
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransfer of photosynthetic NADP+/NADPH recycling activity to a porous metal oxide for highly specific, electrochemically-driven organic synthesis†
Bhavin
Siritanaratkul
a,
Clare F.
Megarity
a,
Thomas G.
Roberts
a,
Thomas O. M.
Samuels
c,
Martin
Winkler
b,
Jamie H.
Warner
 c,
Thomas
Happe
b and
Fraser A.
Armstrong
*a
c,
Thomas
Happe
b and
Fraser A.
Armstrong
*a
aDepartment of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: fraser.armstrong@chem.ox.ac.uk
bAG Photobiotechnologie Ruhr-Universität Bochum, 44801 Bochum, Germany
cDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK
First published on 5th May 2017
Abstract
In a discovery of the transfer of chloroplast biosynthesis activity to an inorganic material, ferredoxin–NADP+ reductase (FNR), the pivotal redox flavoenzyme of photosynthetic CO2 assimilation, binds tightly within the pores of indium tin oxide (ITO) to produce an electrode for direct studies of the redox chemistry of the FAD active site, and fast, reversible and diffusion-controlled interconversion of NADP+ and NADPH in solution. The dynamic electrochemical properties of FNR and NADP(H) are thus revealed in a special way that enables facile coupling of selective, enzyme-catalysed organic synthesis to a controllable power source, as demonstrated by efficient synthesis of L-glutamate from 2-oxoglutarate and NH4+.
Introduction
The small flavoenzyme (45 kDa) ferredoxin–NADP+ reductase (FNR) is the pivotal link between photosynthetic electron transfer and biosynthesis.1,2 Located in the stromal space of chloroplasts, FNR receives the energetic electrons dispensed from photosystem I via ferredoxin, a strongly reducing one-electron carrier protein, and catalyses the two-electron reduction of NADP+ to NADPH, thereby recycling the unique reducing agent for the Calvin cycle that assimilates CO2. Many other biosynthetic processes depend on NADP+/NADPH recycling, for which there is considerable technological interest in achieving a wide range of stereo- and regio-specific organic reactions using enzymes – ease, efficiency and kinetic performance being key considerations.As shown in Fig. 1A, electron transfer between ferredoxin and FNR to produce NADPH is essentially an electrochemical process, the essence of which might be realized at a suitable electrode material, thereby creating an ‘electrochemical leaf’. Fig. 1B and C show the structure of FNR in its complex with ferredoxin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NADP+ has been incorporated using data from another structure of FNR containing bound NADP+, thus forming the ternary complex implicated by kinetic studies.5 The interaction between FNR and ferredoxin arises mainly from electrostatic attractions: ferredoxin is a negatively charged protein with a large excess of acidic amino acids, and the contact surface of FNR is dominated by basic residues. The shortest distance between the [2Fe–2S] cluster of ferredoxin and the FAD group of FNR is approximately 6 Å, which allows rapid, direct electron transfer.3
:NADP+ has been incorporated using data from another structure of FNR containing bound NADP+, thus forming the ternary complex implicated by kinetic studies.5 The interaction between FNR and ferredoxin arises mainly from electrostatic attractions: ferredoxin is a negatively charged protein with a large excess of acidic amino acids, and the contact surface of FNR is dominated by basic residues. The shortest distance between the [2Fe–2S] cluster of ferredoxin and the FAD group of FNR is approximately 6 Å, which allows rapid, direct electron transfer.3
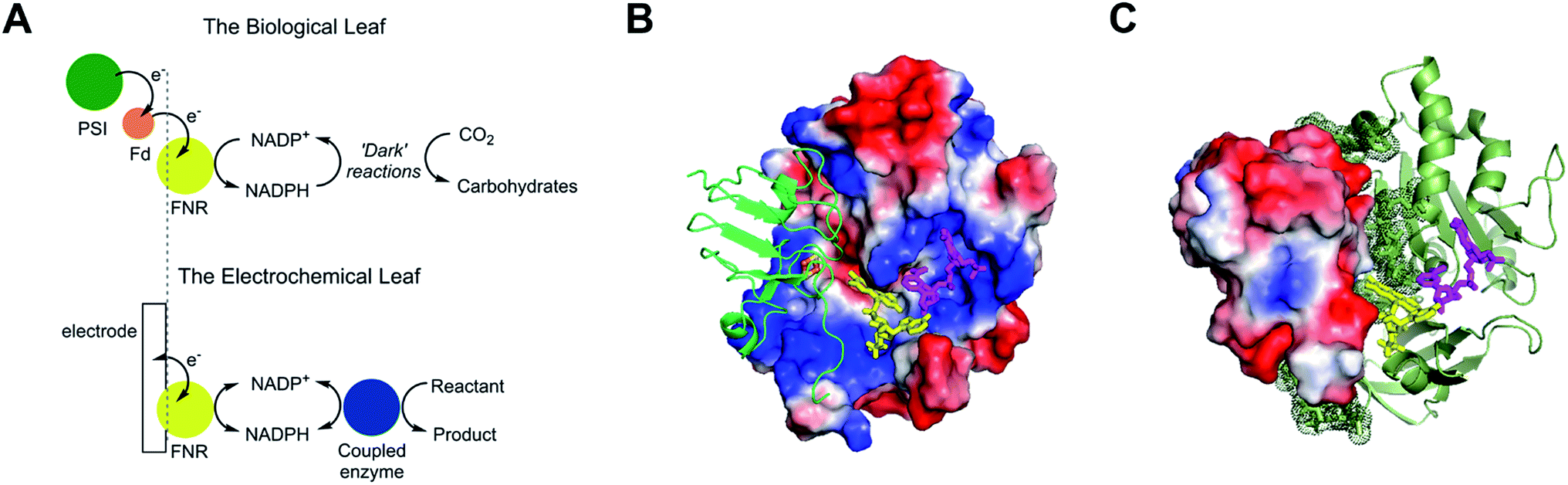 | ||
| Fig. 1 Structure and function of ferredoxin–NADP+ reductase (FNR). (A) Cartoon diagram comparing the concept of the ‘electrochemical leaf’ with the biological leaf. (B) and (C) Show structure of FNR complexed with ferredoxin (pdb 1GAQ).1 The position of bound NADP+ was included within the same structure (Pymol) by aligning the ferredoxin:FNR complex (pdb 1GAQ) with the structure of FNR in complex with NADP+ (pdb 1GJR);4 the protein structure from pdb 1GJR was then deleted to leave only the NADP+ molecule in position within the ferredoxin:FNR complex. (B) Projection in which FNR is shown as a charge-smoothed surface (red and blue represent positive and negative charges, respectively) and bound ferredoxin is shown in green, as cartoon main chain. (C) Projection in which ferredoxin is shown as a charge-smoothed surface and FNR is shown as cartoon main chain in green. In both projections, FAD is represented as yellow sticks, and NADP+ is represented as mauve sticks. | ||
Guided by the natural interaction with ferredoxin, we investigated porous conducting metal oxides as candidates for the incorporation of FNR to reveal, directly, the electrocatalytic properties of the FAD cofactor, and yield a robust material for rapid cofactor recycling. Indium tin oxide (ITO), manufactured on a large scale for electronic displays, has been extensively used as an electrode for protein electrochemistry,6–8 and with an isoelectric point around pH 6,9 its surface should be negatively charged in the range pH 6–9 useful for enzyme-catalysed organic synthesis. In this article, we describe the remarkable electrocatalytic activity of FNR when immobilized on and within porous ITO: the discovery leads to the reversible electrochemistry of NADP+/NADPH and potentially limitless opportunities, either for selective synthesis or sensors, through coupling to enzyme-catalysed organic reactions that use this cofactor system.
Results and discussion
FNR under non-turnover conditions
Fig. 2A shows a scanning electron microscope (SEM) image of an ITO electrode, constructed by electrophoretically depositing ITO nanoparticles onto an ITO glass slide (see Experimental methods). The individual ITO particle sizes were estimated to be 50 nm, the pore apertures were in the order of 100 nm in diameter, and the typical ITO layer thickness was 1–3 μm. The ITO layer was also constructed on a pyrolytic graphite edge (PGE) electrode or on flexible Ti foil (later, wire), giving analogous electrochemical results but greatly increasing technological scope in terms of scalability. In view of the likelihood that numerous porous metal oxides (MO) and support materials will prove to be suitable, we will adopt, wherever generic terminology is most appropriate, the nomenclature ‘FNR@MO/support’ to describe the hybrid electrode materials that we next investigated in more detail. | ||
| Fig. 2 Electrochemistry of ferredoxin–NADP+ reductase (FNR) immobilized at an ITO electrode, demonstrating rapid and reversible interconversion of oxidized and reduced forms. (A) SEM image of an ITO electrode made by electrophoretic deposition of ITO particles onto an ITO glass slide at 9000× magnification. Scale bar is indicated as a black line. (B) Cyclic voltammogram of FNR (black line) and background-subtracted trace (red line, magnified 2×) at pH 8.0. The label indicates the peak width at half maximum height. (C) Background-subtracted signals from pH 5.0 (right) to 9.0 (left). (D) pH dependence of the reduction potential of FNR compared to the pH dependence of the reduction potential of the NADP+/NADPH couple (red). Other conditions: 25 mM MES (2-(N-morpholino)ethane-sulfonic acid), 25 mM TAPS (tris(hydroxymethyl)methylaminopropanesulfonic acid), 20 °C, scan rate 5 mV s−1, stationary electrode, solution purged with Ar before measurements. | ||
Fig. 2B shows a cyclic voltammogram of FNR adsorbed on a porous ITO electrode in the absence of its substrate, NADP+, at pH 8.0 and 20 °C. Briefly, an ITO/PGE electrode was made by electrophoretic deposition, and a small aliquot of FNR solution (typically 0.5–3 μL of 0.2 mM) was spotted and spread across the surface, allowing 3 minutes to soak in before removing excess. Upon placing the electrode into the buffer solution in the electrochemical cell, stable oxidation and reduction peaks are observed at −0.38 V vs. SHE at pH 8, which are due to the FAD group in FNR: the peaks are referred to as ‘non-turnover’ signals.10 As expected for an immobilized redox species, the peak current should vary linearly with scan rate: the plot shown in Fig. S2† shows that this is true up to a scan rate of 20 mV s−1, above which a downward deviation occurs, coincident with some peak broadening.
The ideal peak width at half height for an immobilized redox couple is 89/n mV at 20 °C, where n is a coefficient reflecting the number of electrons transferred cooperatively. A fully cooperative two-electron reaction, where the intermediate radical is extremely unstable should have a peak width of 45 mV for both oxidation and reduction. As evident from Fig. 2A, the peak widths are 57–59 mV in either direction at low scan rates – the fact that these are much smaller than 89 mV confirming that the reaction involves two electrons transferring with considerable cooperativity. The redox chemistry of the FAD cofactor in FNR involves two sequential one-electron transfers, as in the following Latimer scheme.
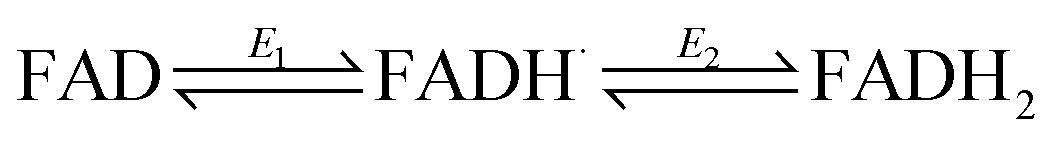
The overall two-electron reduction potential is the average of the two one-electron potentials E1 (oxidized/semiquinone) and E2 (semiquinone/hydroquinone). Our value agrees well with results of potentiometric titrations (−0.376 V vs. SHE at pH 8.0) which showed11 that a semiquinone species does form but at a level <10% of total flavin content. Based on the relationship between E1 − E2 and the peak width12 at low scan rate, and assuming homogeneity (see later) we estimate the difference in E1 and E2 to be approximately −14 mV, i.e. the reduction potential of the first step is slightly more negative than for the second step, reflecting evolved perfection for producing NADPH (hydride) from photosynthetic electrons.1,2
The area under the background-subtracted current represents the total faradaic charge passed and is proportional to the amount of electroactive FNR adsorbed onto the ITO electrode (two electrons per FAD). Using the same PGE electrode as a support, experiments were performed to determine how coverage varies with pH, applying the same amount of FNR in each case. Fig. 2C shows the non-turnover signals of FNR obtained over the pH range 5 to 9. In each case, the peak widths lie in the range 56–64 mV in either direction at a scan rate of 5 mV s−1, showing that ‘soft’ two-electron cooperativity occurs over the entire pH range. Most strikingly, the results demonstrate a strong dependence of coverage on pH, with a much higher coverage being obtained at pH 9 compared to pH 5. The affinity and reversibility of FNR binding to ITO was investigated by varying the pH back and forth starting at pH 9 or pH 5 (ESI Fig. S4 †). The results showed that FNR has a very high affinity for ITO when applied at pH 9, adsorbing to a level exceeding 500 pmol cm−2 (geometric area). The diameter of FNR is approximately 6 nm, therefore an ideal monolayer coverage assuming a planar surface would be approximately 5 pmol cm−2. The high coverage of FNR, more than 100 monolayer equivalents, must be due to the porous nature of the ITO electrode,13,14 as confirmed by electron microscopy (see Fig. 2A). When the electrode was transferred to a solution at lower pH, the coverage decreased but was largely restored upon returning to pH 9. In contrast, when FNR was initially applied at pH 5, the low coverage did not increase when the electrode was transferred to a solution of higher pH. The electrochemical homogeneity is particularly remarkable, recalling that 45 mV is the minimum half-height peak value were the FAD to undergo a fully cooperative 2-electron reaction (E2 ≫ E1).
From the results of the pH-step experiment, we conclude that binding of FNR within the pores involves: (i) general permeation that is more favourable at high pH, (ii) localized binding in an electroactive configuration that is also stronger at high pH. Lowering the pH therefore causes some enzyme molecules to become more loosely bound, but most remain buried in the pores, so that restoring the high pH condition causes the still-entrapped enzyme molecules to re-bind.
Fig. 2D compares the average (two-electron) reduction potential of FNR, obtained from cyclic voltammograms recorded in the pH range 5–9, with the reduction potential of the NADP+/NADPH couple. The latter value is −0.32 V vs. SHE at pH 7, with a slope of −30 mV per pH unit.15 In this pH range, the potential of FNR is always more negative than that of NADP+, the difference increasing with pH; therefore FNR is biased slightly to favour NADP+ reduction, especially in the chloroplast stroma where the pH is approximately 8. As long known, the two-electron potential of FAD in the enzyme is much more negative than in the free state (−0.22 V at pH 7.0);16 accordingly, we also verified that free FAD has a reduction potential of −0.25 V at pH 8.0.
Reversible, diffusion-controlled NADP+/NADPH conversion by FNR@ITO
Fig. 3A shows the electrochemical activity of a stationary FNR@ITO/PGE electrode in the presence of NADP+ at pH 8.0. Upon adding NADP+ to the cell, an enhanced reduction current is observed, and on the return scan, the resulting NADPH is oxidized. The result proves that FNR adsorbed at the ITO electrode is highly active for cofactor regeneration in both directions. Scan rate dependence measurements (ESI†) showed that the peak current varies linearly with the square root of scan rate, which is diagnostic for diffusion-controlled interconversion of NADP+ and NADPH. A diffusion coefficient of 4.2 × 10−6 cm2 s−1 at 20 °C was calculated for NADP+, well within the range reported17–20 for NADH and NADPH in aqueous solution, which is 2.0–6.7 × 10−6 cm2 s−1. The diffusion coefficients for NADH and NADPH in water should be similar.18 The Michaelis–Menten constant (Km) for NADP+, calculated from the reduction current at each NADP+ concentration, was 39 ± 15 μM at −0.375 V vs. SHE, close to the range (7–30 μM)5,21 obtained by solution measurements using reduced ferredoxin. | ||
| Fig. 3 Cyclic voltammetry of NADP+/NADPH. (A) Diffusion-controlled, quasi-reversible interconversion of NADP+ and NADPH at a stationary FNR@ITO/PGE electrode, with increasing amounts of NADP+, scan rate 5 mV s−1. (B) Effect of electrode rotation at scan rate 1 mV s−1: stationary electrode with 50 μM NADP+ (black) and under 3000 rpm rotation with 50 μM NADP+ (blue) or 50 μM NADPH (red). Other conditions: buffer solution containing 25 mM MES, 25 mM TAPS, pH 8.0, 20 °C, purged with Ar before measurement. | ||
A rotating disc FNR@ITO/PGE electrode was used to measure NADP+/NADPH interconversion under hydrodynamic control. In Fig. 3B, a fully sigmoidal curve for reduction of 50 μM NADP+ was only obtained at a high rotation rate (3000 rpm) and a slow scan rate (1 mV s−1). At slower rotation and/or faster scan rates, NADP+ reduction showed a small peak-like feature. Likewise, if the cell solution contained only NADPH (also 50 μM), a sigmoidal oxidation wave became clear at 3000 rpm and 1 mV s−1. Interconversion between NADP+ and NADPH is clearly fast and reversible. Currents for NADP+ reduction over the range −0.51 to −0.61 V vs. SHE gave linear Levich plots over the rotation rate range 400–3000 rpm (i.e. the current was proportional to the square root of the scan rate). The diffusion coefficient was always lower than obtained from cyclic voltammetry, and the origins of this difference, which may relate to surface geometry and uniformity of enzyme coverage, are under investigation.
The turnover frequency (TOF) of FNR, defined as moles of NADPH produced per mole of total electroactive FNR per second, is obtainable from the steady-state catalytic reduction current if the number of electroactive enzyme molecules is known (e.g. from the area of a non-turnover peak).10 An immediate observation was that the TOF with 50 μM NADP+ was typically 0.4 s−1 at pH = 8.0, which is three orders of magnitude lower than expected from steady state experiments.5,22 The discrepancy could be explained as follows, using the fact that the electrochemical reduction of NADP+ is controlled by diffusion through the bulk aqueous solution. In this situation, only those FNR molecules bound closest to the solution interface will contribute to catalysis, and buried FNR molecules could be completely redundant. Assuming, instead, that the amount of FNR accessible to free nicotinamide is equivalent to (at most) one monolayer, this leads to a revised TOF of at least 40 s−1. In support of this model, a shoulder is sometimes clearly visible on the oxidative scan, located negative of the oxidation peak for NADPH (ESI†). From its position, the shoulder coincides with the FNR oxidative non-turnover peak, and it may be assigned to FNR molecules that are buried and thus redundant in regard to catalytic activity. The shoulder fades in comparison to the main peak as the scan rate is decreased (since slow conditions favour oxidation of diffusing NADPH) and is not visible on the reductive scan because its position is more negative than the NADP+ reduction potential, thus it is masked under the NADP+ reduction peak. Redundant FNR likewise accounts, at least in part, for the faint, peak features appearing during electrode rotation. Ferredoxin–NADP+ reductase thus joins an increasing number of enzymes displaying reversible electrocatalysis.23 Most notably, the large, soluble domain of mitochondrial complex I attached to a PGE electrode shows reversible electrocatalysis of NAD+/NADH interconversion, but the current is too small and short-lived to be useful.24
Coupling FNR@ITO to stereo-/regio-selective organic synthesis
We proceeded to exploit the parallel between the activity of FNR in the chloroplast, and at the FNR@MO/support electrode, aptly named the “Electrochemical Leaf”, as shown in Fig. 1A. In the biological leaf, FNR receives electrons from the light harvesting photosensitizer apparatus via ferredoxin to reduce NADP+ to NADPH, which is used by downstream enzymes for CO2 assimilation. In the electrochemical leaf, NADP+ reduction is driven by a small overpotential, and the electrode can be coupled to an NADPH-dependent enzyme of choice. The system can also be driven in reverse, i.e. to oxidize NADPH and regenerate NADP+, in contrast to the natural case.As a test case, we coupled FNR@ITO to glutamate dehydrogenase (GLDH), which catalyses the conversion of 2-oxoglutarate (also known as α-ketoglutarate) to L-glutamate in the presence of NH4+ and NADPH.25 Strong coupling of GLDH to NADPH regeneration is directly observable by voltammetry as shown in Fig. 4A.
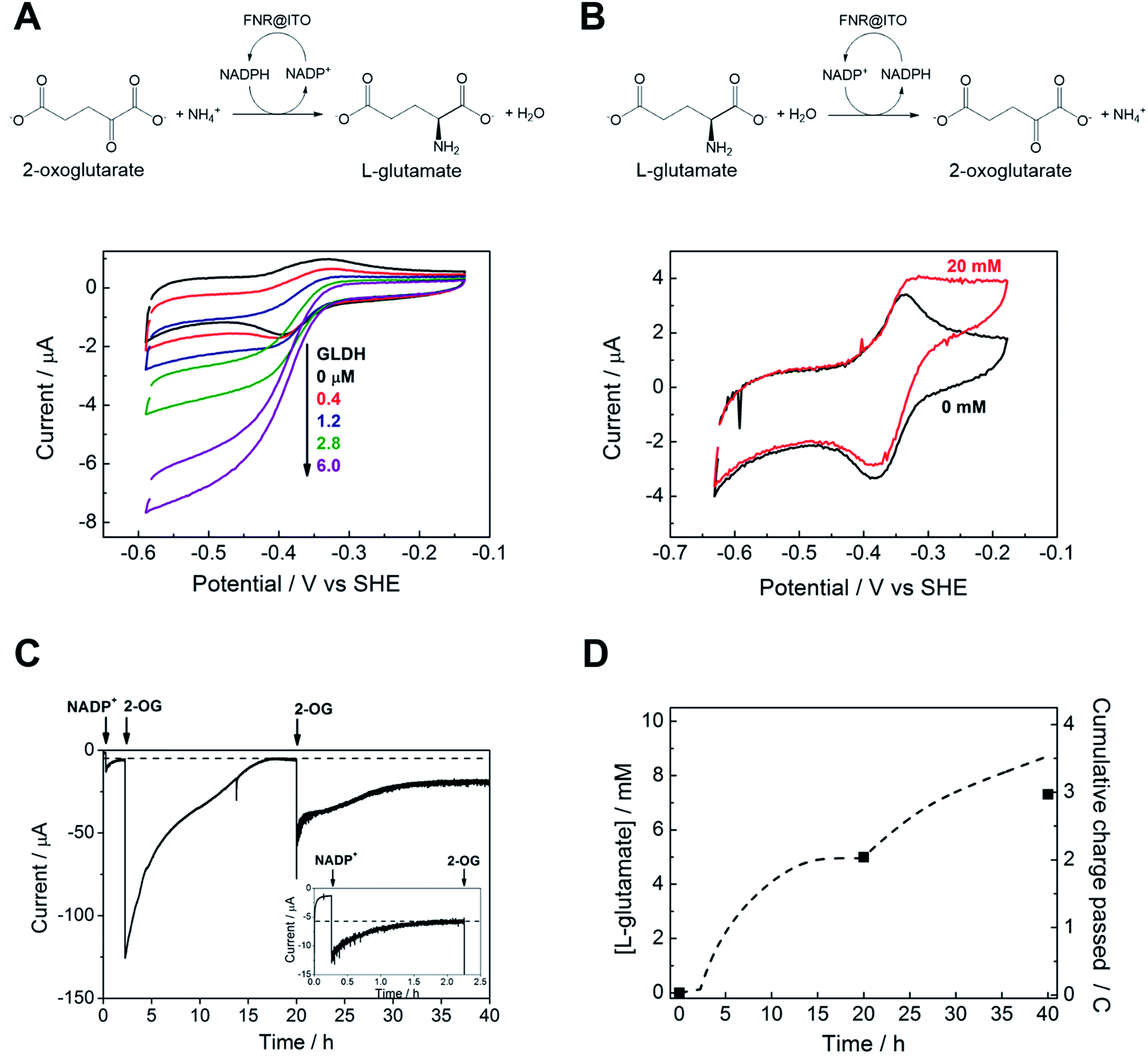 | ||
| Fig. 4 Coupling FNR@ITO to enzyme-catalysed organic synthesis. (A) Reductive amination of 2-oxoglutarate by glutamate dehydrogenase contained in the cell solution. (NH4+ 20 mM, 2-oxoglutarate 10 mM, NADP+ 50 μM, scan rate 5 mV s−1). (B) Oxidative deamination of L-glutamate by glutamate dehydrogenase co-loaded on to FNR@ITO electrode (see text for details), NADPH 50 μM, L-glutamate 20 mM, scan rate 1 mV s−1. Other conditions: 25 mM MES, 25 mM TAPS buffer solution at pH 8.0, 20 °C, stationary electrode, solution purged with Ar before measurement. (C) Electrochemical monitoring of L-glutamate synthesis coupled to FNR@ITO. Electrode: FNR@ITO/ITO glass (3 cm2) conditions: electrode potential −0.46 V vs. SHE, 50 mM borate (pH 8.0), cell volume 2.1 mL, NH4+ 20 mM, glutamate dehydrogenase 4 μM, room temperature, constant Ar bubbling. Injections of NADP+ 20 μM and 2-oxoglutarate 5 mM (twice) are indicated by arrows. Inset shows magnification of the initial 2.5 h. (D) Background-subtracted cumulative charge passed (dashed line) from (C). Black symbols dots refer to direct measurement of L-glutamate concentration by NMR spectroscopy. | ||
First, an FNR@ITO/PGE working electrode (area 0.09 cm2) was prepared as described in the previous section, then NADP+ was added to the cell solution (final concentration 50 μM) to obtain the expected NADP+ reduction and NADPH oxidation currents. The substrates for GLDH were already present (10 mM (NH4)2SO4, 10 mM 2-oxoglutarate). Additions of GLDH (0.4–6.0 μM final concentration) caused the oxidation and reduction peaks to convert to sigmoidal reduction waves, the intensity of which increased with the amount of GLDH added. The coupled catalytic reaction is directly observable by cyclic voltammetry because it is sufficiently fast to regenerate the initial state of the cofactor within the diffusion layer.
Fig. 4B shows the same reaction operated in reverse, where FNR@ITO/ITO glass (geometric area 1.5 cm2) is used as the electrode for regenerating NADP+, which is then used to oxidize L-glutamate to 2-oxoglutarate with release of NH4+. To observe the voltammetric response in this less favourable direction,25 it was necessary to co-immobilize GLDH directly onto the larger FNR@ITO/ITO glass electrode and use a slower scan rate (1 mV s−1). From the beginning, FNR (6 μL of 200 μM) and GLDH (8 μL of 200 μM) were loaded at the same time onto the electrode and incubated for 10 minutes at room temperature before recording the cyclic voltammetry of NADPH (50 μM). Upon injecting L-glutamate (final concentration 20 mM), the oxidation peak converted to a more sigmoidal shape, showing that NADPH is no longer depleted at the electrode surface, and confirming that the FNR@ITO electrode also behaves as a NADP+ regeneration system.
The GLDH-catalysed production of L-glutamate provided an excellent reporting tool for probing cofactor regeneration dynamics. Small-scale bulk conversion experiments were therefore conducted to obtain L-glutamate product for direct detection via NMR analysis (see ESI Fig. S5†), along with simultaneous monitoring of the faradaic charge passed. All experiments were carried out ‘on the bench’ with the solution agitated by bubbling Ar without any further steps being taken to improve anaerobicity. The results of several trials are shown in Table 1. In all cases, the Pt counter electrode was separated in a side arm linked to the main compartment by a glass frit. Fig. 4C corresponds to entry A of Table 1. An FNR@ITO electrode was constructed on an ITO glass support (total active area 3 cm2) and the experiment was initiated with only GLDH (4 μM) and NH4+ (20 mM) in solution, with the potential held at −0.46 V vs. SHE (i.e. approximately 0.11 V of overpotential for NADP+ reduction at pH 8.0). Upon injection of NADP+ (20 μM) a reduction current was observed, which decreased exponentially to a steady level after 1.5 h, indicating exhaustive consumption of NADP+ (see below). At this point, 2-oxoglutarate was injected to give a final concentration of 5 mM, which immediately generated a large current that was monitored until it dropped to a similar low background level. After 20 h, an aliquot of the cell solution was removed for NMR analysis. The charge consumed was also determined (Fig. 4D) taking into account the small residual background current that was observed in all chronoamperometric experiments. We attribute the background current to traces of dissolved O2 entering the cell from the atmosphere or through the glass frit, despite continuous bubbling with Ar.
| Electrode area (cm2) | Support | Average FNR coverage (pmol cm−2) | Duration (h) | L-Glutamate | TTNc (product/NADP+) | Rate (μmoles h−1 cm−2) | |||
|---|---|---|---|---|---|---|---|---|---|
| mM | μmoles | By charge (continuous) | By NMR | ||||||
| a Experiment conditions: electrode potential held at −0.46 V vs. SHE; 50 mM borate, pH 8; 20 μM NADP+; 40 mM NH4+ (for C, D, E and F); 30 mM NH4+ (for A and B); 30 mM 2-oxoglutarate (for, D, E and F); 20 mM 2-oxoglutarate (for A, B and C); 3.2 μM glutamate dehydrogenase (for C, D, E and F); 4 μM glutamate dehydrogenase (for A and B); 20 °C; bubbled with Ar throughout, unless otherwise indicated. The duration is counted from the addition of 2-oxoglutarate to the end of chronoamperometry. b No Ar bubbling up to 22 h (i.e. stationary solution); Ar bubbling from 22 h onwards. c TTN: total turnover number defined as number of moles of L-glutamate made per initial number of moles of NADP+ (TTN = (moles L-glutamate)/(moles of NADP+)); moles L-glutamate estimated from charge passed in chronoamperometry and also as measured by NMR. | |||||||||
| A | 3 | ITO glass | 163 | 37.7 | 7.31 | 15.4 | 436 | 366 | 0.14 |
| B | 2.5 | Ti foil | 19.5 | 16.7 | 4.74 | 9.5 | 207 | 237 | 0.23 |
| Cb | 10 | Ti foil | 90 | 41.7 | 13.3 | 33.4 | 813 | 667 | 0.08 |
| D | 17 | Ti foil and wound wire | 115 | 16.4 | 17.5 | 43.7 | 908 | 873 | 0.16 |
| E | 20 | Ti foil and wound wire | 60 | 19.6 | 20.1 | 50.2 | 1137 | 1004 | 0.13 |
| F | 6 | Ti wire (crumpled) | 136 | 37.8 | 20.5 | 51.2 | 1108 | 1025 | 0.22 |
For Fig. 4C, the amount of charge passed before adding 2-oxoglutarate was 8.9 mC, equivalent to 4.6 × 10−8 moles of NADPH (concentration 22 μM, in 2.1 mL of solution). The charge passed after adding 2-oxoglutarate, corrected for background, was 2.01 C, equivalent to 1.04 × 10−5 moles of L-glutamate, giving a concentration of 4.97 mM in 2.1 mL of solution. From NMR, the L-glutamate concentration after 20 h was 5.2 mM, the increase above the expected concentration (5.0 mM) indicating that some concentration had occurred. An important metric for evaluating a NADPH regeneration system is the total turnover number (TTN), defined as the number of moles of product of the coupled enzyme per mole of starting NADP+, i.e. the average number of times NADP+ has been recycled in the system. Based on the coulometric data, the TTN was 226 after 20 h: this compares with 261 using the expected quantity of NADP and the NMR-derived L-glutamate concentration. Errors in coulometric calculations arise from the uncertainty in subtracting background current, whereas errors in chemical quantities arise from the difficulty in accurately weighing small amounts of nicotinamide cofactor, and reduction of solution volume by evaporation over the course of the experiment, and/or crossover of product into the sidearm.
At 20 h, a further addition of 2-oxoglutarate was made, sufficient to give approximately 5 mM final concentration, and the experiment was resumed. The immediate recovery of at least 50% of the expected reduction current showed that the FNR@ITO/ITO electrode was still active. Fig. 4D includes the product and charge accumulation from this second stage. From NMR, the L-glutamate concentration after 40 h was 7.31 mM, amounting to 73% conversion (of 10 mM 2-oxoglutarate) and a TTN of 366 (entry A of Table 1 shows this final result). After this extended period, when the system is still running, the FNR molecules closest to the ITO surface must have cycled approximately 106 times (assuming the activity stems mainly from these molecules). A similar outcome was achieved using an electrode FNR@ITO/Ti constructed with flexible Ti foil (2.5 cm2 total active area) as support, the trial being shown in ESI Fig. S6,† and included as entry B in Table 1.
The feasibility for achieving simple, efficient and potentially very large scale-up levels was assessed by constructing the ITO electrode on larger areas of Ti foil, around some of which was wound Ti wire, or crumpled Ti wire alone. Trials remained at bench scale using the same small cell as before. Typical performances are included in Table 1 as entries C–F. The results show that it should be feasible to obtain, routinely, TTN's of several thousand in an overnight run.
Experimental methods
Purification of FNR
Competent Escherichia coli BL21-DE3 cells were transformed with a pASK-IBA7 plasmid containing the gene encoding Chlamydomonas reinhardtii FNR, as well as the genes to confer ampicillin resistance. A single colony resulting from this transformation was grown overnight and scaled up, using ampicillin throughout to ensure selection. Over-expression of FNR was induced by the addition of anhydrotetracycline. Cells were disrupted by French press and insoluble material removed by centrifugation. The resulting supernatant was purified using a strep-tactin chromatography column; FNR was eluted from the column using desthiobiotin and the fractions were pooled, concentrated and stored at −80 °C. A His-tagged analogue was also expressed and its activity was comparable to the strep-tagged version. Briefly, the coding sequence for FNR (present in the pASK-IBA7 vector) was amplified by PCR and cloned into a pLATE5 N-terminal Histag/EK vector by ligation-independent cloning. The His-tagged enzyme was expressed in E. coli BL21 cells and purified using nickel-affinity chromatography.Fabrication of ITO electrodes
Indium tin oxide (ITO) electrodes were made by electrophoretic deposition13,14 of ITO powder (Sigma) onto a conductive support, either ITO-glass slides (SPI Supplies), a pyrolytic graphite edge (PGE) electrode, a piece of Ti foil (thickness 00.127 mm, Sigma), Ti wire (diameter 0.25 mm or 0.81 mm, Sigma) or a combination of Ti foil and wire. A suspension of ITO (0.02 g) and I2 (0.005 g) was made in acetone (20 mL) and sonicated for 30 min. Two conductive supports were held in parallel orientation in the ITO suspension with a separation of 1–2 cm, and a voltage of 10 V was applied for 3–6 min. The electrode on which ITO had been deposited was then left to dry in air before use.Electron microscopy
Scanning electron microscope (SEM) images were acquired using a Hitachi S-4300 instrument with a field-emission electron source at an accelerating voltage of 3 kV.Electrochemical measurements
All electrochemical measurements were conducted with a closed glass cell having a 3-electrode configuration, with the reference electrode (SCE or Ag/AgCl) in a side arm connected via a Luggin capillary, and a Pt counter electrode in a side arm separated from the main compartment by a glass frit. Corrections made to conform to the standard hydrogen electrode scale were +0.24 (SCE) and +0.21 (AgCl). The cell solution was purged with Ar to remove dissolved O2 before starting measurements. A potentiostat (Autolab PGSTAT10 or PGSTAT101) controlled by Nova software (EcoChemie) was used to record the measurements. A small amount of FNR solution (typically 0.5–3 μL of 0.2 mM) was drop-cast onto the ITO electrode, spread, left for 3 min to partially dry, then rinsed with water before placing the electrode into the cell solution. Solutions of NADP+ and various substrates for the coupled reaction were made up with the same buffer solution before injecting into the cell.NMR analysis
A sample of the cell solution was mixed with D2O to make a 90:10 (H2O:D2O) mixture, and the 1H spectra were recorded on a Bruker AVIIIHD 400 instrument. The peak area corresponding to the product was compared to a calibration curve obtained from standard solutions of known concentration. See ESI Fig. S5† for NMR spectra.
Conclusions
As a new hybrid material, FNR@ITO allows the catalytic electron-transfer properties of a crucial photosynthetic enzyme to be studied in considerable detail. The resulting electrochemistry of NADP+ is specific, reversible and diffusion-controlled, making it possible to drive coupled enzyme-catalysed organic reactions at an electrode. A large variety of methods have been studied for cofactor regeneration,26 including formate dehydrogenase in solution27 and H2 driven hydrogenases on conductive particles.28,29 The rate and direction of cofactor regeneration in our system are easily controlled by the electrode potential, and the progress of the organic synthesis can be monitored continuously by coulometry. With just a modest overpotential, cofactor regeneration is so fast that the overall reaction is controlled by the dynamics of the coupled reaction that are limited by inherent enzyme rates and/or substrate mass transport. The FNR@MO/support system – the electrochemical leaf – has numerous options for development, particularly electrode and reactor design (ultimately achieving the advantages of dimensionality so obvious in a chloroplast) and exploration of myriad coupling reactions, including monooxygenases. It seems likely that total turnover numbers (product/NADP) of several thousand for a day-long batch will be easily achievable. A large range of complex regio- and stereospecific organic reactions thus becomes accessible to simple electrochemical procedures and continuous monitoring.Acknowledgements
This research was supported by the ERASynBio consortium ‘Sun2Chem’, administered by the Biotechnology and Biological Sciences Research Council (BB/M005720/1) and the Deutsche Forschungsgemeinschaft (DFG) (DIP project cooperation “Nanoengineered optoelectronics with biomaterials and bioinspired assemblies”), and the Volkswagen Foundation (LigH2t). F. A. A. is a Royal Society Wolfson Research Merit Award holder.References
- N. Carrillo and E. A. Ceccarelli, Eur. J. Biochem., 2003, 270, 1900–1915 CrossRef CAS PubMed.
- M. A. Musumeci, E. A. Ceccarelli and D. L. Catalano-Dupuy, in Advances in Photosynthesis - Fundamental Aspects, ed. M. Najafpour, InTech, 2012 Search PubMed.
- G. Kurisu, M. Kusunoki, E. Katoh, T. Yamazaki, K. Teshima, Y. Onda, Y. Kimata-Ariga and T. Hase, Nat. Struct. Biol., 2001, 8, 117–121 CrossRef CAS PubMed.
- J. A. Hermoso, T. Mayoral, M. Faro, C. Gómez-Moreno, J. Sanz-Aparicio and M. Medina, J. Mol. Biol., 2002, 319, 1133–1142 CrossRef CAS PubMed.
- C. J. Batie and H. Kamin, J. Biol. Chem., 1984, 259, 1976–1985 Search PubMed.
- S. Frasca, T. von Graberg, J.-J. Feng, A. Thomas, B. M. Smarsly, I. M. Weidinger, F. W. Scheller, P. Hildebrandt and U. Wollenberger, ChemCatChem, 2010, 2, 839–845 CrossRef CAS.
- A. Bachmeier, B. J. Murphy and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136, 12876–12879 CrossRef CAS PubMed.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2012, 134, 8332–8335 CrossRef CAS PubMed.
- T. Daido and T. Akaike, J. Electroanal. Chem., 1993, 344, 91–106 CrossRef CAS.
- C. Léger, S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653–8662 CrossRef PubMed.
- C. J. Batie and H. Kamin, J. Biol. Chem., 1986, 261, 11214–11223 CAS.
- H. A. Heering, J. H. Weiner and F. A. Armstrong, J. Am. Chem. Soc., 1997, 119, 11628–11638 CrossRef CAS.
- L. Besra and M. Liu, Prog. Mater. Sci., 2007, 52, 1–61 CrossRef CAS.
- H. G. Krüger, A. Knote, U. Schindler, H. Kern and A. R. Boccaccini, J. Mater. Sci., 2004, 39, 839–844 CrossRef.
- F. L. Rodkey and J. A. Donovan, J. Biol. Chem., 1959, 234, 677–680 CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman, New York, 5th edition, 2002 Search PubMed.
- J. Moiroux and P. J. Elving, J. Am. Chem. Soc., 1980, 102, 6533–6538 CrossRef CAS.
- A. Domenech, E. Garcia-Espana, J. A. Ramirez, B. Celda, M. Carmen Martinez, D. Monleon, R. Tejero, A. Bencini and A. Bianchi, J. Chem. Soc., Perkin Trans. 2, 1999, 23–32 RSC.
- Z. Wu, W. Jing and E. Wang, Electrochem. Commun., 1999, 1, 545–549 CrossRef CAS.
- C. E. Banks and R. G. Compton, Analyst, 2005, 130, 1232–1239 RSC.
- M. Shin, in Methods in Enzymology, Academic Press, 1971, vol. 23, pp. 440–447 Search PubMed.
- A. K. Arakaki, E. A. Ceccarelli and N. Carrillo, FASEB J., 1997, 11, 133–140 CAS.
- F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 CrossRef CAS PubMed.
- Y. Zu, R. J. Shannon and J. Hirst, J. Am. Chem. Soc., 2003, 125, 6020–6021 CrossRef CAS PubMed.
- J. E. Rife and W. W. Cleland, Biochemistry, 1980, 19, 2321–2328 CrossRef CAS PubMed.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773 RSC.
- Z. e. Shaked and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 7104–7105 CrossRef CAS.
- L. Greiner, I. Schroder, D. H. Muller and A. Liese, Green Chem., 2003, 5, 697–700 RSC.
- H. A. Reeve, L. Lauterbach, O. Lenz and K. A. Vincent, ChemCatChem, 2015, 7, 3480–3487 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00850c |
| This journal is © The Royal Society of Chemistry 2017 |