
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRiboflavin as a bioorthogonal photocatalyst for the activation of a PtIV prodrug†
Silvia
Alonso-de Castro
a,
Emmanuel
Ruggiero
a,
Ane
Ruiz-de-Angulo
a,
Elixabete
Rezabal
bc,
Juan C.
Mareque-Rivas
ad,
Xabier
Lopez
ce,
Fernando
López-Gallego
ad and
Luca
Salassa
 *acde
*acde
aCIC biomaGUNE, Paseo de Miramón 182, Donostia-San Sebastián, 20014, Spain
bFarmazia Fakultatea, Kimika Fisikoa Departamentua, Euskal Herriko Unibertsitatea, UPV/EHU, 01006, Vitoria-Gasteiz, Spain
cDonostia International Physics Center (DIPC), P.K. 1072, Donostia-San Sebastián, 20080, Spain
dIkerbasque, Basque Foundation for Science, Bilbao, 48011, Spain
eKimika Fakultatea, Euskal Herriko Unibertsitatea, UPV/EHU, Donostia-San Sebastián 20080, Spain. E-mail: lsalassa@dipc.org
First published on 19th April 2017
Abstract
Encouraging developments demonstrate that few transition metal and organometallic catalysts can operate in a bioorthogonal fashion and promote non-natural chemistry in living systems by minimizing undesired side reactions with cellular components. These catalytic processes have potential for applications in medicinal chemistry and chemical biology. However, the stringent conditions of the cell environment severely limit the number of accessible metal catalysts and exogenous reactions. Herein, we report an unorthodox approach and a new type of bioorthogonal catalytic reaction, in which a metal complex is an unconventional substrate and an exogenous biological molecule acts as a catalyst. In this reaction, riboflavin photocatalytically converts a PtIV prodrug into cisplatin within the biological environment. Due to the catalytic activity of riboflavin, cisplatin-like apoptosis is induced in cancer cells under extremely low doses of light, potentially preventing systemic off-target reactions. Photocatalytic and bioorthogonal turnover of PtIV into PtII species is an attractive strategy to amplify the antineoplastic action of metal-based chemotherapeutics with spatio-temporal control.
Introduction
The combination of catalysis and bioorthogonality1–3 promises to impact drug discovery and bioimaging by facilitating the execution of non-natural chemical reactions in living systems. Catalytic turnover can boost the efficiency of bioorthogonal chemical reactions, unveiling new strategies for prodrug activation and uncaging of molecular probes.4–8In this context, transition metal and organometallic catalysts have opened new avenues for the advancement of bioorthogonal catalysis in cells.5,9–16 In the studies carried out in the laboratories of Meggers, Mascareñas, Unciti-Broceta, and Bradley, organoruthenium and palladium catalysts were used to deprotect pro-fluorescent substrates and activate prodrugs in cancer cells or in their compartments and surroundings.11–14,16 Rotello devised biomimetic nanoenzymes for imaging and therapy by encapsulating ruthenium and palladium catalysts into water-soluble gold nanoparticles and controlling their catalytic activity in HeLa cells through supramolecular chemistry.15 These pioneering systems have exploited metal-based catalytic uncaging of allylcarbamate- and propargyl-protected amines as viable strategies for bioorthogonal catalysis; however, new biocompatible transformations are highly needed to further advance this extremely challenging field that is still in its infancy.
Herein, we described an original photocatalysis approach to control the reactivity of transition metal complexes in a bioorthogonal fashion. In a new type of light-driven reaction, the exogenous biological molecule riboflavin (Rf) functions as a bioorthogonal photocatalyst and a metal complex acts as an unconventional substrate (Scheme 1).
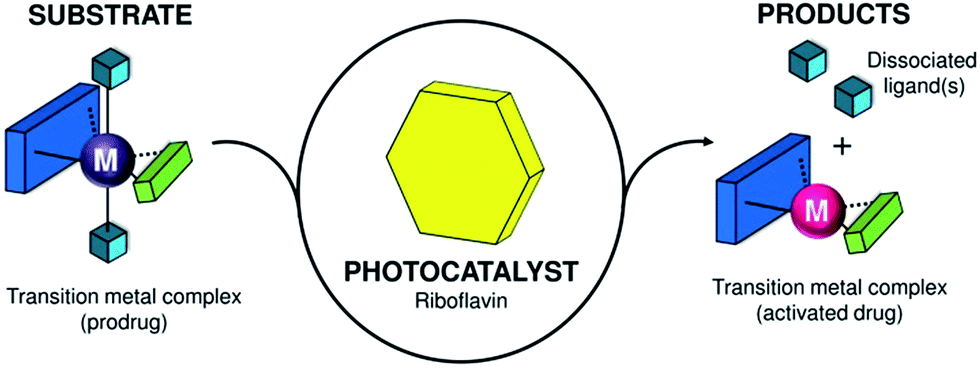 | ||
| Scheme 1 Transition-metal complex acting as a substrate and its bioorthogonal activation by riboflavin that functions as a photocatalyst. | ||
This unusual catalyst/substrate pair relies on the photoredox properties of Rf to enable the selective activation of a PtIV prodrug of cisplatin with exceptionally low doses of blue light and induce apoptotic death in PC-3 human prostate cancer cells.
Metal complexes are typically regarded as catalysts that convert organic substrates into more valuable compounds; however, to date, catalytic transformations of metal complexes are practically unknown and represent a paradigm shift in catalysis.17,18 Their development can expand the scope of bioorthogonal chemical reactions to inorganic substances and metal-based prodrugs, fostering the creation of new inorganic chemistry toolkits for biology and medicine.
Results and discussion
As part of our ongoing efforts to design innovative light-activation modes for anticancer platinum complexes,19 we reasoned that Rf and its rich photochemistry would facilitate the photoreduction of cis,cis,trans-[Pt(NH3)2(Cl)2(O2CCH2CH2CO2)2]2− (1) to cisplatin via light irradiation at 460 nm, a wavelength that is appropriate for use in biological systems (Scheme 2). Complex 1 is a cisplatin prodrug suitable for photochemotherapy because of its high dark stability in aqueous solutions and negligible dark cytotoxicity in several cancer cell lines such as prostate cancer PC-3 cells.20,21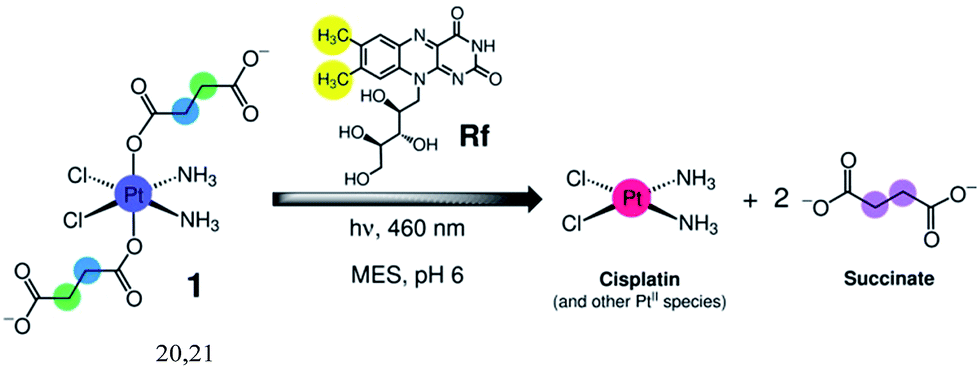 | ||
| Scheme 2 Light-induced reduction of cis,cis,trans-[Pt(NH3)2(Cl)2(O2CCH2CH2CO2)2]2− (1) promoted by riboflavin (Rf) in MES buffer. | ||
Upon UVA light excitation (385 nm), 1 undergoes photochemical activation. However, UVA light is of limited use in therapy, and PtIV complexes such as 1 rarely display satisfactory absorption features at wavelengths longer than 400 nm (Fig. 1a).
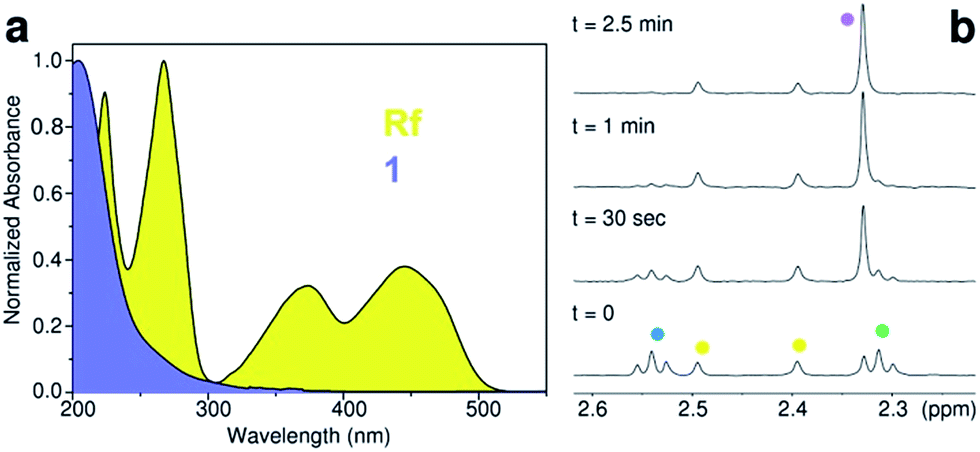 | ||
Fig. 1 (a) Absorption spectrum of riboflavin (Rf) and cis,cis,trans-[Pt(NH3)2(Cl)2(O2CCH2CH2CO2)2]2− (1) in aqueous solution. (b) Rf-catalysed photoreduction of 1 in the MES buffer (18 mM, pH 6) monitored by 1H NMR. Spectra were obtained for a MES/D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution of 120 μM 1 and 50 μM Rf upon t = 0 s, 30 s, 1 min, and 2.5 min of 460 nm light irradiation (2.5 mW cm−2). 1H NMR signal labelling: :1) solution of 120 μM 1 and 50 μM Rf upon t = 0 s, 30 s, 1 min, and 2.5 min of 460 nm light irradiation (2.5 mW cm−2). 1H NMR signal labelling:  Pt–OCOCH2CH2CO2−, Pt–OCOCH2CH2CO2−,  Pt–OCOCH2CH2CO2−, Pt–OCOCH2CH2CO2−,  methyl groups of Rf isoalloxazine ring, and methyl groups of Rf isoalloxazine ring, and  free O2CCH2CH2CO2−. free O2CCH2CH2CO2−. | ||
Rf is vitamin B2 and the precursor of biologically important cofactors such as FMN and FAD, which are essential to humans and animals due to their redox activity.22 The yellow-colored Rf can absorb light with a wavelength as high as ca. 500 nm in aqueous media with good extinction coefficients (ε446 > 104 M−1 cm−1)23 (Fig. 1a) and can promote a great variety of light-induced reactions that depend on its 7,8-dimethyl-10-alkylisoalloxazine fragment.22Rf has been adopted as a photocatalyst in several organic reactions including the photooxidation of benzyl alcohols and alkyl benzenes or the [2 + 2] cycloaddition of styrene dienes and bis(arylenones).24–26
Photocatalytic activation of a PtIV prodrug by riboflavin in solution
After confirming that blue light had no direct effects on 1 (Fig. S1–3†), we investigated the capability of Rf to photoactivate the complex upon 460 nm excitation in MES buffer. Using low excitation power density (2.5 mW cm−2), 120 μM solutions of 1 were photolysed in the presence of Rf at various concentrations (12–120 μM, Fig. S4–7†). The process was monitored via1H NMR by the evolution of diagnostic peaks corresponding to the Pt-bound (triplets) and free (singlet) succinate ligands (Fig. 1b).Sub-stoichiometric quantities of Rf were capable of causing full conversion of 1 into its photoproducts under light excitation, demonstrating that Rf did not act as a simple photosensitizer but was indeed a photocatalyst. The efficiency of this catalytic process was remarkable since 12 μM Rf converted 100% of 120 μM 1 in 5 minutes (light dose 0.75 J cm−2). A Rf concentration as low as 0.13 μM still photocatalysed the transformation of 1 (120 μM); however, more than 2 hours were required to achieve 27% of conversion (Fig. S8†). No reaction between 1 and Rf occurred in the dark within 1 week (Fig. S9†). Interestingly, photoconversion of 1 occurred in pure water (pH 5) or in phosphate buffer (PB, 100 mM, pH 5.5), but never reached completion due to the poor photostability of Rf in these media (vide infra). Thus, MES buffer plays a key role in the catalytic process by preventing Rf from undergoing photodecomposition reactions.
To assess the rate law for the Rf-catalysed photoreduction of 1 to PtII species, we studied the reaction rate at different substrate concentrations (120 μM–1.92 mM, i.e. 2.4–38.4 mol equiv. of 1 compared to Rf) in 18 mM MES buffer during 30 s of irradiation at 298 K (Fig. S10†). The effect of MES on the reaction rate was evaluated in a separate set of experiments, in which MES concentration was varied in the 3–20 mM range (Fig. S11†). Results demonstrated that the rate of the reaction linearly increased with the concentration of 1 and MES, corresponding to a first-order reaction for both species. Importantly, the reaction showed a stronger dependency on the concentration of 1 than on the concentration of MES, suggesting that PtIV reduction is the limiting step of the reaction.
Since our experiments employed a large excess of MES and all the reaction steps were irreversible, the rate constant could be described using the pseudo-first-order model (ESI†). Using 50 μM Rf, we obtained a pseudo-first-order reaction constant (kobs = 10.0 ± 0.05 × 10−3 s−1) that increased with the catalyst concentration (Fig. S12 and S13†) and depended on the MES concentration.
A turnover frequency (TOF) value of 0.22 ± 0.06 s−1 was determined for the conversion of 1 (1.92 mM) by Rf (50 μM) under light irradiation at 298 K in 18 mM MES buffer. Under these reaction conditions, the maximum total turnover number (TTN) value was 38 after 3 minutes of light irradiation, and no decomposition of the catalyst was observed by 1H NMR. The Rf/1 catalyst/substrate pair achieved approx. 70–700 times higher TOF compared to that of ruthenium(II) organometallic catalysts, which catalytically converted NAD+ to NADH or transformed O-allyl carbamates into their respective amines under biologically relevant conditions and in cells.8,9 High TOF is crucial for application in photochemotherapy since it guarantees rapid and sufficient conversion of 1 under short irradiation time and low light dose conditions.
Mechanism of photocatalytic activation
Rf is extremely light sensitive, and its photochemical reactivity strongly depends on the surrounding environment. Electron transfer and proton-coupled reactions or singlet-oxygen generation take place upon light excitation of Rf, depending on the availability of electron donors. In addition, light decomposes Rf into several fragments through intramolecular reactions in which the ribityl chain can be used as an electron source.27Direct energy transfer from Rf to 1 can be ruled out since there is no overlap between the emission band of Rf (λem = 535 nm) and the absorption band of 1.
Hence, the sensitizing and catalytic capacity of Rf in MES buffer reasonably relies on electron transfer processes triggered by light. In the triplet excited state (3Rf*), Rf is a strong oxidant (E° = 1.77 V)22 capable of efficiently extracting electrons from the abundant MES molecules and generates the two-electron reduced RfH2/RfH− species (pKa ∼ 6) together with morpholino radicals,28 which eventually evolve to the oxidized N-oxide form of MES (Fig. S14†).29
Employing ferrioxalate actinometry,30 we determined the photochemical quantum yield for Rf/1 (50 μM/1.0 mM, 18 mM MES) to be 1.4 ± 0.1 (Fig. S15 and S16†).
Yield values >1 are rather common in photoredox catalysis where radical chain propagation cycles form part of the catalytic mechanism.31
MES buffer dramatically improves Rf photostability by preventing the isoalloxazine unit from reacting with the ribityl moiety or with molecular oxygen. NMR and UV-vis spectra showed that MES substantially preserved Rf from decomposition for over 30 min, whereas the catalyst was fully converted to the photoproduct lumichrome in water within 1 min of light irradiation and then to 2,3-butanedione at longer irradiation times (Fig. S17–19†).32
The role of the buffer was confirmed using HEPES (18 mM, pH 6), an analogue zwitterionic buffering agent (Fig. S20 and S21†), in which Rf and 1 behaved similar to MES in terms of photocatalytic activity.
The presence of sodium azide (singlet oxygen scavenger) in water and PB also improved the efficiency of the photocatalytic reaction of Rf with 1 (Fig. S22 and S23†). When added to the MES buffer, sodium azide did not improve the efficiency of the photocatalytic reaction of Rf with 1 (Fig. S24†) and excluded the participation of 1O2 and other oxygen radicals playing major roles in the catalytic mechanism. On the other hand, O2 partially deactivated Rf since under an inert Ar atmosphere, the photoconversion of 1 was faster (Fig. S25†).
Importantly, complex 1 (1.8 mM) does not affect the fluorescence lifetime of Rf in MES (Fig. S26 and S27†), indicating that the active catalyst is not likely to be a Rf excited-state species. Therefore, photooxidation of MES ultimately leads to the formation of reduced (ground-state) RfH2/RfH−, whose low redox potential (ca. −0.2 V)22 cannot directly promote the reduction of 1 (−0.9 V).33
However, as suggested by the density functional theory (DFT) modelling (PBE0/def2-SVP) and in accordance with the results obtained under an Ar atmosphere, 1 is capable of forming adducts with either RfH2 (Fig. 2a) or RfH− (Fig. S28–30†) via H-bonding interactions between its succinate and amino ligands and the isoalloxazine and ribityl groups of Rf. FMN and FAD also photocatalyze the PtIV conversion of 1, displaying an efficiency comparable to that of Rf (Fig. S31–34†). FAD, however, is somewhat less active, possibly due to steric constrains introduced by its adenine moiety, which would disfavour H-bonding between the complex and the flavin.
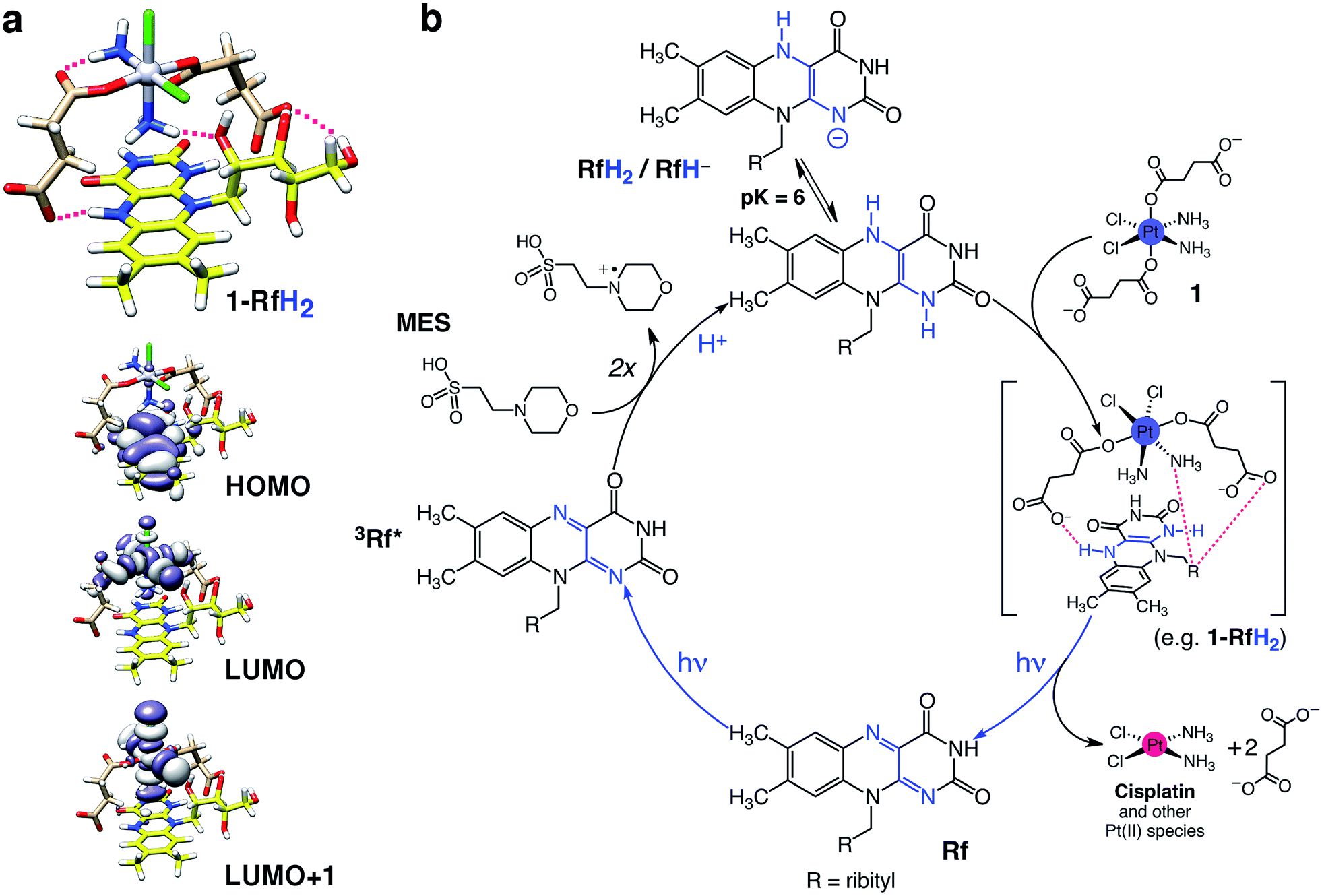 | ||
| Fig. 2 Proposed mechanism for the photocatalytic activation of 1 by Rf. (a) Computed structure and frontier molecular orbitals (DFT:PBE0/def2-SVP) of a selected 1-RfH2 adduct. Intermolecular H-bonds in 1-RfH2 are highlighted with magenta lines (top). Isodensity surfaces are plotted with the isovalue of 0.02 e− bohr−3. Atoms color code: Pt grey, Cl green, O red, N blue, C pale brown (1) or yellow (Rf), H white. (b) Rf absorbs 460 nm photons to generate the triplet excited state (3Rf*), which oxidizes two MES molecules to give the reduced species RfH2/RfH−. Next, complex 1 forms stable adducts with either RfH2 (shown in Fig. 2a) or RfH− and undergoes photoreduction and elimination reactions upon absorption of more photons, liberating cytotoxic PtII species and regenerating the Rf catalyst. | ||
Computed 1-RfH2 and 1-RfH− adducts have the HOMO localized on the Rf isoalloxazine rings, whereas LUMO and LUMO+1 are σ-antibonding orbitals of 1. Absorption of a second photon and subsequent light-induced population of the dissociative LUMO orbitals can trigger photoreduction and ligand elimination reactions,34 ultimately promoting the formation of cisplatin and other PtII species. However, at this stage, we cannot exclude that these strong and specific interactions could significantly lower the redox potential of 1 and cause direct reduction of the prodrug once the Rf-adducts are formed.35,36 Calculated binding energies for 1-RfH2 and 1-RfH− adducts are in the range of 52–69 kcal mol−1, indicating that these transient species are strongly stabilized and may bestow unique selectivity to the Rf/1 catalyst/substrate pair (vide infra).
A pH-dependency profile for the photoreaction at fixed light-irradiation time (2.5 min) showed that complete photoconversion of 1 occured above pH 6 in MES, whereas at lower pHs, the photocatalysis was less efficient (Fig S35†). This finding is in agreement with the prevalence of the RfH2/RfH− forms of reduced Rf at pH values higher than 6.22
On the basis of the described evidence, we propose a tentative photocatalytic mechanism for the Rf/1 catalyst/substrate pair, as shown in Fig. 2b; however, further investigations will be needed for the complete elucidation of this mechanism.
Photocatalysis in the biological environment
To test the potential of Rf photocatalyst as a bioorthogonal tool for photochemotherapy, we studied the activation of 1 by Rf in a cell culture medium and its effects in PC-3 cancer cells (in which 1 has no dark toxicity). At first, photocatalysis experiments and controls (Fig. S36 and S37†) were performed in Ham's F-12K medium supplemented with fetal bovine serum, in which biological components such as growth factors, antibodies, amino acids, vitamins, and inorganic salts were present at concentrations ranging from μM to mM.1H NMR data showed that 3 min of blue light irradiation (light dose 1.08 J cm−2) can fully convert 1.92 mM 1 to PtII species in the presence of 50 μM Rf and 3 mM MES, without any significant side reaction affecting either the medium components or the catalyst (Fig. S38†). Under these conditions, the TOF and TTN for the Rf catalyst were as good as those in pure MES buffer solutions, indicating that the catalytic process was bioorthogonal in cell culture medium.
The antiproliferative activity of Rf/1 against PC-3 cancer cells was investigated in the dark and under 460 nm light irradiation by co-administering the catalyst/substrate pair at the molar ratio of 1:4 and using three different concentrations of complex (40, 80, and 120 μM). In our cell experiments, Rf prevalently activated 1 in the extracellular space since we performed light irradiation after a short pre-incubation period and replaced culture medium after 6 h. Once photocatalytically generated, PtII species could be internalized by cells and exerted their antiproliferative action. MES is well tolerated by cells (Fig. S39†) and was hence employed during cell viability assays as an electron donor. Under these conditions, a short light irradiation period (1 min) and an extremely low light dose (0.36 J cm−2) were sufficient for the full photoconversion of 1 by Rf (Fig. S40 and S41†). In the absence of MES, the photoactivation of 1 still took place, although less efficiently (Fig. S42†), likely because other biological components of the medium acted as electron donors. Against PC-3 cells, the Rf/1 catalyst/substrate pair displayed dose-dependent light-induced toxicity comparable to that of cisplatin in the dark. Remarkably, 120 μM 1 and 30 μM Rf induced a 55 ± 5% reduction in cell biomass under light irradiation, against a 65 ± 5% reduction caused by cisplatin at the same concentration. Control experiments indicated that Rf/1 did not reduce viability of the PC-3 cells when kept in the dark neither did any of the components when individually irradiated (Fig. 3a).
 | ||
| Fig. 3 Antiproliferative activity in human prostate cancer PC-3 cells. (a) Percentage cell viability of PC-3 cells following treatment with Rf, 1, Rf/1 and cisplatin with and/or without light activation (460 nm, 0.36 J cm−2). (b) Fluorescence microscopy images showing the effects of Rf/1 on PC-3 cells upon light irradiation. (A and B) Untreated PC-3 cells, (C and D) Rf/1 (30:120 μM) in the dark, (E and F) Rf/1 (30:120 μM) activated by 460 nm light and (G and H) cisplatin (120 μM) in the dark. Top row: cell nuclei (green); bottom row: apoptotic cells (red). (c) Quantification of early apoptotic PC-3 cells (Annexin V+/SYTOX−) treated by Rf (30 μM), Rf/1 (30:120 μM) and cisplatin (120 μM) with and/or without light activation. Cell viability and flow cytometry data are presented as mean ± SEM of at least three independent measurements. ***P < 0.001, **P < 0.01, ns = non-significant by two-way ANOVA followed by Bonferroni's test (a) or by one-way ANOVA followed by Tukey's test (c). | ||
The antiproliferative action of Rf-activated 1 is associated with the formation of cisplatin as one of the major cytotoxic photoproducts. Initial evidence was gathered from binding experiments with the RNA and DNA base model 5′-guanosine monophosphate (GMP). 1H NMR showed that irradiated Rf/1 solutions incubated with GMP (0–24 h) presented the diagnostic peak, corresponding to the cisplatin mono-GMP–PtII adduct (Fig. S43 and S44†).37 When incubated with pET28b as a model of double-stranded circular DNA (24 h, MES 1.5 mM, pH 6), light-activated Rf/1 (2.5:10 μM) inhibited the polymerase chain reactions (PCR). Thus, thirty seconds of light irradiation was sufficient to stop DNA amplification and reach a PCR inhibition level comparable to that of cisplatin (10 μM), hence confirming the capacity of this biorthogonal system to produce DNA-targeting species (Fig. S45†).38,39
Fluorescence microscopy results on PC-3 cells treated with either irradiated Rf/1 (30:120 μM) or cisplatin (120 μM) were in agreement with this scenario (Fig. 3b and S46†). In both cases, images obtained after 48 h of incubation showed increased percentage of apoptotic versus viable cells, together with changes in the cell morphology that are characteristic of apoptosis, i.e. cell shrinkage and rounding. Non-treated cells and cells treated with Rf/1 in the dark included as controls did not induce appreciable cell death.
Consistently, flow cytometry results confirmed that PC-3 cells exposed to the Rf/1 mixture and light (30:120 μM) died through apoptosis 48 hours after irradiation. Cisplatin induced comparable levels of apoptosis under the same conditions.
Double staining with Pacific Blue™ Annexin V/SYTOX® allowed to differentiate between early-stage and late-stage apoptosis. Upon treatment with irradiated Rf/1, the percentage of early apoptotic cells was 16.7 ± 2.8% against 19.9 ± 1.3% obtained for cisplatin. Rf, 1, and Rf/1 in the dark exhibited no significant population of the cells in either stage of apoptosis after 48 h of incubation (Fig. 3c).
Conclusions
In summary, we described the first photocatalytic activation of a metal-based prodrug in solution and in a biological environment. Co-administration of the Rf/1 as a catalyst/substrate couple enabled photoconversion of 1 into biologically active species by light irradiation at 460 nm, a wavelength that is ineffective when directly applied to the prodrug. In addition, the Rf/1 prodrug activation strategy induced an anticancer activity comparable to that of cisplatin with light doses as low as 0.36 J cm−2; that is ca. 15–35 times lower than what is typically used for UVA and blue light activation of analogue platinum complexes.40,41The photocatalytic turnover of PtIV into PtII species is an attractive prospect to amplify the antineoplastic action of metal-based prodrugs in a locoregional manner. This is particularly relevant for platinum anticancer agents, which have some of the poorest absorption properties amongst photoactivatable metal complexes but are widely tested in preclinical work and nearly indispensable in clinical practice.
In principle, photocatalysis can help expand the therapeutic potential of platinum prodrugs. Efficient light activation of PtIV complexes through catalysis may help localize the cytotoxic effects of Pt drugs, increase their dosing at the tumour target, and reduce their systemic toxicity. Rf (vitamin B2) is a highly biocompatible molecule, and its capacity to function in a bioorthogonal fashion may serve to enhance the selectivity of metal-based drugs by minimizing side reactions.
Experimental section
Methods
NMR, UV-vis, and fluorescence characterization of light-irradiated Rf/1 and controls, PCR and microscopy data, photochemical quantum yield by actinometry, setup for cell work under light irradiation, computational methodology and instrumentation details are described in the ESI.†Light-irradiation experiments in solution
Photocatalysis studies on Rf/1 were performed under different solution conditions (buffers, cell culture medium, and in the presence of co-reactants). All the reactions were carried out in air at 298 K and pH 6, employing an LED light source (λexc = 460 nm, 2.5 mW cm−2). Reaction kinetics, rate laws of the reaction, turnover frequency (TOF), and total turnover number (TTN) were calculated by varying both the reactants and catalyst concentration and quantifying the amount of photoconverted 1via1H NMR.Cell viability studies
The antiproliferative activity of Rf/1 was determined in human prostate cancer cells PC-3 (ATCC) in the dark and under light irradiation by co-administering Rf and 1 at the fixed molar ratio (1:4) and using three different concentrations (Rf/1:10/40 μM, 20/80 μM, and 30/120 μM). MES was employed in all the cell experiments at the final concentration of 2 mM. PC-3 cells were seeded 24 h before the experiment in 96-well plates with a density of 4000 cells per well and grown under standard conditions (Ham's F-12K medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C, 5% CO2 and 90% humidity). Stock solutions of Rf/1 were added and incubated with cells for 1 h, light irradiated for 1 min at 460 nm (light dose 0.36 J cm−2), and then incubated for other 6 h. Finally, cells were washed in fresh medium and grown for other 42 h. The sulforhodamine B (SRB) colorimetric assay was used for cell density determination. As controls, Rf and 1 were tested under identical conditions, whereas parallel experiments were performed with Rf/1 and cisplatin kept in the dark. A home-made LED plate was employed to irradiate cells in 96-well plates (Fig. S47†).
Fluorescence microscopy assays
PC-3 cells exposed to 30:120 μM Rf/1 were viewed using a Zeiss Axio Observer wide field fluorescence microscope (Carl Zeiss). Cells were plated in an ibidi μ-Slide 0.4 (13500 PC-3 cells/channel) and allowed to adhere overnight before they were treated with Rf/1, as described for cell viability experiments (2 mM MES, 6 h of treatment plus 42 h incubation). Analysis of cellular morphological alteration was performed using a cell-permeable green fluorescent dye from the Live and Dead Cell Staining Kit (Abcam) for cell nuclei (green channel) and SYTOX® AADvanced™ (Invitrogen™) for dead cells (red channel). Cells were stained following commercial protocols and using binding or 10 mM PBS buffer at the end of the incubation period (48 h). Images were acquired using a Plan Apochromat 20× objective and a multi-band pass Colibri filter to obtain the fluorescence emission signals. Control experiments in the dark (no light irradiation) were performed on untreated cells and on the cells exposed to 30:120 μM Rf/1 or 120 μM cisplatin.
Flow cytometry analysis
PC-3 cells were seeded in 96-well plates (2000 cells per well) and treated with Rf/1 (30:120 μM) and MES (2 mM) as described above. After 48 hours of incubation, 7 wells for each sample were pooled into cytometer tubes; cells were washed with 10 mM PBS and stained using 100 μL per tube of the Pacific Blue™ Annexin V/SYTOX® AADvanced™ flow cytometry kit (Invitrogen™). Early and late apoptosis were measured using a FACS Canto II (BD Biosciences) and results were analysed using the FlowJo, LCC software. The PC-3 population was electronically gated based on the forward and side scatter parameters and the non-single events left out based on the forward area and height scatter parameters. Inside this final population, live cells were gated as negative for both dyes; early apoptotic cells were defined as Pacific Blue-Annexin V positive cells and SYTOX® negative, whereas late apoptotic cells were gated as double positive for both dyes. Non-labelled and singly labelled samples were included as a control and as compensation samples, respectively. Experiments were repeated three times. In addition to untreated cells, control experiments included cells treated with 30:120 μM Rf/1 and 120 μM cisplatin in the dark (no light irradiation) and light-irradiated Rf and 1 alone.
Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness (grant CTQ2016-80844-R, BES-2013-065642, BIO2014-61838-EXP and BIO2015-69887-R), the Department of Industry of the Basque Country (grant ETORTEK). L. S. thanks the MICINN of Spain for the Ramón y Cajal Fellowship RYC-2011-07787 and the MC CIG fellowship UCnanomat4iPACT (grant no. 321791). L. S., F. L. G. and J. M. R. thank Ikerbasque for funding. We are also grateful to the members of the European COST Actions CM1105, CM103 and CM1403 for stimulating discussions.References
- C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 651–653 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- A. Unciti-Broceta, Nat. Chem., 2015, 7, 538–539 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS PubMed.
- S. V. Chankeshwara, E. Indrigo and M. Bradley, Curr. Opin. Chem. Biol., 2014, 21, 128–135 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Angew. Chem., Int. Ed., 2014, 53, 3941–3946 CrossRef CAS PubMed.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- P. K. Sasmal, S. Carregal-Romero, W. J. Parak and E. Meggers, Organometallics, 2012, 31, 5968–5970 CrossRef CAS.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 Search PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- L. Gong, Z. Lin, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2010, 49, 7955–7957 CrossRef CAS PubMed.
- M. Fontecave, ChemCatChem, 2010, 2, 1533–1534 CrossRef CAS.
- E. Ruggiero, J. Hernández-Gil, J. C. Mareque-Rivas and L. Salassa, Chem. Commun., 2015, 51, 2091–2094 RSC.
- I. Infante, J. M. Azpiroz, N. G. Blanco, E. Ruggiero, J. M. Ugalde, J. C. Mareque-Rivas and L. Salassa, J. Phys. Chem. C, 2014, 118, 8712–8721 CAS.
- C. R. Maldonado, N. Gómez-Blanco, M. Jauregui-Osoro, V. G. Brunton, L. Yate and J. C. Mareque-Rivas, Chem. Commun., 2013, 49, 3985–3987 RSC.
- Flavins and Flavoproteins, ed. S. Weber and E. Schleicher, Springer New York, New York, NY, 2014, vol. 1146 Search PubMed.
- P. F. Heelis, Chem. Soc. Rev., 1982, 11, 15–39 RSC.
- G. de Gonzalo and M. W. Fraaije, ChemCatChem, 2013, 5, 403–415 CrossRef CAS.
- C. Feldmeier, H. Bartling, K. Magerl and R. M. Gschwind, Angew. Chem., Int. Ed., 2015, 54, 1347–1351 CrossRef CAS PubMed.
- B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427–430 CrossRef PubMed.
- M. Insińska-Rak and M. Sikorski, Chem.–Eur. J., 2014, 20, 15280–15291 CrossRef PubMed.
- C. J. Baker, N. M. Mock, D. P. Roberts, K. L. Deahl, C. J. Hapeman, W. F. Schmidt and J. Kochansky, Free Radicals Biol. Med., 2007, 43, 1322–1327 CrossRef CAS PubMed.
- G. Zhao and N. D. Chasteen, Anal. Biochem., 2006, 349, 262–267 CrossRef CAS PubMed.
- S. L. Hopkins, B. Siewert, S. H. C. Askes, P. Veldhuizen, R. Zwier, M. Heger and S. Bonnet, Photochem. Photobiol. Sci., 2016, 15, 644–653 CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 6019 RSC.
- M. A. Sheraz, S. H. Kazi, S. Ahmed, Z. Anwar and I. Ahmad, Beilstein J. Org. Chem., 2014, 10, 1999–2012 CrossRef PubMed.
- P. Gramatica, E. Papa, M. Luini, E. Monti, M. B. Gariboldi, M. Ravera, E. Gabano, L. Gaviglio and D. Osella, J. Biol. Inorg Chem., 2010, 15, 1157–1169 CrossRef CAS PubMed.
- C. Garino and L. Salassa, Philos. Trans. R. Soc., A, 2013, 371, 20120134 CrossRef PubMed.
- G. Thiabaud, R. McCall, G. He, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Angew. Chem., Int. Ed., 2016, 55, 12626–12631 CrossRef CAS PubMed.
- A. Garaikoetxea Arguinzoniz, N. Gómez Blanco, P. Ansorena Legarra and J. C. Mareque-Rivas, Dalton Trans., 2015, 44, 7135–7138 RSC.
- F. J. Dijt, G. W. Canters, J. H. J. Den Hartog, A. T. M. Marcelis and J. Reedijk, J. Am. Chem. Soc., 1984, 106, 3644–3647 CrossRef CAS.
- C. Ducani, A. Leczkowska, N. J. Hodges and M. J. Hannon, Angew. Chem., Int. Ed., 2010, 49, 8942–8945 CrossRef CAS PubMed.
- A. Terenzi, C. Ducani, L. Male, G. Barone and M. J. Hannon, Dalton Trans., 2013, 42, 11220–11226 RSC.
- J. Kasparkova, H. Kostrhunova, O. Novakova, R. Křikavová, J. Vančo, Z. Trávníček and V. Brabec, Angew. Chem., Int. Ed., 2015, 54, 14478–14482 CrossRef CAS PubMed.
- Y. Zhao, J. A. Woods, N. J. Farrer, K. S. Robinson, J. Pracharova, J. Kasparkova, O. Novakova, H. Li, L. Salassa, A. M. Pizarro, G. J. Clarkson, L. Song, V. Brabec and P. J. Sadler, Chem.–Eur. J., 2013, 19, 9578–9591 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the methods employed, photocatalysis studies in solution and in biological environments, DFT calculations. See DOI: 10.1039/c7sc01109a |
| This journal is © The Royal Society of Chemistry 2017 |