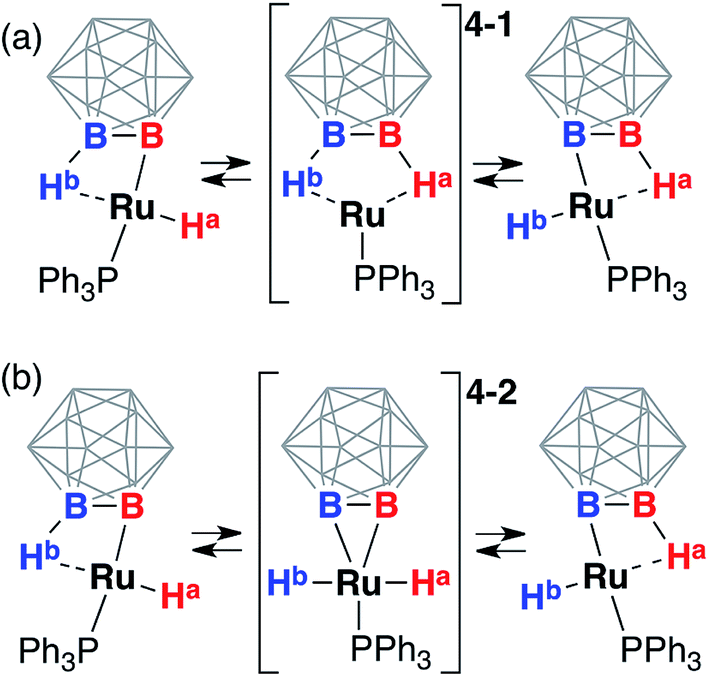 | ||
| Scheme 3 Possible reaction sequences responsible for the rapid exchange of the ruthenium hydride and coordinated borane groups in (POBOP)Ru(H)(PPh3). Chelating ligand arms are not shown for clarity. (a) The pathway involving the Ru(0) diborane complex 4-1 as an intermediate. (b) The pathway involving the Ru(IV) BB–carboryne dihydride complex 4-2 as an intermediate. Note “walking” of the metal center on the carborane cage in both cases. | ||
A relevant fluxional behavior has been observed by Heinekey and co-workers for the putative iridium(III) dihydride POCOP complex featuring the σ-coordinated borane HBPin.55 The exchange of hydrogen atoms of the metal hydride and the borane has been determined to occur with an activation barrier of 14 kcal mol−1 based on the coalescence temperature of 31 °C in 1H NMR spectra. Notably, this transformation does not involve the formation of a 2c-2e metal–boryl bond. Baker, Marder, and co-workers reported an example of an intramolecular hydrogen atom exchange between a metal hydride and a coordinated borane likely also proceeding through a hydroborate intermediate with an activation barrier of 15 kcal mol−1 for the ruthenium complex and 13 kcal mol−1 for the osmium congener.56 Sabo-Etienne and co-workers have extensively studied structural motifs of metal hydride borane complexes with the particular focus on the distinction between borane–hydride and dihydroborate coordination modes.57–59 The complex (POBOP)Ru(H)(PPh3) reported herein, featuring metal boryl, metal hydride, and metal–borane moieties, may be the first example of the rapid transformation between these configurations through the exchange of not only hydrogen atoms but also boron atoms at the metal center. Importantly, the exchange reported herein is likely to occur through (borane)–(boryl/hydride) conversion, as the anionic borate configuration is not normally attainable for boron clusters due to the lack of accessible p-orbitals on boron vertices.
Theoretical calculations
Theoretical calculations at the PBE/TZ2P level were employed in order to obtain better understanding of the possible exchange mechanism in 3 (see ESI† for details). Two reaction pathways with comparable barriers were identified during the 2D scan of a potential energy surface vs. B2–Ru1–H2 and B1–Ru1–H1 angle coordinates (Fig. 5a). The first pathway with an activation barrier of 12.9 kcal mol−1 proceeded through an intermediate complex 4-1 that can be best described as a five-coordinate Ru(0) complex with two B–H⋯Ru coordinated borane groups, two phosphinite pincer arms, and the triphenylphosphine ligand. As discussed above, the formation of 4-1 from 3 can be represented as the reductive elimination of the boryl and hydride ligands from the divalent ruthenium center. The second pathway with an activation barrier of 13.5 kcal mol−1 proceeded through an intermediate complex 4-2 that can be described as a six-coordinate (BB)–carboryne complex of Ru(IV) with two B–Ru bonds, two Ru–H hydrides, two phosphinite pincer arms, and the triphenylphosphine ligand. Close values of energy barriers corresponding to these two pathways suggest similar probability of these two processes leading to the exchange transformation in 3.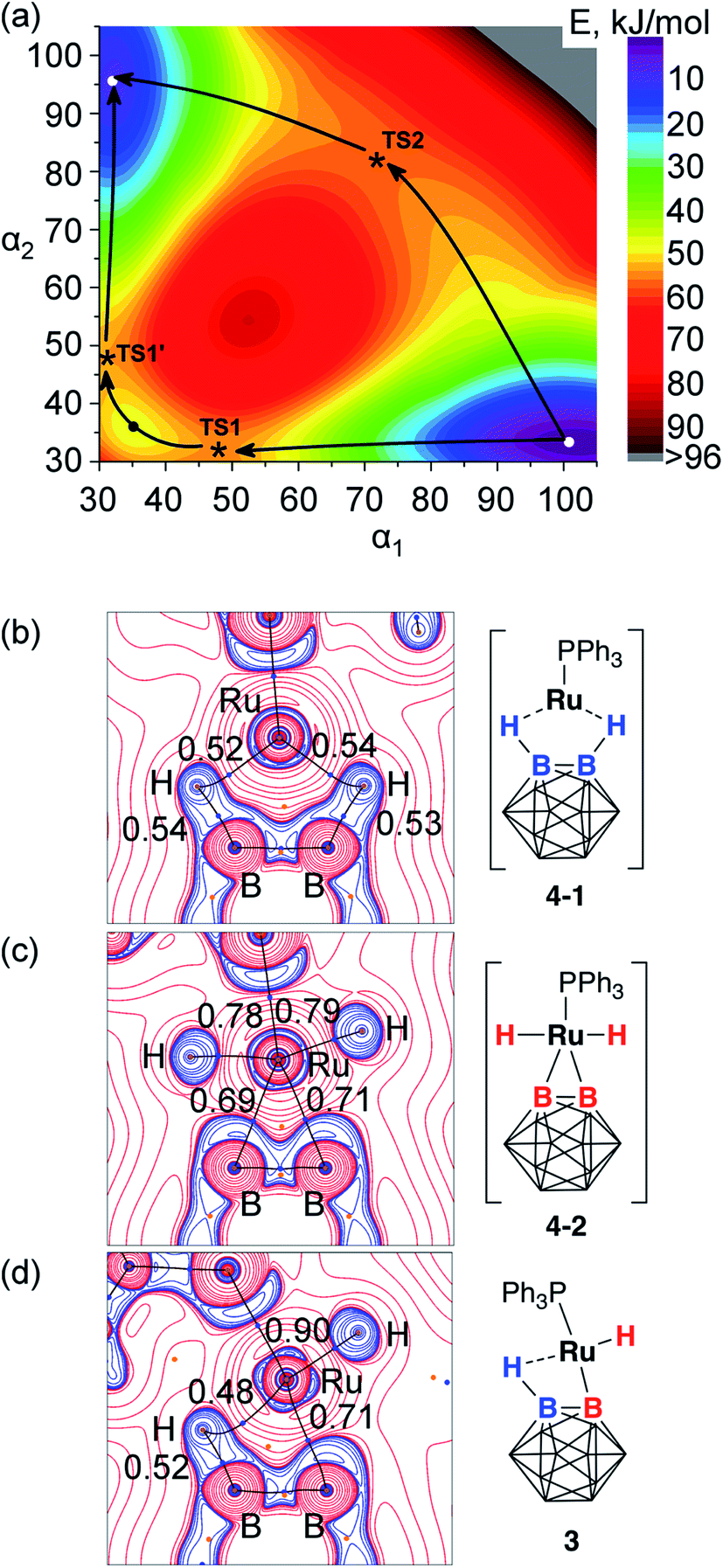 | ||
| Fig. 5 (a) 2D potential energy surface scan along B–Ru–H angle coordinates (α1: B1–Ru–H1, α2: B2–Ru–H2). Transition states TS1/TS1′ and a shallow minimum between them correspond to 4-1, TS2 corresponds to 4-2. (b–d) Laplacian maps and bond path in the B1–B2–Ru1 plane in 4-1 (b), 4-2 (c), and 3 (d). QTAIM delocalization indices are shown for the selected B–H, Ru–H, and Ru–B bonds. | ||
Bonding situation in the complex 3, and the proposed intermediates 4-1 and 4-2 was further analyzed using the analysis of the electron density in the framework of the quantum theory of atoms-in-molecules (QTAIM)60,61 for the electron density computed at the PBE0/def-TZVP level. Fig. 5b–d shows Laplacian maps and bond paths for the three structures plotted in the B1–Ru1–B2 plane. In accordance with the description of 4-2 as a Ru(IV) diboryl dihydride complex, its optimized structure contained two Ru–H and two Ru–B bond paths with delocalization indices (DI, the number of electron pairs shared between two atoms, QTAIM analogs of bond orders) of 0.78 and 0.79 (Ru–H bonds) and 0.69 and 0.71 (Ru–B bonds). The bonding between the ruthenium center and the boron atoms B1 and B2 with outward-bent density concentration is similar to that in the related BB–carboryne complex (POBOP)Ru(CO)2.37 In 4-1, direct Ru–B bond paths are absent. Instead, hydrogen atoms are connected by curved bond paths to both B1/B2 and Ru, representing neutral borane ligand coordination to the formally Ru(0) metal center. Delocalization indices are 0.52 and 0.54 for the Ru–H bonds and 0.53 and 0.54 for the B–H bonds, thus demonstrating that bridging hydrogen atoms have the same bond order with both boron atoms and the metal center. The Ru–B bond delocalization indices of 0.34 and 0.37 in 4-1 are considerably smaller than in 4-2, but not negligible despite the absence of direct bond paths.
In the complex 3, there is only one Ru–B bond path with a delocalization index value 0.71 while the second metal–boron interaction does not have a bond path but has the DI value 0.23. The terminal Ru–H bond has the DI value 0.90 while bridging B–H⋯Ru interaction possesses DI = 0.48 for the Ru–B bond and DI = 0.52 for the B–H bond. These values of delocalization indices are consistent with the complex 3 formulation as the Ru(II) boryl hydride with a coordinated borane ligand, thus exhibiting bonding features of both 4-1 and 4-2 intermediates.
Fluxional behavior related to dissociation/coordination of neutral ligand arms in pincer complexes have been documented while reports of room temperature fluxionality in anionic backbone/arms are rare.11,62–68 The POBOP pincer framework features the three-dimensional carborane backbone that allows the metal center to be in proximity of two cage vertices simultaneously. This geometric arrangement accompanied by the flexibility of boryl/borane coordination modes predisposes the metal center to the rapid reductive elimination/oxidative addition sequence resulting in unique rapid metal center shuttling between two boron atoms of the cage and exchange of hydrogen atoms belonging to B–H and Ru–H bonds. Notably, this process involves breaking and formation of strong covalent metal–boron and metal–hydrogen bonds.
Catalytic transfer dehydrogenation of cyclooctane promoted by (POBOP)Ru(H)(PPh3)
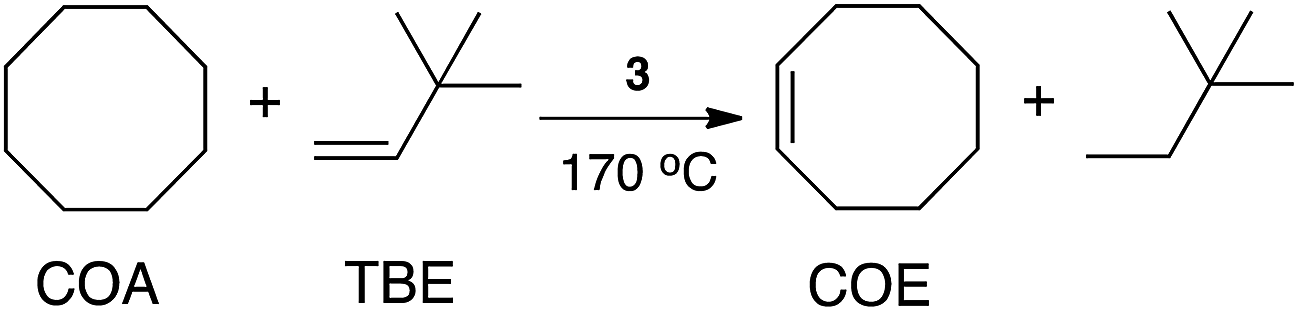
The rapid shuttling of the metal center between boron atoms on the surface of the carborane cage in (POBOP)Ru(H)(PPh3), which, according to theoretical calculations, may proceed through thermally accessible Ru(0) five-coordinate intermediate (see above), and the extreme thermal stability of icosahedral carborane clusters, prompted us to investigate the reactivity of this complex in alkane dehydrogenation. Dehydrogenation of alkanes is an attractive strategy for conversion of readily available but inert saturated hydrocarbons into alkenes, which can serve as versatile building blocks for further transformations. High-temperature (400–600 °C) dehydrogenation of alkanes proceeds with the use of heterogeneous catalysts, thus, an active well-defined homogeneous catalyst operating at lower temperatures would be desirable.69 Transfer dehydrogenation of alkanes promoted by iridium pincer complexes receives continuing attention.12,70–76 Phosphinite-containing POCOP iridium pincers have been reported as effective catalysts with high longevity in the reaction mixture leading to turnover numbers as high as 6000. Many iridium(I)-based systems for transfer hydrogenation operate under strictly inert conditions, including absence of oxygen, water, and dinitrogen. Recently, examples of ruthenium(II) pincer complexes that are competent in alkane dehydrogenation emerged, starting with the prominent example of Roddick's π-accepting pincer system.77,78 A subsequent report by Huang indicated that optimization of reaction conditions and the use of POCOP-based ruthenium(II) hydride complexes can lead to turnover numbers as high as 370 under Ar and 294 under N2.79We found that (POBOP)Ru(H)(PPh3) efficiently promotes catalytic dehydrogenation of cyclooctane (COA) to cyclooctene (COE) with the use of tert-butylethylene (TBE) as a hydrogen acceptor under dinitrogen atmosphere. The results are summarized in Table 1. A turnover number (TON) of 77 ± 5 was achieved with the initial COA:TBE:catalyst molar ratio of 3000:3000:1 (0.03 mol% of catalyst loading) at 170 °C within 2 h in a sealed glass vessel. Longer reaction times (>2 h) did not result in an increased conversion. It has been previously noted that large excess of TBE can impede the reaction, possibly through the formation of a relatively stable complex with the catalyst.79 Lowering the amount of TBE in the reaction mixture to COA:TBE:catalyst molar ratio of 5700:1000:1 with the effective catalyst loading of 0.1 mol% relative to the amount of TBE led to the increase of the observed TON to 400 ± 8 under N2. Transfer dehydrogenation experiments were also carried out under air at 170 °C leading to the appreciable TON of 288 ± 8 after 2 h with 0.1 mol% catalyst loading relative to TBE and 5700:1000:1 COA:TBE:catalyst molar ratio. The dehydrogenation of n-octane (OA) under analogous conditions (5700:1000:1 OA:TBE:catalyst, 170 °C, 1 h) resulted in the TON of 85 ± 10. Regioselectivity of formation of 1-octene was low (less than 5% according to GC-MS analysis). In the light of these results, we conclude that the catalytic activity of the (POBOP)Ru(H)(PPh3) complex in the dehydrogenation of cyclooctane is comparable, if not higher, to the most active ruthenium-based pincer systems reported to date.
|
|
||||
|---|---|---|---|---|
| COA, equiv. | TBE, equiv. | 3, equiv. | Atmosphere | TONa |
| a Experiments were carried out at 170 °C for 2 h in sealed tubes. TONs were determined from 1H NMR spectra of reaction mixtures with the use of naphthalene as an internal standard. Each entry corresponds to three independent experiments. | ||||
| 3000 | 3000 | 1 | N2 | 77 ± 5 |
| 5700 | 1000 | 1 | N2 | 400 ± 8 |
| 5700 | 1000 | 1 | Air | 288 ± 8 |
Transfer dehydrogenation of deuterated n-octane-d18 was studied to probe the mechanism of the transformation. Cycling between Ir(III) and Ir(I) species has been proposed to be an operating mechanism for iridium pincer catalysts.70 In the case of the (POBOP)Ru(H)(PPh3) complex, both transient low-valent Ru(0) and high-valent Ru(IV) species may be accessible during the exchange process according to the theoretical calculations above. Transfer dehydrogenation of n-octane-d18 (d-OA) with TBE as a hydrogen acceptor at 170 °C for 1 h and d-OA:TBE:catalyst molar ratio of 14:10:1 led to the complete consumption of TBE. The 31P NMR spectrum of the reaction mixture indicated the presence of the starting complex 3 as well as a new unidentified pincer complex in a 3:1 ratio. The 1H NMR spectrum of the reaction mixture did not contain signals in the range from 0 ppm to −15 ppm. On the other hand, the 2H NMR spectrum of the mixture after evaporation of volatiles contained a broadened signal at −8.9 ppm corresponding to the partially deuterated complex 3-d2 (see above). These results suggest that the metal hydride exchanges with hydrogen atoms of an alkane substrate either upon dehydrogenation through σ-bond metathesis or during possible isomerization of an alkene product.
Conclusions
In summary, the carboranyl pincer POBOP framework serves as both an anionic (boryl) and a neutral (borane) ligand at the same time causing significant distortion of the metal–boron covalent bond rendering it more reactive. This unique coordination environment results in the rapid metal center “cage-walking” between two adjacent boron atoms of the carborane cage at room temperature representing the “pendulum clock”-type fluxional behavior. This facile B–H bond activation and re-formation observed in the (POBOP)Ru(H)(PPh3) complex highlights an importance of the bridging vicinal B–H⋯M interactions and provides an insight to the possible mechanism of the isomerization during metal-promoted coupling reactions of boron clusters involving initial activation of boron–hydrogen or boron–halogen bonds. Furthermore, the B–H⋯M interaction served as a hemilabile ligand protecting a latent coordination site in the (POBOP)Ru(H)(PPh3) complex, which is a competent catalyst of the transfer dehydrogenation of cyclooctane.Acknowledgements
Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund (Award 54504-DNI3 to D.V.P.) and to the National Science Foundation (Award CHE-1654301 to D.V.P.). We are grateful to Dr Perry. J. Pelechia (Director of NMR Services, University of South Carolina) for immense help with variable temperature NMR spectroscopy studies. A. A. P. acknowledges funding by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 648295 “GraM3”). Computational resources were provided by the Center for Information Services and High Performance Computing (ZIH) in TU Dresden. The authors thank Ulrike Nitzsche for technical assistance with computational resources in IFW Dresden and Ekaterina Dolgopolova (University of South Carolina) for help with the production of the TOC graphic.References
- S. Bontemps, G. Bouhadir, W. Gu, M. Mercy, C.-H. Chen, B. M. Foxman, L. Maron, O. V. Ozerov and D. Bourissou, Angew. Chem., Int. Ed., 2008, 47, 1481–1484 CrossRef CAS PubMed.
- A. F. Hill, S. B. Lee, J. Park, R. Shang and A. C. Willis, Organometallics, 2010, 29, 5661–5669 CrossRef CAS.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS PubMed.
- G. R. Owen, Chem. Commun., 2016, 52, 10712–10726 RSC.
- Y. Segawa, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 9201–9203 CrossRef CAS PubMed.
- M. Hasegawa, Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2012, 51, 6956–6960 CrossRef CAS PubMed.
- T. Miyada, E. Huang Kwan and M. Yamashita, Organometallics, 2014, 33, 6760–6770 CrossRef CAS.
- A. F. Hill and C. M. A. McQueen, Organometallics, 2014, 33, 1977–1985 CrossRef CAS.
- C. M. A. McQueen, A. F. Hill, M. Sharma, S. K. Singh, J. S. Ward, A. C. Willis and R. D. Young, Polyhedron, 2016, 120, 185–195 CrossRef CAS.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310–15313 CrossRef CAS PubMed.
- W.-C. Shih, W. Gu, M. C. MacInnis, S. D. Timpa, N. Bhuvanesh, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2016, 138, 2086–2089 CrossRef CAS PubMed.
- W. C. Shih and O. V. Ozerov, Organometallics, 2017, 36, 228–233 CrossRef CAS.
- A. M. Spokoyny, M. G. Reuter, C. L. Stern, M. A. Ratner, T. Seideman and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 9482–9483 CrossRef CAS PubMed.
- M. Y. Tsang, C. Viñas, F. Teixidor, J. G. Planas, N. Conde, R. SanMartin, M. T. Herrero, E. Domínguez, A. Lledós, P. Vidossich and D. Choquesillo-Lazarte, Inorg. Chem., 2014, 53, 9284–9295 CrossRef CAS PubMed.
- B. J. Eleazer, M. D. Smith and D. V. Peryshkov, Organometallics, 2016, 35, 106–112 CrossRef CAS.
- B. J. Eleazer, M. D. Smith and D. V. Peryshkov, J. Organomet. Chem., 2017, 829, 42–47 CrossRef CAS.
- M. E. El-Zaria, H. Arii and H. Nakamura, Inorg. Chem., 2011, 50, 4149–4161 CrossRef CAS PubMed.
- M. Yamashita, Bull. Chem. Soc. Jpn., 2016, 89, 269–281 CrossRef CAS.
- W.-C. Shih, W. Gu, M. C. MacInnis, D. E. Herbert and O. V. Ozerov, Organometallics, 2017, 36, 1718–1726 CrossRef CAS.
- R. N. Grimes, Dalton Trans., 2015, 44, 5939–5956 RSC.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CrossRef CAS PubMed.
- C. Selg, W. Neumann, P. Lönnecke, E. Hey-Hawkins and K. Zeitler, Chem.–Eur. J., 2017 DOI:10.1002/chem.201701037.
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC.
- A. R. Popescu, F. Teixidor and C. Viñas, Coord. Chem. Rev., 2014, 269, 54–84 CrossRef CAS.
- O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680–12681 CrossRef CAS PubMed.
- J. C. Axtell, K. O. Kirlikovali, P. I. Djurovich, D. Jung, V. T. Nguyen, B. Munekiyo, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 15758–15765 CrossRef CAS PubMed.
- E. A. Qian, A. I. Wixtrom, J. C. Axtell, A. Saebi, D. Jung, P. Rehak, Y. Han, E. H. Moully, D. Mosallaei, S. Chow, M. S. Messina, J. Y. Wang, A. T. Royappa, A. L. Rheingold, H. D. Maynard, P. Král and A. M. Spokoyny, Nat. Chem., 2017, 9, 333–340 CrossRef CAS PubMed.
- V. Lavallo, J. H. Wright, F. S. Tham and S. Quinlivan, Angew. Chem., Int. Ed., 2013, 52, 3172–3176 CrossRef CAS PubMed.
- A. L. Chan, J. Estrada, C. E. Kefalidis and V. Lavallo, Organometallics, 2016, 35, 3257–3260 CrossRef CAS.
- T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173–3177 CrossRef CAS PubMed.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed.
- F. Teixidor, M. A. Flores, C. Viñas, R. Sillanpää and R. Kivekäs, J. Am. Chem. Soc., 2000, 122, 1963–1973 CrossRef CAS.
- Y. H. Lee, J. Park, J. Lee, S. U. Lee and M. H. Lee, J. Am. Chem. Soc., 2015, 137, 8018–8021 CrossRef CAS PubMed.
- J. J. Schwartz, A. M. Mendoza, N. Wattanatorn, Y. Zhao, V. T. Nguyen, A. M. Spokoyny, C. A. Mirkin, T. Baše and P. S. Weiss, J. Am. Chem. Soc., 2016, 138, 5957–5967 CrossRef CAS PubMed.
- K. O. Kirlikovali, J. C. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyny, Chem. Sci., 2016, 7, 5132–5138 RSC.
- B. J. Eleazer, M. D. Smith, A. A. Popov and D. V. Peryshkov, J. Am. Chem. Soc., 2016, 138, 10531–10538 CrossRef CAS PubMed.
- H. Lyu, Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 10623–10626 CrossRef CAS PubMed.
- Y. Quan and Z. Xie, J. Am. Chem. Soc., 2015, 137, 3502–3505 CrossRef CAS PubMed.
- Y. Zhang, Y. Sun, F. Lin, J. Liu and S. Duttwyler, Angew. Chem., Int. Ed., 2016, 55, 15609–15614 CrossRef CAS PubMed.
- R. Cheng, Z. Qiu and Z. Xie, Nat. Commun., 2017, 8, 14827 CrossRef CAS PubMed.
- H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2016, 138, 12727–12730 CrossRef CAS PubMed.
- Y. Shen, Y. Pan, J. Liu, T. Sattasathuchana, K. K. Baldridge and S. Duttwyler, Chem. Commun., 2016, 53, 176–179 RSC.
- Y. Quan, C. Tang and Z. Xie, Chem. Sci., 2016, 7, 5838–5845 RSC.
- Z.-J. Yao, W.-B. Yu, Y.-J. Lin, S.-L. Huang, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2014, 136, 2825–2832 CrossRef CAS PubMed.
- C. Vinas, R. Nunez, M. A. Flores, F. Teixidor, R. Kivekas and R. Sillanpaea, Organometallics, 1995, 14, 3952–3957 CrossRef CAS.
- S. Du, J. A. Kautz, T. D. McGrath and F. G. A. Stone, Angew. Chem., 2003, 115, 5906–5908 CrossRef.
- E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Dalton Trans., 2007, 4829–4844 RSC.
- A. El-Hellani, C. E. Kefalidis, F. S. Tham, L. Maron and V. Lavallo, Organometallics, 2013, 32, 6887–6890 CrossRef CAS.
- C. Viñas, R. Nuñez, F. Teixidor, R. Kivekäs and R. Sillanpää, Organometallics, 1996, 15, 3850–3858 CrossRef.
- F. Teixidor, C. Vinas, J. Casabo, A. M. Romerosa, J. Rius and C. Miravitlles, Organometallics, 1994, 13, 914–919 CrossRef CAS.
- F. Teixidor, J. A. Ayllon, C. Vinas, R. Kivekas, R. Sillanpaa and J. Casabo, Organometallics, 1994, 13, 2751–2760 CrossRef CAS.
- D. D. Ellis, P. A. Jelliss and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 2000, 2113–2122 RSC.
- D. Liu, L. Dang, Y. Sun, H.-S. Chan, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2008, 130, 16103–16110 CrossRef CAS PubMed.
- T. J. Hebden, M. C. Denney, V. Pons, P. M. B. Piccoli, T. F. Koetzle, A. J. Schultz, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, J. Am. Chem. Soc., 2008, 130, 10812–10820 CrossRef CAS PubMed.
- R. T. Baker, J. C. Calabrese, S. A. Westcott and T. B. Marder, J. Am. Chem. Soc., 1995, 117, 8777–8784 CrossRef CAS.
- V. Montiel-Palma, M. Lumbierres, B. Donnadieu, S. Sabo-Etienne and B. Chaudret, J. Am. Chem. Soc., 2002, 124, 5624–5625 CrossRef CAS PubMed.
- S. Lachaize, K. Essalah, V. Montiel-Palma, L. Vendier, B. Chaudret, J.-C. Barthelat and S. Sabo-Etienne, Organometallics, 2005, 24, 2935–2943 CrossRef CAS.
- Y. Gloaguen, G. Bénac-Lestrille, L. Vendier, U. Helmstedt, E. Clot, G. Alcaraz and S. Sabo-Etienne, Organometallics, 2013, 32, 4868–4877 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, New York, 1994 Search PubMed.
- R. F. W. Bader and D. A. Legare, Can. J. Chem., 1992, 70, 657–676 CrossRef CAS.
- A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2015, 6, 2060–2067 RSC.
- A. Y. Verat, M. Pink, H. Fan, J. Tomaszewski and K. G. Caulton, Organometallics, 2008, 27, 166–168 CrossRef CAS.
- S. J. Connelly, A. G. Chanez, W. Kaminsky and D. M. Heinekey, Angew. Chem., Int. Ed., 2015, 54, 5915–5918 CrossRef CAS PubMed.
- S. D. T. Cherry, W. Kaminsky and D. M. Heinekey, Organometallics, 2016, 35, 2165–2169 CrossRef CAS.
- A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Organometallics, 2016, 35, 2600–2608 CrossRef CAS.
- D. E. Herbert, A. D. Miller and O. V. Ozerov, Chem.–Eur. J., 2012, 18, 7696–7704 CrossRef CAS PubMed.
- J. Hyvl, W. Y. Yoshida, A. L. Rheingold, R. P. Hughes and M. F. Cain, Chem.–Eur. J., 2016, 22, 17562–17565 CrossRef CAS PubMed.
- S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem.–Eur. J., 2015, 21, 12226–12250 CrossRef CAS PubMed.
- K. Zhu, P. D. Achord, X. Zhang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13044–13053 CrossRef CAS PubMed.
- I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed.
- W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 1390–1394 CrossRef CAS PubMed.
- A. Kumar, T. Zhou, T. J. Emge, O. Mironov, R. J. Saxton, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2015, 137, 9894–9911 CrossRef CAS PubMed.
- O. O. Kovalenko and O. F. Wendt, Dalton Trans., 2016, 45, 15963–15969 RSC.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, e1501591 Search PubMed.
- X. Jia and Z. Huang, Nat. Chem., 2016, 8, 157–161 CrossRef CAS PubMed.
- B. C. Gruver, J. J. Adams, S. J. Warner, N. Arulsamy and D. M. Roddick, Organometallics, 2011, 30, 5133–5140 CrossRef CAS.
- B. C. Gruver, J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2013, 32, 6468–6475 CrossRef CAS.
- Y. Zhang, H. Fang, W. Yao, X. Leng and Z. Huang, Organometallics, 2016, 35, 181–188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1545735 and 1545736. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01846k |
| This journal is © The Royal Society of Chemistry 2017 |