
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diversity-oriented synthesis of heterocycles and macrocycles by controlled reactions of oxetanes with α-iminocarbenes†
Alejandro
Guarnieri-Ibáñez
a,
Florian
Medina
a,
Céline
Besnard
b,
Sarah L.
Kidd
c,
David R.
Spring
 c and
Jérôme
Lacour
*a
c and
Jérôme
Lacour
*a
aDepartment of Organic Chemistry, University of Geneva, quai Ernest Ansermet 30, CH-1211 Geneva 4, Switzerland
bLaboratory of Crystallography, University of Geneva, quai Ernest Ansermet 24, CH-1211 Geneva 4, Switzerland
cDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 12th June 2017
Abstract
Using N-sulfonyl triazoles as substrates, compounds as diverse as 2-imino tetrahydrofurans, 13- and 15-membered ring aza-macrocycles can be prepared selectively via formal [1 + 4], [5 + 4 + 4] and [3 + 4 + 4 + 4] condensations of α-imino carbenes and oxetanes under Rh(II)-catalysis or thermal activation. Spirocyclic N-heterocycles are also accessible by means of Buchwald–Hartwig and Pictet–Spengler cyclizations. By reaction control, substrate selection or further derivatization, a large variety of chemical structures is thus achievable. Finally, using triazoles reacting under thermal activation, interesting mechanistic insight was obtained.
N-Sulfonyl-1,2,3-triazoles, 1, are important building blocks used routinely in synthetic, biological and medicinal chemistry.1 These compounds are readily accessible through the Cu(I)-catalyzed azide alkyne cycloadditions (CuAACs) of alkynes and sulfonyl azides.2 Importantly, N-sulfonyl-triazoles, 1, are in equilibrium with α-diazo imines of type 1′ that decompose under metal-catalyzed conditions to afford electrophilic α-imino carbenes, 2 (Scheme 1, top).3 These intermediates have received much attention in recent years as they afford synthetically useful and original processes, from migrations to ylide forming reactions and subsequent transformations.4,5 In terms of oxonium ylide chemistry, under dirhodium catalysis, classical cyclic ethers like THF exhibit only moderate reactivity when used as substrates.6 It is necessary to utilize activated nucleophiles such as acetals (1,3-dioxolanes, 1,3-dioxanes and 1,3,5-trioxane)7,8 or epoxides9 to yield condensation products (6 to 9-membered rings, Scheme 1, top). Herein, the reactions of sulfonyl triazoles, 1, with oxetanes, 3, are reported (Scheme 1, bottom). Under dirhodium catalysis (Rh2(S-TCPTTL)4, 1–2 mol%) and at high temperature (100 °C), either 5-membered or 15-membered ring heterocycles are generated. In fact, using regular concentration conditions (0.1 M in CH2Cl2), 2-iminotetrahydrofurans, 4, are formed preferentially while unsaturated aza-macrocycles, 5, are isolated when using oxetanes as the solvent and with high concentration (1.0 M). These two sets of conditions favor the condensations of one and three oxetane moieties respectively, and hence the formation of compounds 4 and 5 occurs in the [1 + 4] and [3 + 4 + 4 + 4] processes respectively.
 | ||
| Scheme 1 Diversity-oriented synthesis of heterocycles and macrocycles by reaction of N-sulfonyl-1,2,3-triazoles, 1, with cyclic ethers, 3. | ||
Additionally and interestingly, using alkyl instead of aryl sulfonyl triazoles, interesting 13-membered heterocycles, 6, are generated by the reaction of two oxetanes with the involvement of a S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond as an intramolecular nucleophile. Favorable structural diversity is hence created through control of the reaction conditions or substrate selection. Insight into the mechanistic pathways was also obtained using triazoles reacting under thermal activation specifically.10,11 Finally, the straightforward syntheses of spiro N-heterocycles, such as indoline 7 and tetrahydroquinoline 8, were achieved by means of Buchwald–Hartwig and Pictet–Spengler cyclizations. Compounds 7 and 8 complete effectively the product diversity.12
O bond as an intramolecular nucleophile. Favorable structural diversity is hence created through control of the reaction conditions or substrate selection. Insight into the mechanistic pathways was also obtained using triazoles reacting under thermal activation specifically.10,11 Finally, the straightforward syntheses of spiro N-heterocycles, such as indoline 7 and tetrahydroquinoline 8, were achieved by means of Buchwald–Hartwig and Pictet–Spengler cyclizations. Compounds 7 and 8 complete effectively the product diversity.12
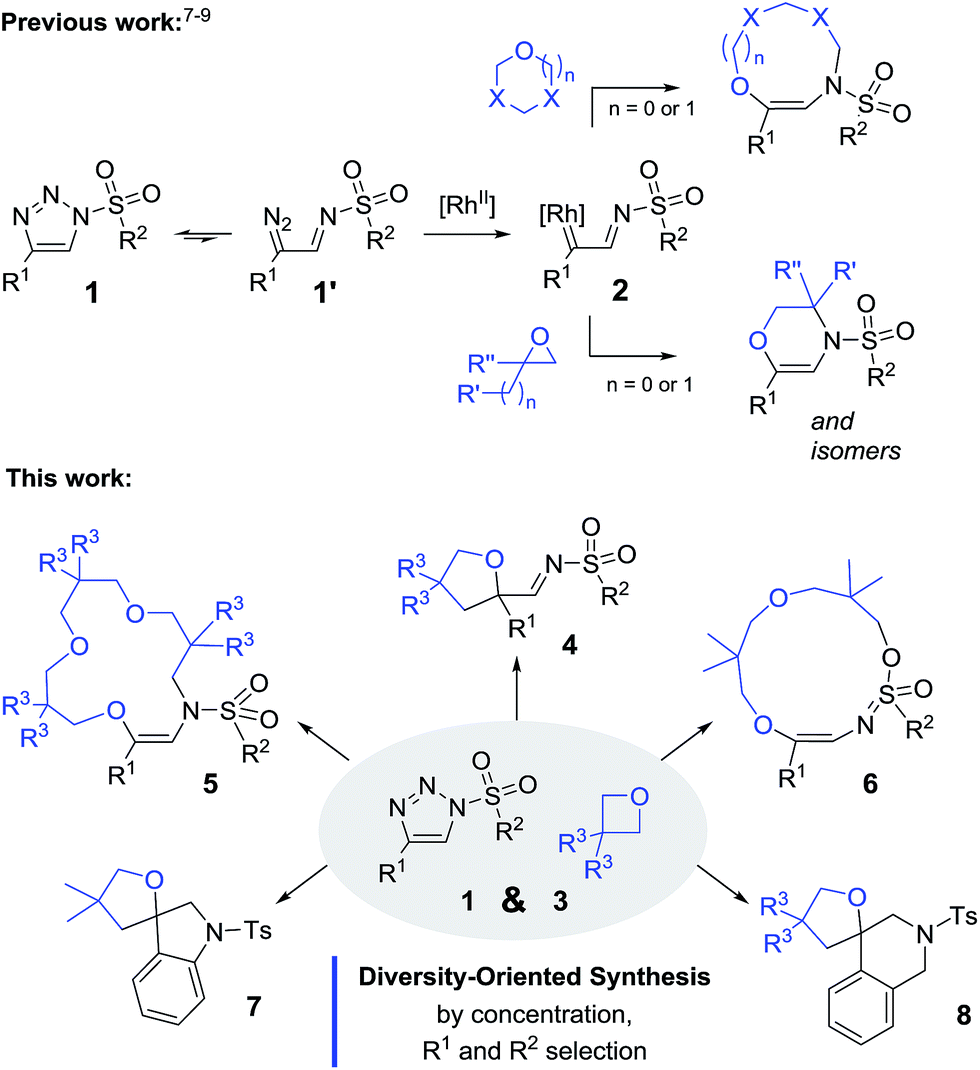
The transformation of oxetanes, 3, into tetrahydrofurans by reaction with α-keto carbenes was first reported by Noyori and Nozaki, with the reactive carbene intermediates generated in situ by the Cu-catalyzed decomposition of diazo ester reagents.13 This interesting ring expansion reaction has generated sustained interest in the synthetic organic community.14 Drawing from the direct analogy that exists between α-keto carbenes and α-imino intermediates, 2, it was desirable to test the reactivity of oxetanes in the presence of N-sulfonyl-1,2,3-triazoles and dirhodium catalysts. A series of reagents, 1a–1i, was prepared using CuAAC.15 Initial experiments were performed using N-tosyl-4-phenyltriazole, 1a (R1 = Ph, R2 = pTol). The results of the optimization studies are reported in Table S1.† Rh2(S-TCPTTL)4 was found to be generally effective (1 mol%); complete consumption of 1a was reached after 3 h at 100 °C using a slight excess of 3,3-dimethyloxetane, 3A (1.5 equiv.), and CH2Cl2 as the solvent (0.1 M). 1H-NMR spectroscopic analysis of the crude reaction mixture indicated the clean formation of imino products of type 4. However, and not surprisingly, compound 4aA was found to be unstable on silica gel or alumina. Product isolation was achieved by reduction with LiAlH4 (1.5 equiv., 20 °C). The resulting amino tetrahydrofuran, 9aA, was isolated in 80% yield over two steps, the structure of which was confirmed by X-ray diffraction analysis (Scheme 2, top).16 With these conditions in hand, triazoles 1b–h were tested with oxetane 3A to form the corresponding tetrahydrofurans. Substitutions at the ortho and para positions of the aromatic groups (R1) led to the formation of 9bA–9dA with moderate yields (58–68%). Other R1 substituents such as 6-methoxynaphthalene (1e) and thiophene (1f) were also compatible, leading to the expected products 9eA and 9fA in 74% and 75% yields respectively. The mesyl and nosyl derivatives afforded products 9gA and 9hA in 71% and 58% yields (R2 = Me and pNO2C6H4 respectively). Unsubstituted oxetane 3B, 3,3-bis(chloromethyl) oxetane 3C, and 2,6-dioxaspiro[3,3]heptane 3D were found to be reactive with 1a to afford products 9aB–9aD in moderate to good yields (44–75%).17 Finally, using Davies’ N-tosyl-4-ethoxytriazole, 1i, (R1 = OEt, R2 = pTol), we could probe the reaction under metal-free conditions.10b In fact, and in contrast to the previous derivatives, 1a–1h, triazole 1i decomposes by thermal activation only, with the strong π-donor ethoxy group (R1) stabilizing the transient donor/acceptor carbene. To our satisfaction, in the absence of Rh2(S-TCPTTL)4, product 9iA was isolated in 44% yield. Decreasing the thermal activation temperature to 60 °C improved the yield to 50% but at the expense of the reactivity (6 h reaction time).
 | ||
| Scheme 2 Reaction scope and isolated yields of 9. (a) Stick view of the crystal structure of 9aA; part of the tolyl group was removed for clarity. (b) Performed over 6 h. (c) Performed with 2 mol% of Rh2(S-TCPTTL)4 over 14 h. (d) Reduction with LiAlH4 at 0 °C over 30 min. (e) Metal-free at 60 °C over 6 h. | ||
While optimizing the reaction conditions for the formation of products 9 (Table S1†), it was noticed that the concentration of 1 and the ratio between 1 and 3 were strongly influencing the formation of the oxolane product. For instance, using oxetane 3A as the solvent, we observed a variety of other products in the crude reaction mixtures, including a large proportion of compounds 5 while these derivatives were absent under 0.1 M conditions; the nature of the byproducts will be discussed later in the manuscript.18 NMR spectroscopic analysis of 5 was compatible only with a macrocyclic structure made by the addition of three oxetane units onto one α-imino carbene moiety.19,20
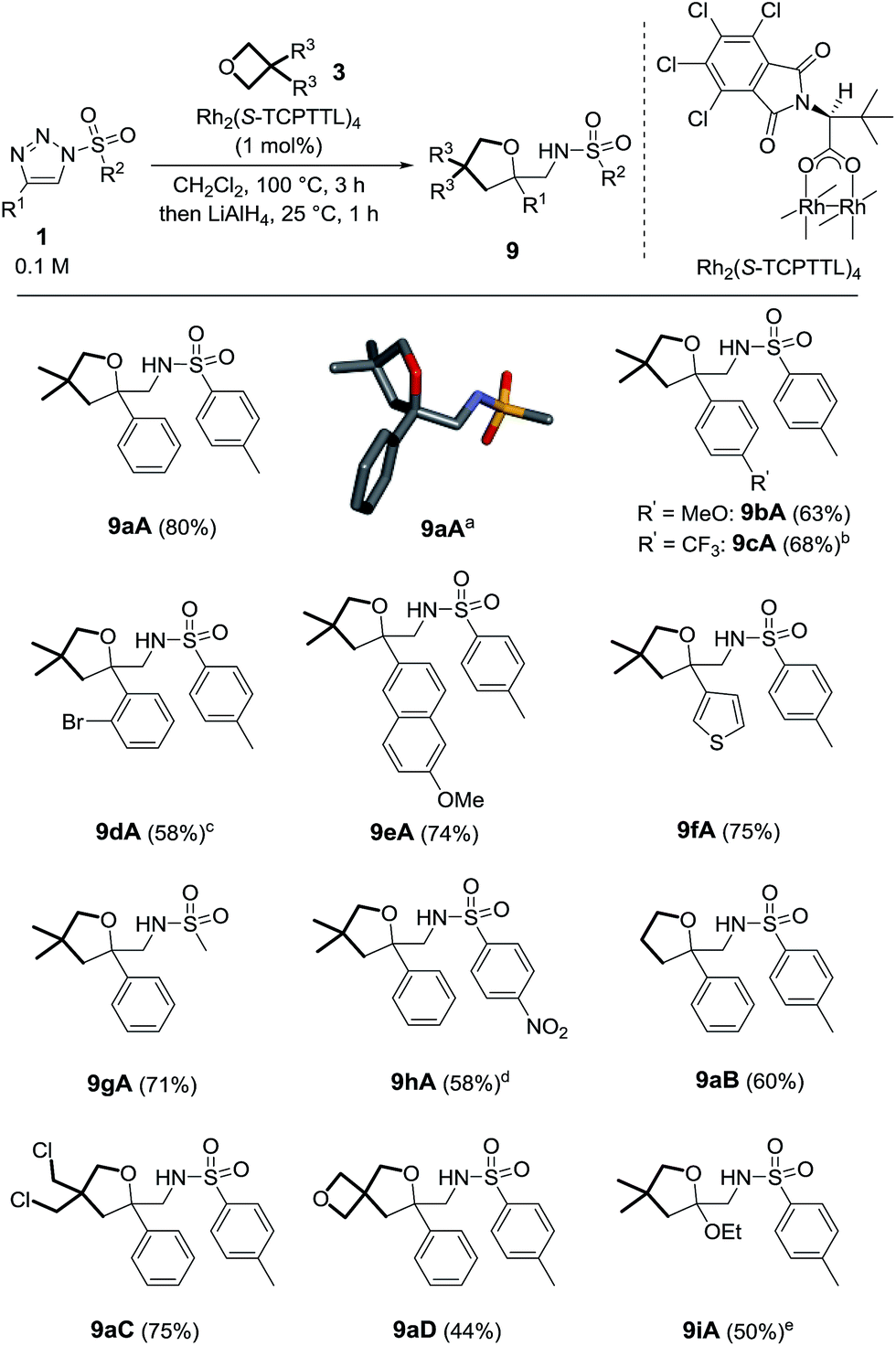
The exact structure of compounds 5 was confirmed later by X-ray diffraction analysis (Scheme 3).21 Considering that the synthesis of functionalized macrocycles remains a challenge, particularly under high concentration conditions,22 it was deemed interesting to further study the one-pot synthesis of such 15C4 derivatives. Optimization experiments were performed using N-bromophenylsulfonyl-4-phenyltriazole 1k (R1 = Ph, R2 = pBrC6H4) and the results are reported in Table S2.† With Rh2(OAc)4 as the catalyst (1 mol%) and 3,3-dimethyloxetane 3A as the solvent, complete consumption of triazole 1k (0.5 M) was reached after 5 h at 100 °C to form 5kA in 28% yield. Higher concentration conditions afforded better catalytic activity, higher conversions and similar yields for the same amount of time. With 1k, the highest yield (46%) was afforded using Rh2(S-TCPTTL)4 with a loading of 1 mol% in pure 3,3-dimethyloxetane.23 Such a yield could appear low in comparison to those obtained in the previous transformation (1 → 9, Scheme 2) but, in regard to the interesting poly-condensation [3 + 4 + 4 + 4] mechanism, it is quite remarkable.
 | ||
| Scheme 3 Reaction scope and yields of 5. (a) Stick view of the crystal structure of 5kA; part of the pBrC6H4 group was removed for clarity. (b) Performed with 2 mol% of Rh2(S-TCPTTL)4. | ||
With these conditions in hand, 5aA was obtained in 34% yield (Scheme 3). Substitutions at the para position of the aromatic group R1 led to the formation of 5bA24 and 5cA in 19% and 38% yields respectively. Ester 1j was also compatible with this transformation, leading to the formation of 5jA in 39% yield. Nosyl derivatives afforded 5hA in 57% yield. Increasing the catalyst loading to 2 mol% did not change the reaction time but did improve the results (64% yield). Slower reactions and lower yields were observed for macrocycles 5lA and 5mA (37% and 34% respectively). With unsubstituted oxetane 3B, similar observations were made. Prolonged reaction times were necessary for making 5aB, 5lB and 5mB (24 h, 10 h and 15 h), with the macrocycles being obtained in moderate yields (33–50%). 5hB and 5kB were afforded in 50% and 40% yields respectively.
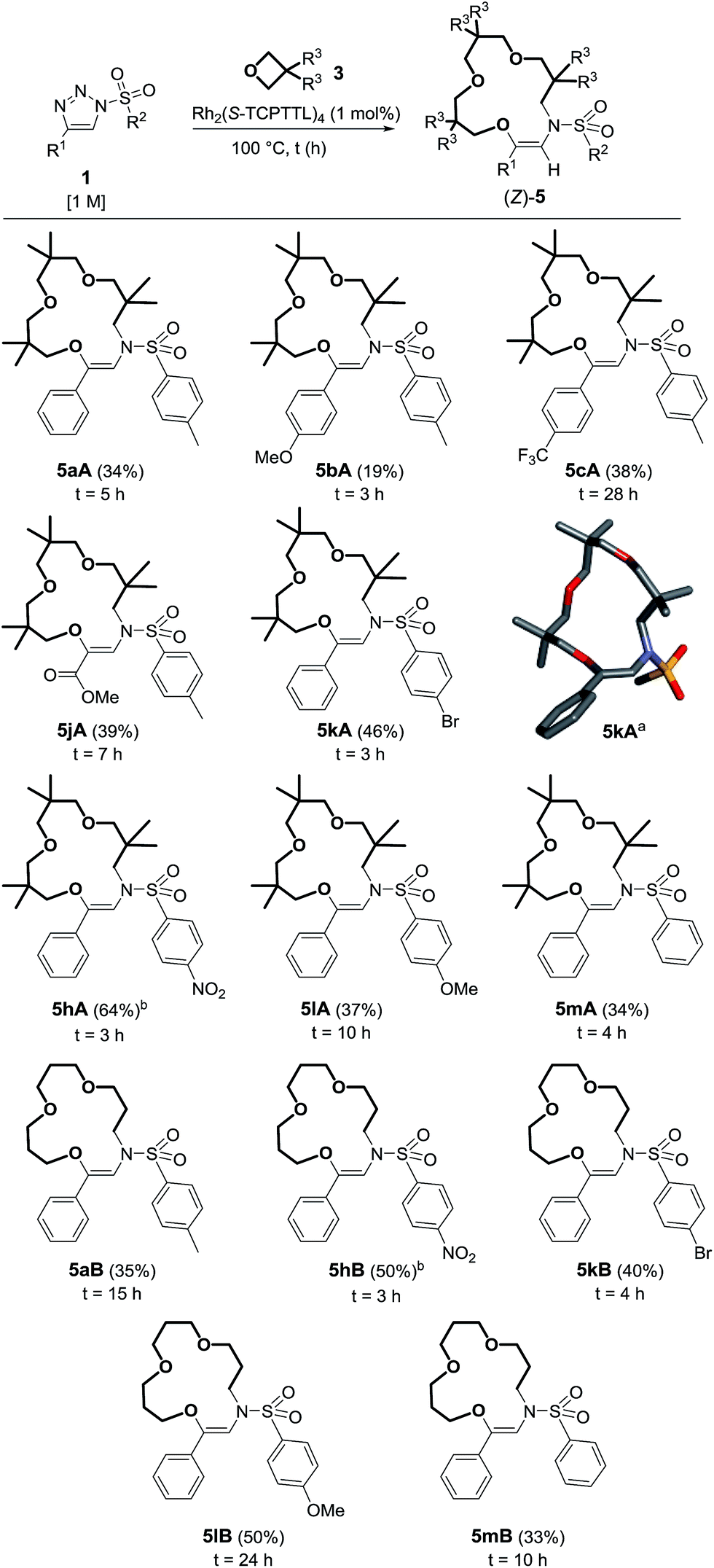
With reagents 1a, 1l and 1m (R2 = pTol, pMeOC6H4 or Ph), 1H-NMR spectroscopic and GC-MS analysis of the crude reaction mixtures revealed the additional presence of imines, 4, and two (by)products. Care was taken to isolate these moieties as detailed in Scheme 4 for the reaction of 1a with 3A. After careful analysis, 11- and 13-membered rings 10aA and 6aA from double oxetane addition were identified. They were generated in 5% and 11% yields respectively, while imine 4aA was present in low quantities (8% NMR yield).
 | ||
| Scheme 4 Observed products from the reaction of 1a and 3A. (a) NMR yield determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as a reference. (b) Isolated yields. | ||
The occurrence of compounds of type 6 was intriguing, particularly the presence of the intracyclic sulfonimidate group. To our knowledge, there have been only a few reports detailing the direct involvement of sulfonyl groups in α-imino carbene chemistry.25 An effort was therefore made to improve the synthesis of this type of macrocycle. As already observed in the literature,8 triazoles bearing methyl (alkyl) sulfonyl groups may afford different reactivity. This was also fortunately the case as triazole 1g yielded the 13-membered heterocycle 6gA predominantly (57% yield, Scheme 5). The exact structure of 6gA was confirmed by X-ray diffraction analysis. The reaction was performed with 1n and 1o to afford 6nA and 6oA in 48% and 61% yields respectively. In the case of trifluoromethylated 1n, it was necessary to use 2 mol% of catalyst. In all cases, macrocycles of type 5 were present in the crude reaction mixture but in low yields (10–20%).26,27
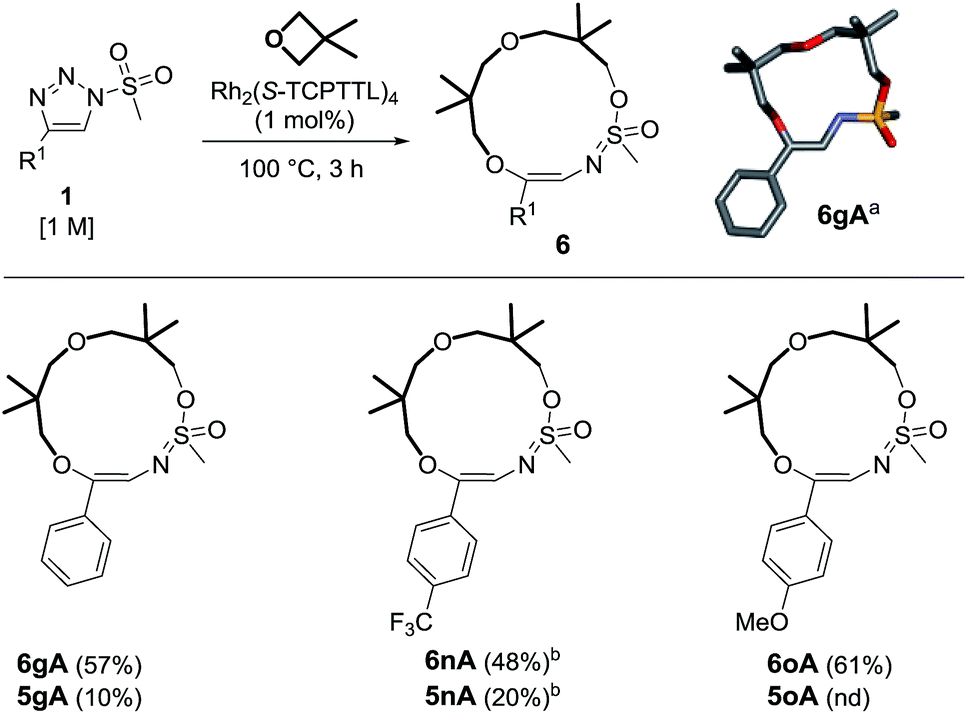 | ||
| Scheme 5 Reactivity of mesyl triazoles and 3,3-dimethyloxetane. Reagents and conditions: (a) stick view of the crystal structure of 6gA. (b) Performed with 2 mol% of Rh2(S-TCPTTL)4 over 7 h. | ||
At this stage, to gain some insight into the reaction mechanism, and into the influence (or lack thereof) of the metal catalyst on the macrocyclization steps, reactions were performed under thermal activation. Following the report of Alford and Davies on metal-free decompositions,10aN-sulfonyl-triazoles 1p and 1q bearing a phthalimido R1 group were prepared (R2 = C6H5CH2 or pMeOC6H4). These activated substrates were heated at 60 °C (1 M concentration in 3,3-dimethyloxetane). Full conversion was achieved after only 2 hours and the macrocyclic products 5p and 5q were isolated (Scheme 6).28 Surprisingly however, mixtures of Z and E-isomers were obtained, the configurations of which were ascertained by X-ray structural analysis (Fig. S1 and further details in the ESI†). The Z-configured products were found to be predominant (ratio ∼ 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The minor products, (E)-5p and (E)-5q, exhibit a trans orientation of the O and N-atoms across the double bond. Remarkably, preliminary calculations (DFT/BP86) indicate that the E-isomers of 5p and 5q are more stable than the Z-analogues by ∼2.1 kcal mol−1.29 This trend is attributed to a lower ring strain in the E-configured 15-membered cycles than in the Z geometries. A series of experiments was performed to possibly pinpoint the origin of this disparity. Firstly, the macrocyclization reaction of triazole 1p was performed in the presence of 1 mol% of Rh2(S-TCPTTL)4, and neither the yield nor the (Z):(E) ratio were modified by the presence of the metal complex.30 The pure isolated products, (Z)-5p and (E)-5p, were also heated at 60 °C in 3,3-dimethyloxetane for 15 h, in the absence or presence of Rh2(S-TCPTTL)4 (1 mol%). In these reactions, and those performed under the same conditions at 100 °C, evidence of the isomerization of either the (Z) or (E)-isomers of 5p could not be found. These results, combined with our calculations, indicate that products 5p and 5q are produced under kinetic control (no equilibration was observed).
:1). The minor products, (E)-5p and (E)-5q, exhibit a trans orientation of the O and N-atoms across the double bond. Remarkably, preliminary calculations (DFT/BP86) indicate that the E-isomers of 5p and 5q are more stable than the Z-analogues by ∼2.1 kcal mol−1.29 This trend is attributed to a lower ring strain in the E-configured 15-membered cycles than in the Z geometries. A series of experiments was performed to possibly pinpoint the origin of this disparity. Firstly, the macrocyclization reaction of triazole 1p was performed in the presence of 1 mol% of Rh2(S-TCPTTL)4, and neither the yield nor the (Z):(E) ratio were modified by the presence of the metal complex.30 The pure isolated products, (Z)-5p and (E)-5p, were also heated at 60 °C in 3,3-dimethyloxetane for 15 h, in the absence or presence of Rh2(S-TCPTTL)4 (1 mol%). In these reactions, and those performed under the same conditions at 100 °C, evidence of the isomerization of either the (Z) or (E)-isomers of 5p could not be found. These results, combined with our calculations, indicate that products 5p and 5q are produced under kinetic control (no equilibration was observed).
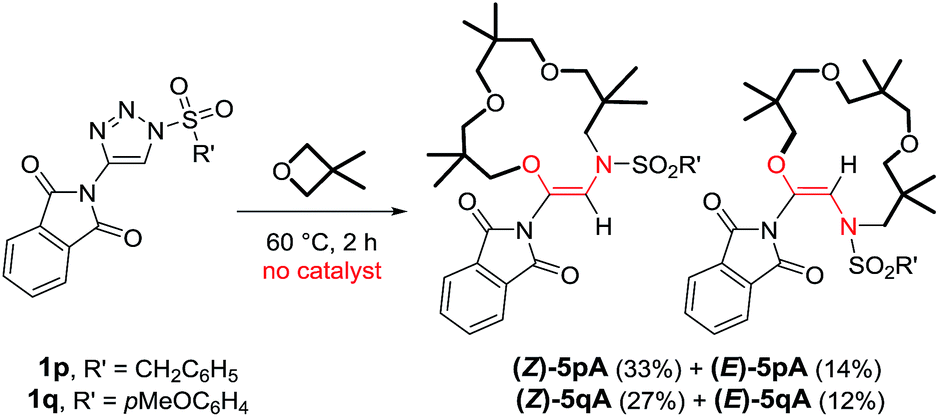 | ||
| Scheme 6 Reactivity of N-sulfonyl-4-phthalimido-1,2,3-triazoles 1p and 1q with 3,3-dimethyloxetane. | ||
The mechanistic rationales are outlined in Schemes 7 and 8. As discussed, in the presence of Rh(II) catalysts, N-sulfonyl triazoles, 1, generate α-imino metal carbenes, 2, by nitrogen extrusion. These electrophilic intermediates react with one molecule of oxetane to form oxonium ylides of type 11. The high nucleophilicity of the O-atom in both regular oxetane and 3,3-dimethyloxetane facilitates this step.31 Currently, the lack of stereoinduction in the reactions performed with Rh2(S-TCPTTL)4 tends to indicate the formation of metal-free ylides, rather than metal-bound species.
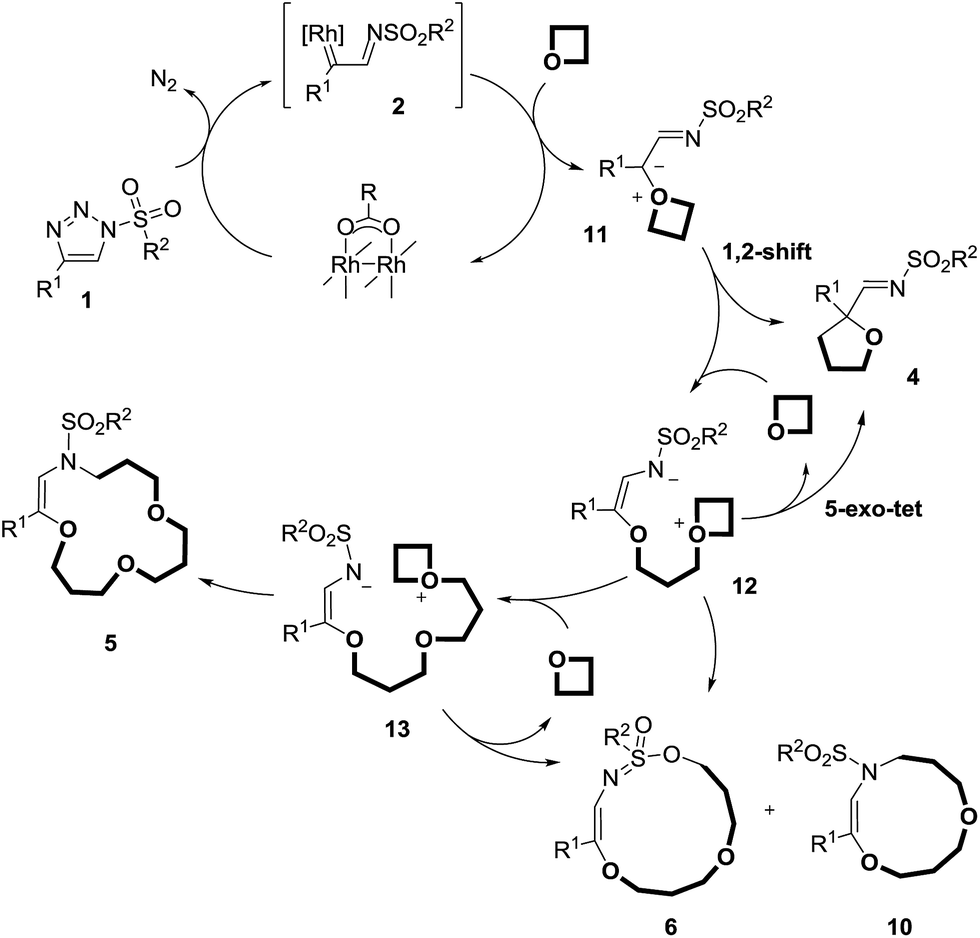 | ||
| Scheme 7 Rationale for the metal catalyzed pathway. | ||
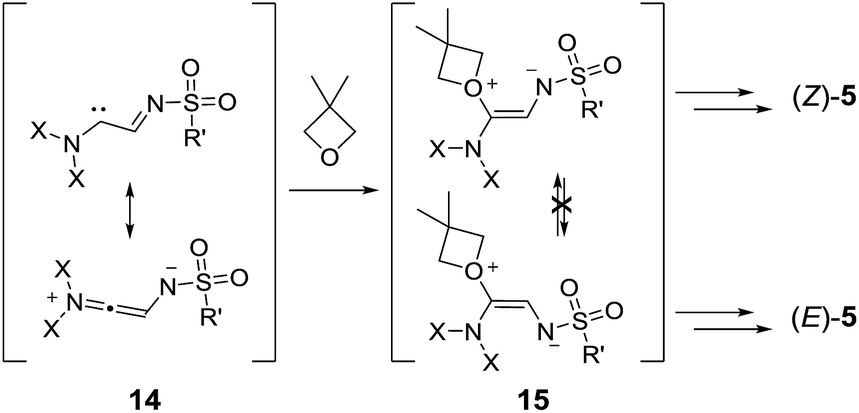 | ||
| Scheme 8 Formation of (Z)-5 and (E)-5. NX2 represents the phthalimido group. | ||
This highly reactive (ring-strained) intermediate can undergo a 1,2-shift and form adducts, 4; the intramolecular process is favored by “dilute” conditions (0.1 M). At high concentration (1 M conditions), 11 reacts with the best nucleophile in the medium, the oxetane itself,31a generating 12. This intermediate can generate products of type 4 again by means of a 5-exo-tet cyclization, or products 6 and 10via 11-endo-tet or 9-endo-tet cyclizations respectively. A third oxetane addition affords species 13, which are then trapped intramolecularly by the terminal nitrogen atom, leading to the final products, 5.20d The reasons for the sequential addition of three oxetanes is due primarily to the need for SN2-like transition states and/or trans-annular strain.20d
In thermally activated processes (1p and 1q), the situation is somewhat different. Resonance-stabilized carbene intermediates, 14, are formed by direct nitrogen extrusion (Scheme 8).10a Reactions with one equivalent of oxetane form ylide intermediates, 15, that are probably configurationally stable and exist in non-interconvertible (Z) and (E) forms. Further reactions with oxetanes and ring closures led to the formation of the macrocycles; the ratio between products 5pA and 5qA reflected the (Z):(E) ratio among the intermediates, 15.
With compounds 5 in hand, hydrogenation conditions were also developed. Not surprisingly, it was necessary to use more forcing conditions with sterically hindered 5A than with 5B (Scheme 9). In effect, Pd(OH)2 on activated charcoal, Pearlman’s catalyst, was required with adducts made from 3,3-dimethyloxetane, and derivatives of 16A were obtained in good to excellent yields (92–97%). While compounds carrying methoxy and methyl groups reacted without interference from the functional groups, derivative 5hA, substituted with a nitro moiety, afforded the amino compound, 16hA. With 16B, heterogeneous catalysis with Pd/C was possible and nitro group reduction was again observed. Treatment of N-macrocycles 16aA and 16aB with LiAlH4 in Bu2O at 120 °C resulted in the cleavage of the sulfonyl group,32 and free-amino building blocks 17aA and 17aB were isolated in 70% and 90% yields (Scheme 9).
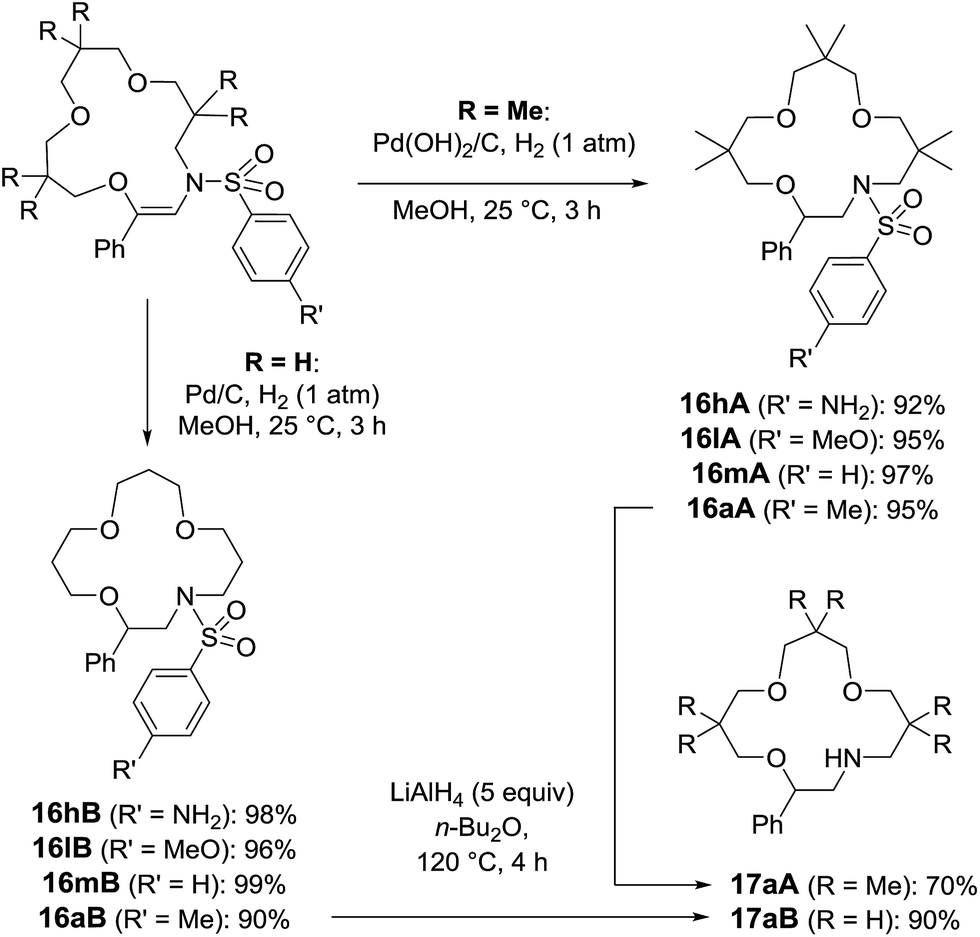 | ||
| Scheme 9 Hydrogenation and tosyl group removal conditions. | ||
Furthermore, care was taken to extend the structural diversity of the products made in this study by examining different trapping protocols for oxolane 4aA or derivatizations of compounds 9 (Scheme 10). Firstly, it was possible to hydrolyze imine 4aA to aldehyde 18 or transform it into dithiane3319 in 69% and 70% yields respectively (in two steps, with no isolation of 4aA required). In another set of experiments, the nosyl group of 9hA was successfully removed, leading to the formation of free amine 20 (65% yield).34 Direct construction of spiro N-heterocycles was achieved by intramolecular C–N bond formations. For instance, spiroindoline 7 was prepared from 9dAvia a Buchwald–Hartwig reaction (86% yield).35 2-Amino tetrahydrofurans 9aA, 9aB and 9aC were reacted with paraformaldehyde under acidic conditions to afford spirotetrahydroquinolines 8A, 8B and 8C in moderate to good yields (65–90%) via Pictet–Spengler cyclizations.5g,36
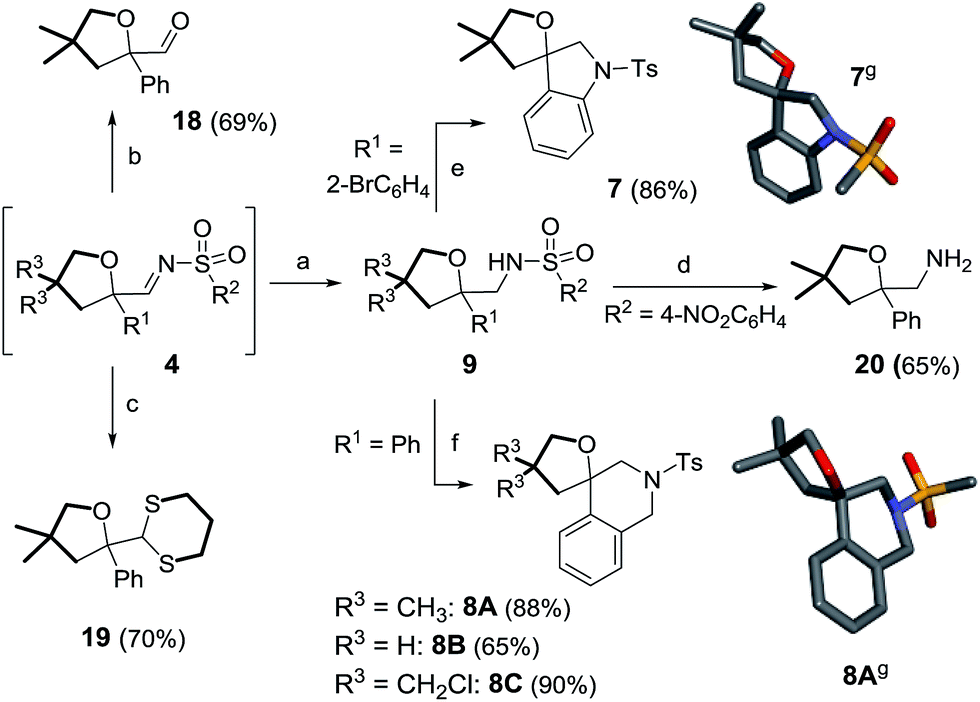 | ||
| Scheme 10 Post-functionalization of 4 or 9. Reagents and conditions: (a) LiAlH4, 25 °C, 1 h. (b) Silica gel, 25 °C, 24 h. (c) HS(CH2)3SH, ClSiMe3, Zn(OTf)2, 25 °C, 1 h. (d) PhSH, K2CO3, CH3CN/DMSO (49:1), 50 °C, 2 h. (e) Pd(OAc)2, ±BINAP, K2CO3, toluene, 115 °C, 14 h. (f) (HCHO)n, TFAA, MsOH, 1,2-DCE, 0 °C, 25 min. (g) Stick view of the crystal structures of 7 and 8A; part of the tolyl groups has been removed for clarity. | ||
Finally, with compounds 5–9 and 16–20 in hand, care was taken to evaluate the structural diversity generated by these derivatives. In fact, geometrically constrained macrocycles and spiro derivatives are attractive and possibly underrepresented targets in drug discovery.37 Due to the conformational pre-organization associated with these skeletons, such moieties often display higher affinity and selectivity for biological targets. Herein, using the classical tools of “Diversity-Oriented Synthesis”,12,38 we report the chemoinformatic analysis of the diversity of the library, in terms of (i) the molecular shape distribution via principal-moment of inertia (PMI) analysis (Fig. 1) and (ii) 15 chemo-physical properties via principal component analysis (PCA) (Fig. S3†), in comparison to two reference sets constituting 40 top-selling drugs and 60 natural products (see the ESI† for details). PMI analysis suggested that the DOS library exhibited a broad shape distribution, comparable to that of the natural product reference set, with decreased rod-like features compared to the drug reference set. Notably, macrocycle 5jA and spirocycle 7 displayed significant ‘spherical’ characteristics, populating the far right-hand side of the plot. Pleasingly, the DOS library also had considerable overlap with both reference sets in the PCA analysis, suggesting that the DOS library accessed attractive areas of chemical space and contained compounds with drug-like properties (Fig. S3†).
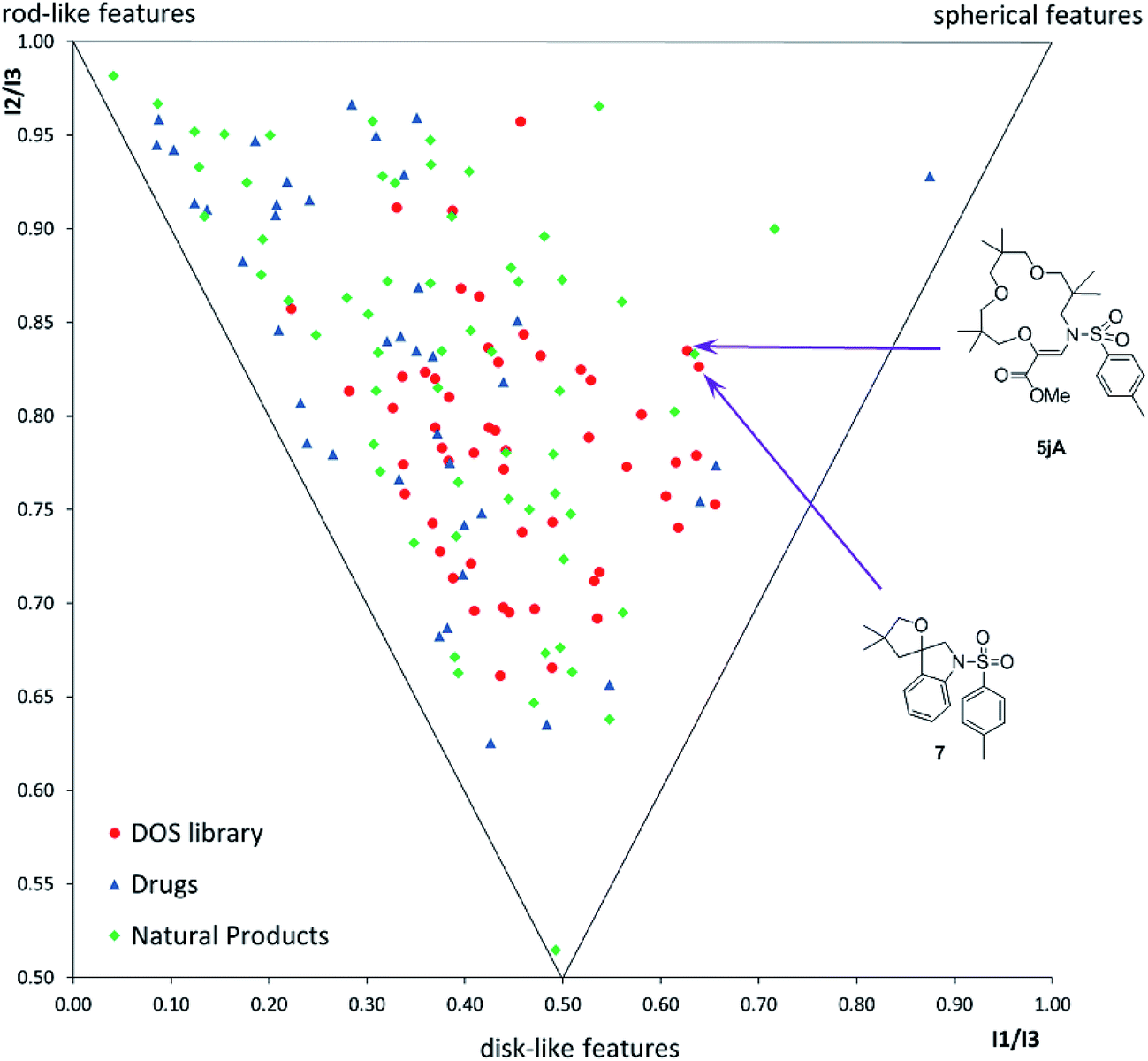 | ||
| Fig. 1 Comparative PMI plot of fifty-three DOS library compounds (red dots), 40 top-selling brand-name drugs (blue triangles) and 60 diverse-natural products (green rhombi). | ||
Conclusions
In summary, 2-iminotetrahydrofurans, 4, are readily formed by [1 + 4] condensations of α-imino carbenes and oxetanes under “dilute” conditions. These compounds possess a rather large range of reactivity, from reductions and hydrolyses to conversions into heterocycles such as spiroindolines, 7, or spirotetrahydroquinolines, 8. Under high concentration conditions (1 M), however, 13- and 15-azamacrocycles can be obtained via formal [5 + 4 + 4] and [3 + 4 + 4 + 4] condensations of α-imino carbenes and two/three oxetanes. In terms of mechanistic information and in the case of macrocycles 5 in particular, the E or Z-geometry of the unsaturated products was found to depend on the mode of carbene initiation – thermal activation vs. metal-catalysis. This afforded an interesting insight into the configurational stability of the subsequent ylide intermediates. Finally, chemoinformatic analysis suggested that the resulting library exhibited a broad molecular shape distribution, with significant spherical shape, covering attractive areas of chemical space.Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation for financial support. We also acknowledge the contributions of the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences, University of Geneva. We also thank Dr A. I. Poblador-Bahamonde for advice concerning the computational experiments.Notes and references
-
(a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
; (b) W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radić, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CrossRef CAS
-
(a) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed
-
(a) R. E. Harmon, F. Stanley, S. K. Gupta and J. Johnson, J. Org. Chem., 1970, 35, 3444–3448 CrossRef CAS
-
(a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS PubMed
- For selected Rh-catalyzed reactions of triazoles, see the following articles on C–H, C–O, C–S, N–H or O–H bond insertions:
(a) S. Chuprakov, J. A. Malik, M. Zibinsky and V. V. Fokin, J. Am. Chem. Soc., 2011, 133, 10352–10355 CrossRef CAS PubMed
- T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed
- F. Medina, C. Besnard and J. Lacour, Org. Lett., 2014, 16, 3232–3235 CrossRef CAS PubMed
- J. Pospech, R. Ferraccioli, H. Neumann and M. Beller, Chem. – Asian J., 2015, 10, 2624–2630 CrossRef CAS PubMed
-
(a) X. Ma, S. Pan, H. Wang and W. Chen, Org. Lett., 2014, 16, 4554–4557 CrossRef CAS PubMed
-
(a) J. S. Alford and H. M. L. Davies, Org. Lett., 2012, 14, 6020–6023 CrossRef CAS PubMed
- The presence of strong π-donor groups at position R1 stabilizes the transient donor/acceptor carbenes favoring their thermal decomposition.
-
(a) T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48–56 CrossRef CAS PubMed
- H. Nozaki, S. Moriuti, H. Takaya and R. Noyori, Tetrahedron Lett., 1966, 7, 5239–5244 CrossRef
-
(a) K. Friedrich, U. Jansen and W. Kirmse, Tetrahedron Lett., 1985, 26, 193–196 CrossRef CAS
- J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS PubMed
- Despite the use of an enantiopure catalyst, product 9aA was obtained in racemic form only. Reactions at 60 and 80 °C also yielded racemic materials.
- 2-Substituted oxetanes led to complex crude mixtures from which separated products could not be isolated by chromatography.
- Using oxetanes as the solvent, crude reaction mixtures usually contain products 4, 5, 6 and 10 in various proportions.
- Such a reactivity matches previously reported reactions of oxetanes with α-keto carbenes that yield polyoxa macrocycles, and 15-membered ring derivatives in particular. See ref. 20.
-
(a) W. Kirmse, R. Lelgemann and K. Friedrich, Chem. Ber., 1991, 124, 1853–1863 CrossRef CAS
- Products 5 are quite sensitive to acidic conditions decomposing upon standing over silica gel and after a few hours (days) in CDCl3. Linear keto-alcohols are then obtained.
-
(a) W. Zeghida, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2010, 49, 7253–7256 CrossRef CAS PubMed
- Increasing the catalyst loading did not improve the reaction significantly. Lower yields were obtained when co-solvents were used in 1:1 mixtures with 3,3-dimethyloxetane;
these conditions correspond to 5 equivalents of 3,3-dimethyloxetane. See the ESI† for details.
- Careful examination of the crude reaction mixture revealed the presence of imine 4 (18%, NMR yield).
-
(a) D. Frank, G. Himbert and M. Regitz, Chem. Ber., 1978, 111, 183–188 CrossRef CAS
- In the case of 5oA, this compound was detected in the crude mixture but its isolation was not possible.
- N-Mesyl triazole derivatives substituted either by ethoxy or phthalimido groups as R1 substituents react, under thermal activation (see Scheme 6 and associated paragraphs), to give products different to 13-membered rings 6. In fact, the ethoxy derivative favors the formation of a THF derivative of type 4 while the phthalimido substrate leads to the formation of 15-membered 5 predominantly.
- Activated triazole 1i reacts under these reaction conditions to afford a tetrahydrofuran skeleton (9i after reductive workup) and not the corresponding macrocycles. In all likelihood, the better donating group ability of the ethoxy substituent facilitates the 1,2-shift process.
- Using the same in silico conditions, the energies of the observed (Z) and putative (E) isomers of 5kA have been calculated. A lower difference of 0.6 kcal mol−1 is observed, still in favor of the (E) configuration.
- This result is in line with previous observations of the Davies’ group that chiral catalysts have no stereochemical influence on the reactions of triazole of type 1p. See ref. 10.
-
(a) S. Searles, M. Tamres and E. R. Lippincott, J. Am. Chem. Soc., 1953, 75, 2775–2777 CrossRef CAS
- D. Klamann, Monatsh. Chem., 1953, 84, 651–654 CrossRef CAS
- A. Boyer, Org. Lett., 2014, 16, 5878–5881 CrossRef CAS PubMed
-
(a) T. Fukuyama, C.-K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373–6374 CrossRef CAS
-
(a) J. P. Wolfe, S. Wagaw and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 7215–7216 CrossRef CAS
-
(a) O. O. Orazi, R. A. Corral and H. Giaccio, J. Chem. Soc., Perkin Trans. 1, 1986, 1977–1982 RSC
-
(a) H. S. G. Beckmann, F. Nie, C. E. Hagerman, H. Johansson, Y. S. Tan, D. Wilcke and D. R. Spring, Nat. Chem., 2013, 5, 861–867 CrossRef CAS PubMed
-
(a) G. L. Thomas, R. J. Spandl, F. G. Glansdorp, M. Welch, A. Bender, J. Cockfield, J. A. Lindsay, C. Bryant, D. F. J. Brown, O. Loiseleur, H. Rudyk, M. Ladlow and D. R. Spring, Angew. Chem., Int. Ed., 2008, 47, 2808–2812 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, 1H/13C NMR and HR mass spectra are provided. CCDC 1443618–1443620 and 1534812–1534815. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00964j |
| This journal is © The Royal Society of Chemistry 2017 |