
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted antigen delivery by an anti-class II MHC VHH elicits focused αMUC1(Tn) immunity†
Tao
Fang‡
a,
Catharina H. M. J.
Van Elssen§
a,
Joao N.
Duarte
a,
Jonathan S.
Guzman
ab,
Jasdave S.
Chahal¶
a,
Jingjing
Ling‡
ac and
Hidde L.
Ploegh‡
 *ab
*ab
aWhitehead Institute for Biomedical Research, 9 Cambridge Center, Cambridge, MA 02142, USA. E-mail: hidde.ploegh@childrens.harvard.edu
bDepartment of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 26th May 2017
Abstract
Unusual patterns of glycosylation on the surface of transformed cells contribute to immune modulation and metastasis of malignant tumors. Active immunization against them requires effective antigen presentation, which is complicated by a lack of access to tumor-specific posttranslational modifications through standard genetic approaches and by the low efficiency of passive antigen sampling. We found that antigen targeted to antigen presenting cells via class II MHC products can elicit a robust immune response against MUC1(Tn) bearing a defined tumor-associated glycoform, Tn. The two-component vaccine construct was prepared by sortase-mediated protein ligation of a synthetic MUC1(Tn) fragment to a class II MHC-binding single-domain antibody fragment (VHH7) as targeting moiety. We show that VHH7 targets antigen presenting cells in vivo, and when conjugated to MUC1(Tn) can elicit a strong αMUC1(Tn) immune response in mice. The resulting sera preferentially recognized the MUC1 epitope with the tumor-associated carbohydrate antigen Tn and were capable of killing cancer cells in a complement-mediated cytotoxicity assay. Immunoglobulin isotype analysis and cytokine release assays suggested a favorable Th1 response. A single boost 12 months after primary immunization triggered a recall response of the same quality, suggesting that long-term αMUC1(Tn) memory had been achieved.
Introduction
Mucin MUC1, a heavily glycosylated, polymorphic type-I transmembrane glycoprotein, contains an extracellular domain with a variable (10–100) number of tandem repeats (VNTRs), each composed of 20 amino acids (PD![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) RPAPG
RPAPG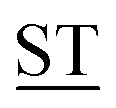 APPAHGV
APPAHGV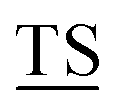 A) with five potential sites (underlined) for O-glycosylation.1 The high density of branched O-linked glycans forces the protein backbone into an extended conformation, so that it protrudes from the apical surface of epithelial cells to form a protective mucous barrier.2 In human adenocarcinomas such as breast, prostate, ovary and pancreatic cancers, MUC1 is overexpressed with loss of apical distribution, displaying truncated and immature glycosylation patterns due to the misregulation of core 1,3-galactosyltransferase (T-synthase) and α-N-acetylgalactosaminide α-2,6-sialyltransferase-1 (ST6GalNAc-1).3 MUC1 promotes tumor dissemination by limiting E-cadherin-mediated cell–cell and integrin-mediated cancer-matrix interactions.4,5 The extracellular domain of MUC1 also serves as a ligand for co-stimulatory intercellular adhesion molecule-1 (ICAM-1) and interferes with T cell activation and proliferation. Based on its surface availability, its widespread expression-including on cancer stem cells- and its intriguing function in driving immune tolerance, the National Cancer Institute has prioritized MUC1 as second among 75 tumor-associated antigens to be targeted for cancer vaccine development.6
A) with five potential sites (underlined) for O-glycosylation.1 The high density of branched O-linked glycans forces the protein backbone into an extended conformation, so that it protrudes from the apical surface of epithelial cells to form a protective mucous barrier.2 In human adenocarcinomas such as breast, prostate, ovary and pancreatic cancers, MUC1 is overexpressed with loss of apical distribution, displaying truncated and immature glycosylation patterns due to the misregulation of core 1,3-galactosyltransferase (T-synthase) and α-N-acetylgalactosaminide α-2,6-sialyltransferase-1 (ST6GalNAc-1).3 MUC1 promotes tumor dissemination by limiting E-cadherin-mediated cell–cell and integrin-mediated cancer-matrix interactions.4,5 The extracellular domain of MUC1 also serves as a ligand for co-stimulatory intercellular adhesion molecule-1 (ICAM-1) and interferes with T cell activation and proliferation. Based on its surface availability, its widespread expression-including on cancer stem cells- and its intriguing function in driving immune tolerance, the National Cancer Institute has prioritized MUC1 as second among 75 tumor-associated antigens to be targeted for cancer vaccine development.6
Approaches to create MUC1 vaccines have included epitope–protein conjugates; multicomponent synthetic MUC1 peptide vaccines; MUC1 peptide-pulsed autologous dendritic cells; full or partially truncated MUC1 proteins; or MUC1-encoding plasmids, synthetic mRNA and viral vectors.7–10 Tumor epitope-based immunization is attractive as a means of reducing the risk of induced autoimmunity. As of today, it may be the only feasible strategy to induce a focused immune response against defined tumor-associated posttranslational modifications. However, small peptides are poorly immunogenic: to elicit an immune response they require conjugation to carrier proteins such as tetanus or diphtheria toxins. The size difference between carrier and conjugated peptide antigens usually causes a substantial portion of the response to be directed towards the carrier. Fully synthetic vaccines can provide solutions to these challenges, by integrating the minimal essential elements for eliciting stronger MUC1-specific immune responses in a defined and predictable manner. Such methods generally converge on final steps that require the doping9,11 or self-assembly12,13 of defined vaccine constructs into macromolecular structures capable of multivalent presentation and accumulation in lymphoid organs.14
The low efficiency of passive antigen sampling poses yet an additional challenge for effective antigen presentation, which is required for CD8 T cell priming and critical to anti-tumor protection. Antigen delivery via antibodies that target antigen-presenting cells (APCs), especially via surface receptors that are endocytosed, enhances antigen loading and presentation, and therefore the efficacy of vaccines. It avoids the use of infectious or replicating materials, and provides a pure protein-based vaccination strategy with minimal amounts of antigen. Full-sized antibodies that target class II major histocompatibility complex (MHC) products,15 CD11c,16 C-type lectin DEC205 and Dectin-1,17,18 pattern recognition receptors (PRR)19 or other APC surface protein,20 conjugated chemically or fused genetically to antigens, have all been reported to induce an immune response to the attached antigen. Epitopes with precise posttranslational modifications are still excluded from this collection because a simple genetic fusion strategy cannot introduce the relevant specific glycoforms into the portions that comprise the relevant target antigen. The conserved and possibly heterogeneous N-glycan modifications of the Fc domain of full-sized antibodies can also complicate the induction of a desired carbohydrate-specific response.
We reasoned that delivery of synthetic antigen via camelid-derived heavy chain-only antibody fragments (VHHs) could address these drawbacks. VHHs, amenable to humanization,21 are the smallest known antibody fragments that retain their antigen binding capacity. Individual VHHs (2.5 nm × 2.5 nm × 4 nm, 15 kDa) are almost ∼10 times smaller than a full-sized mAb (14.2 nm × 8.5 nm × 3.8 nm, 150 kDa),22 facilitating penetration of lymphoid organs. Their single-domain configuration enables high-yield expression in bacterial systems in the absence of N- and O-glycosylation.23 The C-terminus of a VHH is located opposite the complementarity-determining regions (CDRs) that confer binding specificity, making VHHs ideal substrates for installation of a sortase-recognition pentapeptide (LPXTG) motif to enable site-specific protein modification.24–26
We have screened and verified VHHs against various leukocyte surface markers such as class II MHC products, CD11b and CD36. Different antigenic payloads derived from GFP, ubiquitin, OVA peptide, and influenza hemagglutinin's stem, at least when conjugated to these VHHs, elicited strong and characteristic immune responses.27 In particular, VHH7, a VHH that recognizes murine class II MHC products with nanomolar affinity, stains class II MHC+ populations such as dendritic cells (DCs), macrophages and B cells as analyzed by flow cytometry on excised organs. In vivo PET and NIR imaging confirmed the ability of VHH7 to target lymphoid organs.28 Confocal microscopy showed internalization of VHH7 upon binding to class II MHC products.29 Conjugation of a synthetic MUC1(Tn) epitope to VHH7 by means of a sortase reaction could thus provide a route to access structurally defined MUC1 vaccines with defined tumor-associated glycoforms. Directly targeting APCs would then drive a potent immune response against the attached payload. Fully synthetic payloads that cannot be encoded genetically would thus benefit from the type of approach described here.
Here we report the preparation and in vivo application of a structurally defined MUC1(Tn) vaccine that preserves the immunogenic domain of MUC1 VNTR and a tumor-associated carbohydrate antigen Tn (αGalNAc-Thr/Ser). The Tn-antigen is a cancer-specific marker that can affect the conformation of the underlying peptide backbone and subsequent cancer-specific immune response.30 To obtain the desired conjugate, a triglycine moiety was introduced at the N-terminus of the MUC1–VNTR-derived epitope to serve as a nucleophile and facilitate a sortase A (SrtA)-mediated transpeptidase reaction to obtain a structurally defined MUC1(Tn) epitope vaccine. Administration of these vaccine constructs to mice elicited strong humoral and cellular responses and was sufficient to establish long-term memory.
Results and discussion
Generate glycoform-defined VHH–MUC1(Tn) vaccine constructs
We chose the MUC1-derived epitope (TSAPDTRPAP) and extended it at the N-terminus with a triglycine moiety and a cysteine to allow bioconjugation through a sortase reaction and a thiol Michael addition, respectively. TSAPDRPAP is the immunodominant segment of MUC1–VNTR.9,31,32 We installed a tumor-associated carbohydrate antigen Tn at Thr6 (underlined). The carbohydrate moiety structurally influences the underlying peptide backbone and can survive antigen processing. The processed glycoconjugate binds to class II MHC products via its peptide backbone, and is recognized by a specific T cell receptor.33 This provides the rationale for inducing a tumor-specific immune response through T cell recognition of tumor-associated carbohydrate or glycopeptide antigens. The proper extent of glycosylation is critical for an optimal response. The O-glycosylation at a tumor-specific MUC1 epitope PDTRP enhanced the binding by available MUC1-specific monoclonal antibodies due to saccharide-induced conformational changes imposed on the peptide backbone.34,35 However, occupying all possible O-glycosylation sites impairs the antibody response, presumably because a heavily glycosylated MUC1 glycopeptide is processed less efficiently by APCs.36,37
Although solid-phase peptide synthesis is a well established technology, the preparation of glycopeptides remains non-trivial.38,39 Our approach to the synthesis of oligoglycine-modified tumor-associated MUC1(Tn) epitope followed standard Fmoc-based solid phase peptide synthesis on resin until the glycosylation site was reached, at which point the Fmoc-Thr-Tn(Ac)4 building block with acetyl protected hydroxyl groups was coupled manually (Fig. 1a). After coupling, the remaining free N-terminal amine was capped by excess acetic anhydride. The remainder of the sequence was then completed on a synthesizer, followed by cleavage off the resin and side-chain deprotection. O-glycans, in this case the Tn antigen are prone to β-elimination under standard Zemplén deacetylation conditions, resulting in low yields. For this reason, the remaining acetyl groups of the Tn antigen were removed by using a modified hydrazine protocol.39,40 After TFA deprotection and purification, the product was redissolved in 5% hydrazine aqueous solution for 1 h at room temperature, giving complete deacetylation without detectible elimination side products.
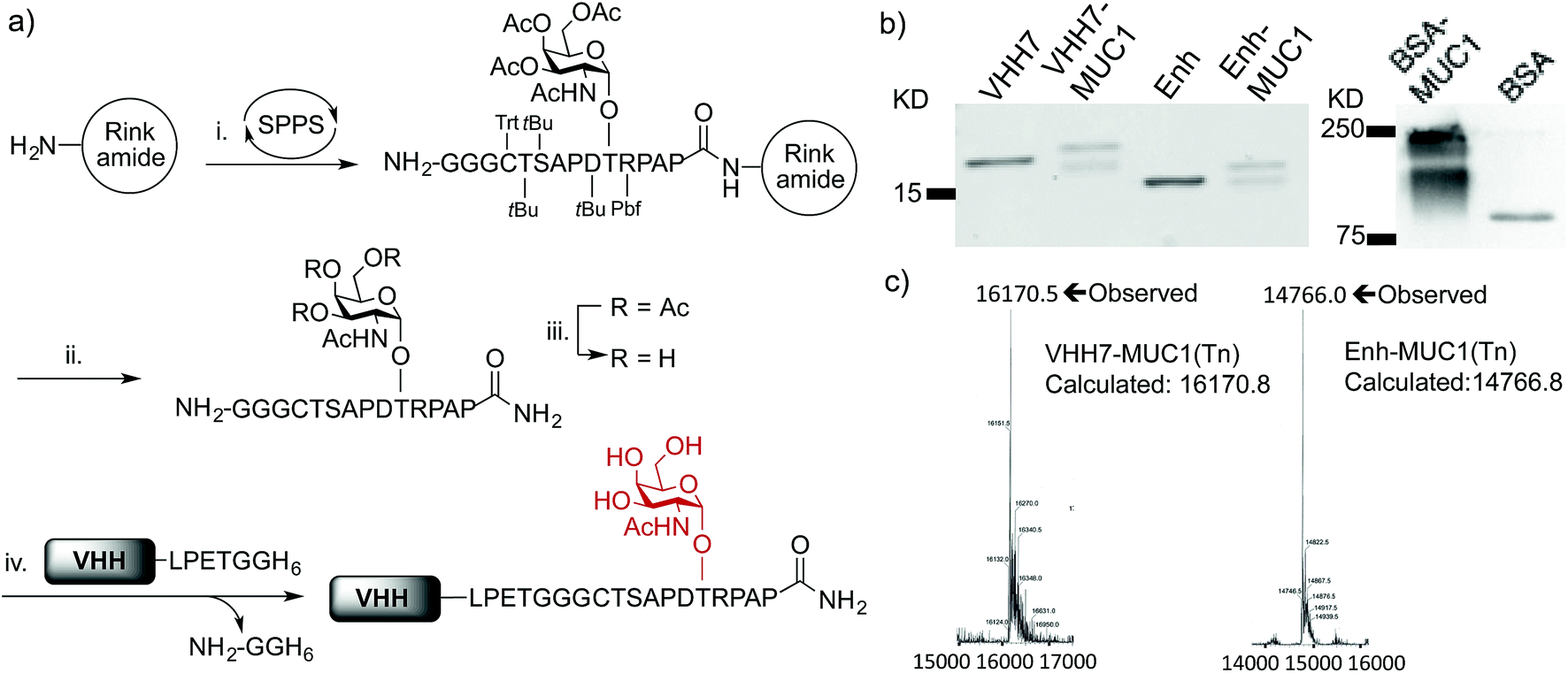 | ||
| Fig. 1 Preparation and characterization of VHH–MUC1 conjugate. (a) Solid phase synthesis of sortaggable MUC1(Tn) glycopeptide and generation of VHH conjugates by sortagging. Reagents and conditions: (i) coupling: HATU, DIPEA, DMF, 5 eq., 2 h (For Thr-Tn(3Ac), 2 eq., overnight); deprotection: 20% piperidine in DMF; (ii) TFA/TIPS/EDT/H2O (90/5/2.5/2.5, v/v); (iii) 5% hydrazine in H2O, 1 h, then direct HPLC; (iv) SrtA (pentamutant), 10 mM CaCl2, 50 mM Tris, pH = 7.4, 12 °C, 2–4 h, >95% conversion based on LC-MS, ∼70% recovery at 1 mg scale after Ni-NTA bead adsorption and PD-10 column purification; (b) SDS-PAGE analysis of VHH–MUC1 conjugates. The integrity of synthesized conjugates was further confirmed by LC-MS, sequence analysis by LC/MS/MS and immunoblot (two independent syntheses gave comparable results); the reason for the doublet seen on SDS-PAGE remains unexplained, as a single mass was observed and both species are immunoreactive (c) LC-MS analysis shows intact mass of conjugates. | ||
We next conjugated the (Gly)3-modified MUC1(Tn) to VHHs through a SrtA-mediated transpeptidase reaction. Besides VHH7, we used a VHH (Enh) specific for Enhanced Green Fluorescent Protein (EGFP)41 as our specificity control. Both VHHs contain a C-terminal His6-tag preceded by a sortase recognition motif, LPETG. The sortase reactions were carried out following established protocols.29 Purification of the desired product is simplified by adsorption of the reaction mixture onto Ni-NTA beads, which depletes His6-tagged sortase and any unreacted VHH that retained its His6 tag, followed by size exclusion chromatography to remove excess nucleophile. The final product yields a single peak on LC-MS. The expected sequence was confirmed by LC/MS/MS (Fig. 1c, S1e and f†). Site-specific conjugation using sortase has the advantage of convenient and unambiguous quality control. In contrast, our efforts to prepare BSA–MUC1(Tn) conjugates from commercially available maleimide-activated bovine serum albumin (BSA) resulted in a heterogeneous mixture, characterized by the presence of high-molecular weight material, presumably due to self-crosslinking and variable quantities of attached peptide (Fig. 1b, S1a and b†).
Optimization of evoked humoral responses
With both VHH–MUC1(Tn) constructs in hand, we immunized mice and identified suitable adjuvants. Quillaja saponaria saponin (QS21),42 polyinosinic-polycytidylic acid [p(I:C)]43 and monophosphoryl lipid A (MPLA)44 are all effective adjuvants that have been used clinically. Mice were immunized with a formulation composed of one of these adjuvants, VHH7–MUC1(Tn) conjugate, and a mAb against CD40. The latter is a potent B cell activator. Five days after the final immunization, blood was collected and tested for antibodies against MUC1(Tn) by ELISA. Inclusion of the adjuvant p(I:C) elicited the strongest immune response: the IgG response was five-fold higher than for other adjuvants (compared at a 1/000 dilution). A lower IgM response was observed in the final serum samples from the p(I:C) cohort, presumably owing to successful class switch recombination to other Ig isotypes, while QS21 and MPLA only provided modest anti-MUC1 responses, with no distinct preference for IgG (Fig. 2a and b). The endpoint titer of IgG1 in the p(I:C) cohort was >1/105 dilution, with a comparable response for IgG2a (Fig. 2b). A weak IgG3 response was observed for all formulations tested. This is likely determined by the steric conformation related to the combination of the carbohydrate portion and the polypeptide chain of MUC1(Tn), since in mice IgG3 is made exclusively in response to carbohydrate antigens.45 All further immunizations were conducted using the optimized adjuvant combination of anti-CD40 and p(I:C).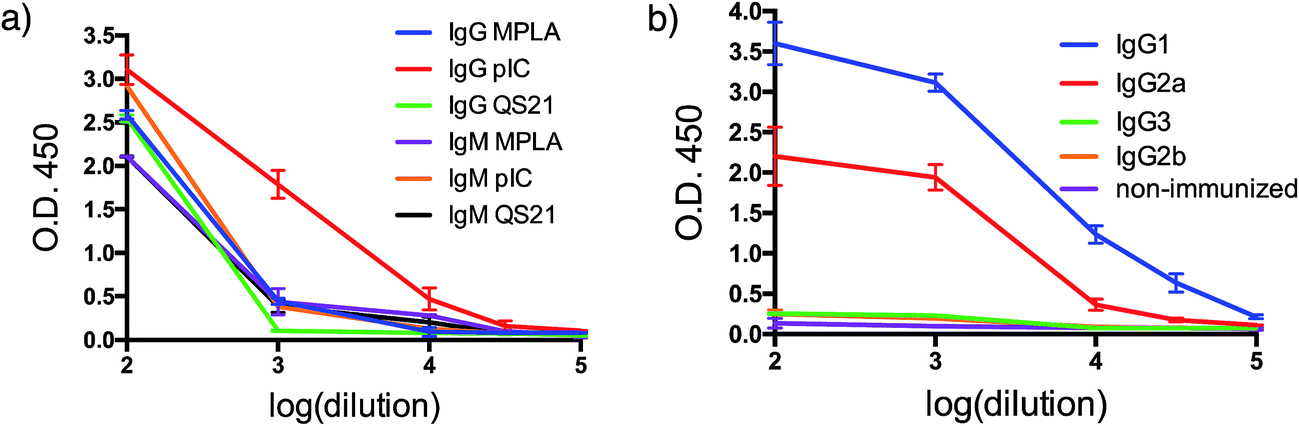 | ||
| Fig. 2 Humoral response. (a) Adjuvant screening based on antibody response; (b) IgG isotyping for VHH7-MUC1(Tn) adjuvanted by p(I:C) (error bars: mean ± SD, n = 3). | ||
MUC1(Tn) is preferentially recognized
Since both VHH7– and Enh–MUC1(Tn) conjugates involve the use of small proteins (VHHs), we examined reactivity towards the VHH carriers, as distinct from that directed against the payload MUC1(Tn). Immunoblots (Fig. 3a) showed that the serum response was weak towards VHH7 with little cross reactivity to Enh, a llama-derived VHH used as fusion tag for immunoblot. The intense serum reactivity against Enh–MUC1(Tn) conjugate therefore must represent a strong response to its appended MUC1(Tn) epitope. In contrast, when mice were immunized with Enh–MUC1(Tn) conjugate (Fig. 3b), the serum response was directed predominantly against Enh instead of MUC1(Tn), with similar signal intensities observed for Enh and Enh–MUC1(Tn), and only a weak signal for VHH7–MUC1(Tn). We attribute this difference to multivalent presentation of the VHH7-conjugated MUC1 epitope on the surface of APCs; however, the exact mechanism will be worthy of further investigation. In silico analysis of potential B epitopes by the Bepipred linear epitope prediction algorithm (Fig. S2†) showed that the C-terminal MUC1 peptide is the dominant epitope predicted for B cell recognition in both VHH7– and Enh–MUC1 conjugates. Targeting of other antigenic payloads via VHH7 is likewise superior to the use of non-targeting VHHs.27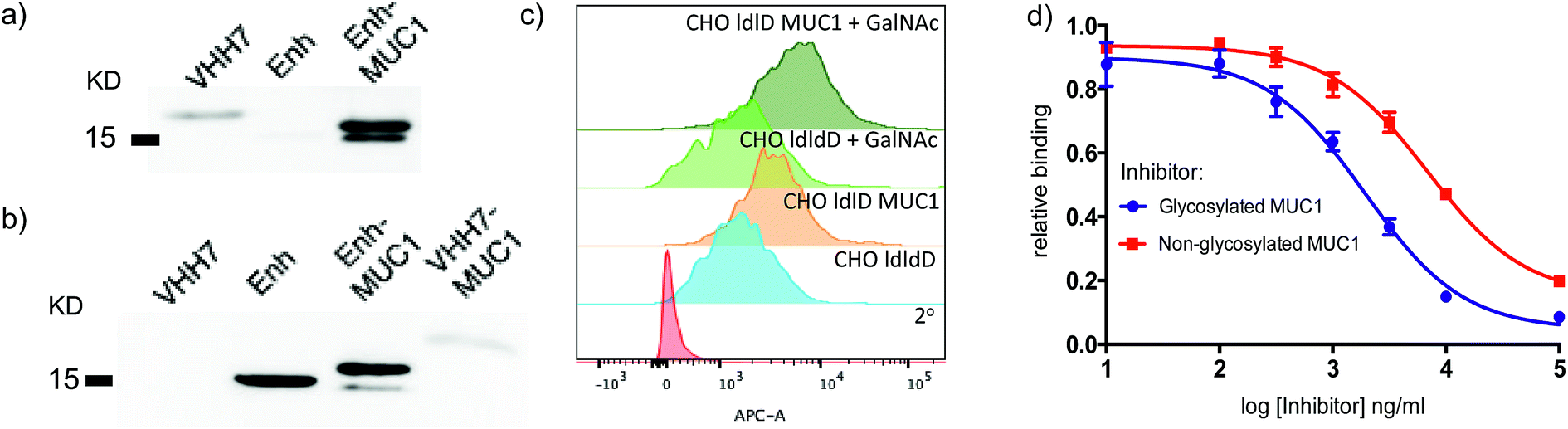 | ||
| Fig. 3 VHH7 elicits a focused immune response directed against its payload. (a) A strong humoral response to the payload conjugated to VHH7 (immunoblot: 1° Ab: sera from mice immunized with VHH7–MUC1; 2° Ab: anti-mIgG-HRP); (b) no immune response was directed to the conjugated epitope with a non-APC targeting VHH (immunoblot: 1° Ab: sera from mice immunized with Enh–MUC1; 2° Ab: anti-mIgG-HRP); (c) CHO-ldlD cell lines transfected with MUC1 cDNA and provided with an exogenous source of GalNAc and their controls either express or lack the desired MUC1 were stained with the elicited sera for flow cytometric analysis; (d) inhibition ELISA revealed stronger binding to Tn-modified MUC1 compared to the peptide backbone alone [serum dilution 1/200, IC50 (red) ∼6800 ng ml−1, IC50 (blue): ∼1900 ng ml−1] (error bars: mean ± SD, n = 3). | ||
We next examined to what extent the appended Tn moiety is recognized by the immune sera generated. A Chinese Hamster Ovary (CHO) ldlD cell line with defective UDP-galactose/UDP-N-acetylgalactosamine 4-epimerase cannot convert UDP-Glc/GlcNAc to UDP-Gal/GalNAc, thereby rendering these mutant cells defective in protein O-/N-glycosylation reactions. When CHO-ldlD was stably transfected with a full-length MUC1 cDNA, the Tn epitope was generated only upon exogenous provision of GalNAc.46 We cultured CHO-ldlD and CHO-ldlD-MUC1 with or without exogenously added GalNAc and tested sera from immunized mice for cell surface binding by flow cytometry. We detected MUC1-specific staining only in cells transfected with MUC1 cDNA. Cells supplemented with GalNAc displayed a further increase in staining intensity, consistent with better antibody recognition of Tn glycosylated MUC1 (Fig. 3c). As expected, cells treated with GalNAc but not transfected with MUC1 were not recognized by the immune sera.
We further characterized the polyclonal response directed against synthetic MUC1 and MUC1(Tn) epitope by inhibition ELISA (Fig. 3d). Reactivity of sera (dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200) from VHH7–MUC1(Tn)-immunized mice could be blocked with various concentrations of the MUC1(Tn) or the MUC1 epitope. An IC50 of ∼1900 ng ml−1 was observed for the MUC1(Tn) epitope and an IC50 of ∼6800 ng ml−1 for MUC1, which demonstrates a preference for the MUC1(Tn) epitope over the peptide backbone alone. Based on the chemical composition of the conjugate, only 0.5 μg carbohydrate antigen was injected to achieve this response, demonstrating the efficiency of directly targeting the antigen presentation pathway.
:200) from VHH7–MUC1(Tn)-immunized mice could be blocked with various concentrations of the MUC1(Tn) or the MUC1 epitope. An IC50 of ∼1900 ng ml−1 was observed for the MUC1(Tn) epitope and an IC50 of ∼6800 ng ml−1 for MUC1, which demonstrates a preference for the MUC1(Tn) epitope over the peptide backbone alone. Based on the chemical composition of the conjugate, only 0.5 μg carbohydrate antigen was injected to achieve this response, demonstrating the efficiency of directly targeting the antigen presentation pathway.
MUC1(Tn) positive cancer cells are recognized and killed by elicited sera
We tested serum reactivity in vitro against established cell lines known to express MUC1(Tn). Human breast and prostate cancer cell lines, MCF-7 and DU-145 respectively, were incubated with sera from VHH7–MUC1(Tn)-immunized mice at dilutions of 1/200 and 1/500, followed by secondary antibody staining with anti-mouse IgG-Alexa Fluor 647 (Fig. 4a). Compared to control sera, flow cytometric analysis demonstrated strong reactivity of immune sera toward these cell lines. We explored antibody-dependent complement-mediated lysis10 and found that both cancer cell lines were efficiently and specifically lysed (Fig. 4b). Sera from control did not show cytolytic activity.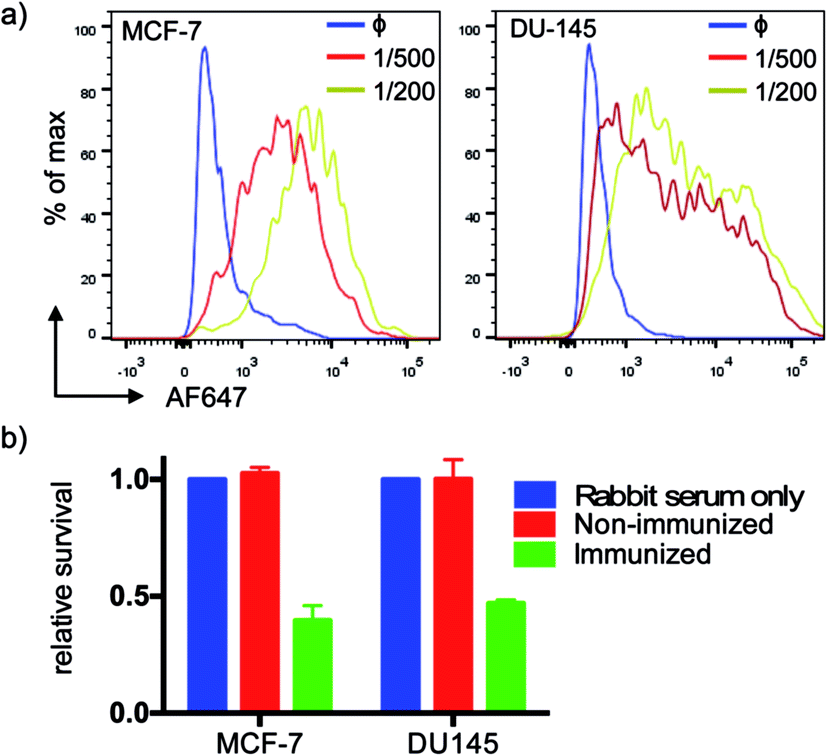 | ||
| Fig. 4 Serum recognition of MUC1 expressing breast and prostate cancer cell lines and complement-mediated killing. (a) Elicited MUC1 anti-sera show high affinity at high dilution (1° Ab, anti-serum; blue: non-immunized mice; red: 1/500 dilution; yellow: 1/200 dilution; 2°: anti-murine IgG-AF647); (b) anti-MUC1 serum mediates effective complement-directed killing of MUC1-positive cancer cells (cells were incubated with serum (1:10) for 30 min, then medium was changed to 10% rabbit serum as a source of complement, followed by incubation for another 2 h. Cell viability was tested by MTT assay. Error bars: mean ± SD, n = 3). | ||
The VHH–MUC1(Tn) conjugate can activate both CD4 and CD8 T cells
To achieve effective tumor eradication, help from CD4 T cells is required to prime and induce memory cytotoxic CD8 T cells. To assess the CD4 and CD8 T cell responses evoked by VHH7-directed antigen delivery, we conducted ex vivo stimulation of T lymphocytes from immunized mice. Antigen presenting cells were isolated from the spleens of naïve mice, pulsed with VHH7 or VHH7–MUC1(Tn) conjugates for antigen processing and presentation, then mixed in a ratio of 1:10 with T cells purified from previously immunized mice. MUC1-specific T cell activation was followed for two days by flow cytometric analysis of intracellular cytokine expression (Fig. 5).
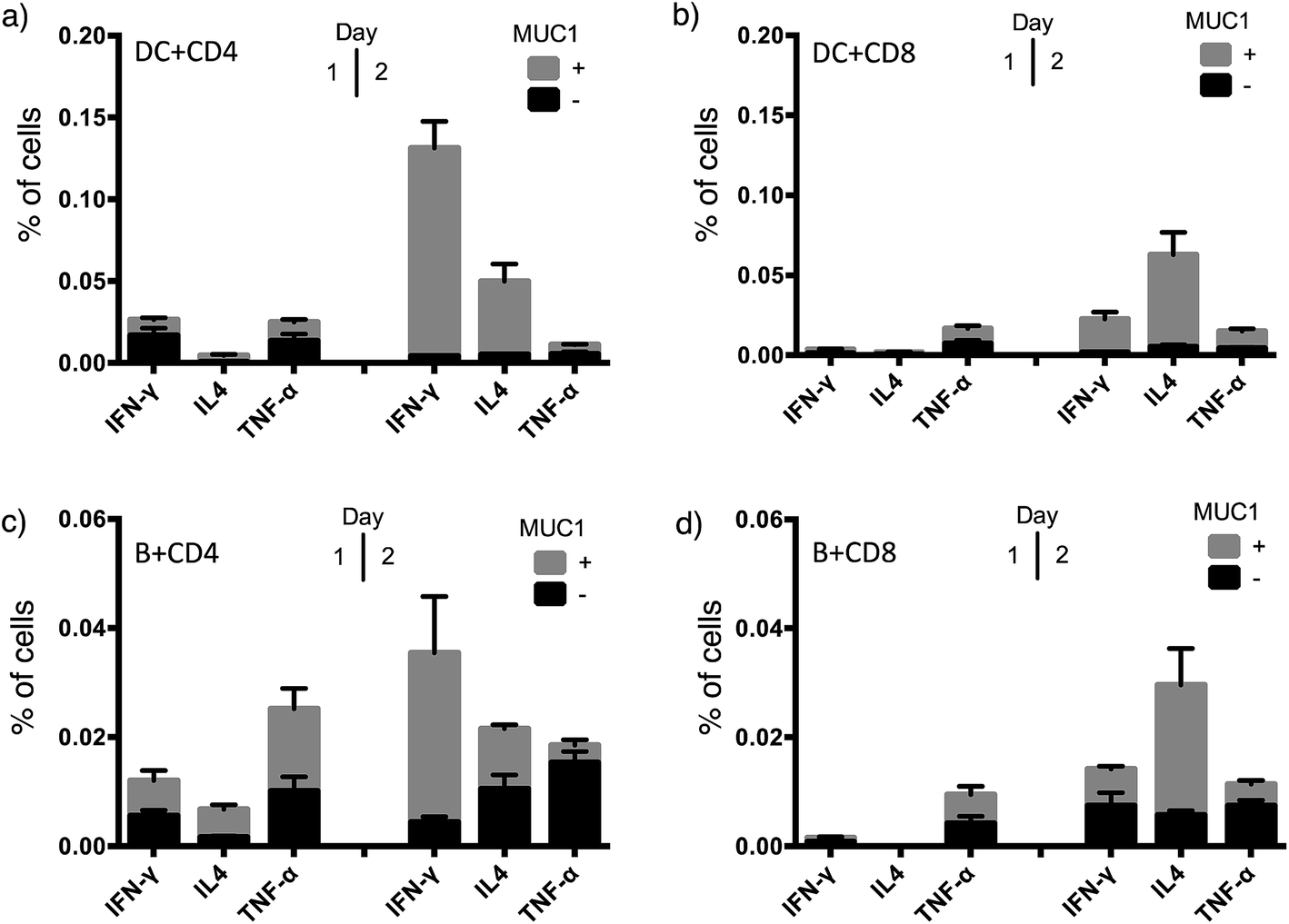 | ||
| Fig. 5 T cell activation and cytokine profile after co-culture with VHH7 or VHH7–MUC1-pulsed naive APCs. (a) CD4 T cell cytokine profile with splenic DCs as APCs; (b) CD8 T cell cytokine profile with splenic DCs as APCs; (c) CD4 T cell cytokine profile with B cells as APCs; (d) CD8 T cell cytokine profile with B cells as APCs. DC and B cells from naïve mice and T cells from immunized mice were purified by immunomagnetic selection. DC and B-cells were pulsed with VHH7 or VHH7–MUC1(Tn) conjugates, and then co-cultured with T cells for 24 h or 48 h before flow cytometric analysis. Error bars: mean ± SD, n = 3. Brefeldin A (BFA) was added 12 h prior to analysis to ensure intracellular retention of the indicated cytokines. | ||
Production of Th1 (IFN-γ, TNF-α) and Th2 (IL4) signature cytokines was examined. Antigen-specific CD4 and CD8 T cells were primed by incubation with antigen presenting cells (DCs or B cells) pulsed with the VHH7–MUC1(Tn) conjugate. Activation was observed within 24 h, and was accompanied by a 4-fold increase in the expression of the early activation marker CD69 (Fig. S3†). Even though less abundant, not surprisingly DCs were more effective than B cells at activating T cells, providing ∼5 times more cytokine induction. CD4 T cells from immunized animals produced high levels of IFN-γ, a hallmark of Th1-polarized T cell activation.47 As both CD4 and CD8 T cell responses were induced by VHH7–MUC1(Tn)-pulsed DCs, we conclude that cross-presentation of the antigen to class I MHC must have occurred.48
Memory response
The generation of long-term memory would be a highly desirable attribute for cancer vaccines. Although the exact requirements for generating and maintaining immune memory remain elusive, a strong primary immune response will generate a larger pool of cells from which to recruit memory cells. We analyzed the antibody response (Fig. S4†) after one or two boosts, using mice immunized with VHH7–MUC1(Tn) conjugate (4 injections) twelve months prior to the boost. A single boost effectively recalled the IgG1 response, as assayed by MUC1(Tn) ELISA compared to mice having completed only the initial immunization schedule. A second boost 7 days later did not further increase serum IgG1 levels.Conclusions
The MUC1 epitope targeted here has been used both as a B cell9 and a T cell31 epitope. Improved design of vaccine constructs is required to overcome poor immunogenicity and obtain a strong humoral and cytotoxic immune response against the MUC1 epitope. The class II MHC-specific single-domain antibody fragment VHH7 is an effective vector to target a larger population of APCs, such as DCs, B cells and macrophages. When VHH7 is conjugated to a tumor-associated antigen, it binds to the surface of targeted cells, is processed and presented for B cell activation. Endocytosis of the VHH-class II MHC complex allows efficient loading of APCs with exogenous antigen and subsequent (cross) presentation.49,50 A strong and lasting immune response against MUC1(Tn) could thus be established. By targeting of antigen to different leukocyte surface markers responses can be skewed towards the B cell or T cell compartment,27 the mechanistic details of which are worthy of further investigation.The current vaccine constructs-completely structurally defined small protein conjugates-relied on the combined strengths of antibody engineering and chemical synthesis. The small, non-glycosylated, but fully functional VHHs that target APCs with low inherent immunogenicity minimized the response to the VHH carrier in the course of immunization, which is critical to obtain a focused immune response to tumor-specific epitopes. The sortase-mediated conjugation strategy conveniently modifies protein vectors with synthetic payloads of choice. Chemical synthesis of glycosylated tumor-associated epitopes provides a gateway to access antigens with defined posttranslational modifications. This strategy is a promising and straightforward method to generate focused immune responses towards epitopes with unique glycophenotypes.
Acknowledgements
This research was supported by NIH (R01AI087879) and NIH DP1. We thank the Whitehead Institute flow cytometry facility for instrument usage, and Eric Spooner for assistance with mass spectrometric analysis. J. N. D. is a Calouste Gulbenkian scholar, funded by the Calouste Gulbenkian Foundation, Champalimaud Foundation, Portuguese Science and Technology Foundation and Portuguese Ministry of Health.Notes and references
- S. J. Gendler, J. Mammary Gland Biol. Neoplasia, 2001, 6, 339–353 CrossRef CAS PubMed.
- C. L. Hattrup and S. J. Gendler, Annu. Rev. Physiol., 2008, 70, 431–457 CrossRef CAS PubMed.
- T. Ju, G. S. Lanneau, T. Gautam, Y. Wang, B. Xia, S. R. Stowell, M. T. Willard, W. Wang, J. Y. Xia, R. E. Zuna, Z. Laszik, D. M. Benbrook, M. H. Hanigan and R. D. Cummings, Cancer Res., 2008, 68, 1636–1646 CrossRef CAS PubMed.
- J. Wesseling, S. W. van der Valk and J. Hilkens, Mol. Biol. Cell, 1996, 7, 565–577 CrossRef CAS PubMed.
- J. Wesseling, J. Cell Biol., 1995, 129, 255–265 CrossRef CAS PubMed.
- M. A. Cheever, J. P. Allison, A. S. Ferris, O. J. Finn, B. M. Hastings, T. T. Hecht, I. Mellman, S. A. Prindiville, J. L. Viner, L. M. Weiner and L. M. Matrisian, Clin. Cancer Res., 2009, 15, 5323–5337 CrossRef PubMed.
- T. Kimura and O. J. Finn, Expert Opin. Biol. Ther., 2013, 13, 35–49 CrossRef CAS PubMed.
- S. Dziadek, D. Kowalczyk and H. Kunz, Angew. Chem., Int. Ed., 2005, 44, 7624–7630 CrossRef CAS PubMed.
- V. Lakshminarayanan, P. Thompson, M. a. Wolfert, T. Buskas, J. M. Bradley, L. B. Pathangey, C. S. Madsen, P. a. Cohen, S. J. Gendler and G.-J. Boons, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 261–266 CrossRef CAS PubMed.
- H. Cai, M.-S. Chen, Z.-Y. Sun, Y.-F. Zhao, H. Kunz and Y.-M. Li, Angew. Chem., Int. Ed., 2013, 52, 6106–6110 CrossRef CAS PubMed.
- S. Ingale, M. a. Wolfert, J. Gaekwad, T. Buskas and G.-J. Boons, Nat. Chem. Biol., 2007, 3, 663–667 CrossRef CAS PubMed.
- Z.-H. Huang, L. Shi, J.-W. Ma, Z.-Y. Sun, H. Cai, Y.-X. Chen, Y.-F. Zhao and Y.-M. Li, J. Am. Chem. Soc., 2012, 134, 8730–8733 CrossRef CAS PubMed.
- Y. Gao, Z.-Y. Sun, Z.-H. Huang, P.-G. Chen, Y.-X. Chen, Y.-F. Zhao and Y.-M. Li, Chemistry, 2014, 20, 13541–13546 CrossRef CAS PubMed.
- H. Liu and D. J. Irvine, Bioconjugate Chem., 2015, 26, 791–801 CrossRef CAS PubMed.
- T. R. Hoover, A. D. Robertson, R. L. Cerny, R. N. Hayes, J. Imperial, V. K. Shah and P. W. Ludden, Nature, 1987, 329, 855–857 CrossRef CAS PubMed.
- F. V. V Castro, A. L. Tutt, A. L. White, J. L. Teeling, S. James, R. R. French and M. J. Glennie, Eur. J. Immunol., 2008, 38, 2263–2273 CrossRef PubMed.
- L. K. Swee, C. P. Guimaraes, S. Sehrawat, E. Spooner, M. I. Barrasa and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1428–1433 CrossRef PubMed.
- R. W. Carter, C. Thompson, D. M. Reid, S. Y. C. Wong and D. F. Tough, J. Immunol., 2006, 177, 2276–2284 CrossRef CAS.
- L.-Z. He, A. Crocker, J. Lee, J. Mendoza-Ramirez, X.-T. Wang, L. A. Vitale, T. O'Neill, C. Petromilli, H.-F. Zhang, J. Lopez, D. Rohrer, T. Keler and R. Clynes, J. Immunol., 2007, 178, 6259–6267 CrossRef CAS.
- I. Caminschi and K. Shortman, Trends Immunol., 2012, 33, 71–77 CrossRef CAS PubMed.
- C. Vincke, R. Loris, D. Saerens, S. Martinez-Rodriguez, S. Muyldermans and K. Conrath, J. Biol. Chem., 2009, 284, 3273–3284 CrossRef CAS PubMed.
- R. Heukers, P. M. P. van Bergen en Henegouwen and S. Oliveira, Nanomedicine, 2014, 10, 1441–1451 CrossRef CAS PubMed.
- S. Muyldermans and V. V. Smider, Curr. Opin. Immunol., 2016, 40, 7–13 CrossRef CAS PubMed.
- M. W. Popp, S. K. Dougan, T.-Y. Chuang, E. Spooner and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3169–3174 CrossRef CAS PubMed.
- M. D. Witte, J. J. Cragnolini, S. K. Dougan, N. C. Yoder, M. W. Popp and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11993–11998 CrossRef CAS PubMed.
- Z. Li, C. S. Theile, G.-Y. Chen, A. M. Bilate, J. N. Duarte, A. M. Avalos, T. Fang, R. Barberena, S. Sato and H. L. Ploegh, Angew. Chem., 2015, 127, 11872–11876 CrossRef.
- J. N. Duarte, J. J. Cragnolini, L. K. Swee, A. M. Bilate, J. Bader, J. R. Ingram, A. Rashidfarrokhi, T. Fang, A. Schiepers, L. Hanke and H. L. Ploegh, J. Immunol., 2016, 197, 4838–4847 CrossRef CAS PubMed.
- M. Rashidian, E. J. Keliher, A. M. Bilate, J. N. Duarte, G. R. Wojtkiewicz, J. T. Jacobsen, J. Cragnolini, L. K. Swee, G. D. Victora, R. Weissleder and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6146–6151 CrossRef CAS PubMed.
- T. Fang, J. N. Duarte, J. Ling, Z. Li, J. S. Guzman and H. L. Ploegh, Angew. Chem., Int. Ed., 2016, 55, 2416–2420 CrossRef CAS PubMed.
- N. Martínez-Sáez, N. T. Supekar, M. A. Wolfert, I. A. Bermejo, R. Hurtado-Guerrero, J. L. Asensio, J. Jiménez-Barbero, J. H. Busto, A. Avenoza, G.-J. Boons, J. M. Peregrina and F. Corzana, Chem. Sci., 2016, 7, 2294–2301 RSC.
- V. Apostolopoulos, J. S. Haurum and I. F. C. McKenzie, Eur. J. Immunol., 1997, 27, 2579–2587 CrossRef CAS PubMed.
- V. Apostolopoulos, V. Karanikas, J. S. Haurum and I. F. McKenzie, J. Immunol., 1997, 159, 5211–5218 CAS.
- F. Y. Avci, X. Li, M. Tsuji and D. L. Kasper, Nat. Med., 2011, 17, 1602–1609 CrossRef CAS PubMed.
- D. M. Coltart, A. K. Royyuru, L. J. Williams, P. W. Glunz, D. Sames, S. D. Kuduk, J. B. Schwarz, X.-T. Chen, S. J. Danishefsky and D. H. Live, J. Am. Chem. Soc., 2002, 124, 9833–9844 CrossRef CAS PubMed.
- U. Karsten, Glycobiology, 2004, 14, 681–692 CrossRef CAS PubMed.
- T. Ninkovic and F.-G. Hanisch, J. Immunol., 2007, 179, 2380–2388 CrossRef CAS.
- A. L. Sørensen, C. A. Reis, M. A. Tarp, U. Mandel, K. Ramachandran, V. Sankaranarayanan, T. Schwientek, R. Graham, J. Taylor-Papadimitriou, M. A. Hollingsworth, J. Burchell and H. Clausen, Glycobiology, 2006, 16, 96–107 CrossRef PubMed.
- M. R. Pratt and C. R. Bertozzi, Chem. Soc. Rev., 2005, 34, 58–68 RSC.
- K.-F. Mo, T. Fang, S. H. Stalnaker, P. S. Kirby, M. Liu, L. Wells, M. Pierce, D. H. Live and G.-J. Boons, J. Am. Chem. Soc., 2011, 133, 14418–14430 CrossRef CAS PubMed.
- H. Kunz and S. Birnbach, Angew. Chem., Int. Ed. Engl., 1986, 25, 360–362 CrossRef.
- A. Kirchhofer, J. Helma, K. Schmidthals, C. Frauer, S. Cui, A. Karcher, M. Pellis, S. Muyldermans, C. S. Casas-Delucchi, M. C. Cardoso, H. Leonhardt, K.-P. Hopfner and U. Rothbauer, Nat. Struct. Mol. Biol., 2010, 17, 133–138 CAS.
- T. Gilewski, S. Adluri, G. Ragupathi, S. Zhang, T. J. Yao, K. Panageas, M. Moynahan, A. Houghton, L. Norton and P. O. Livingston, Clin. Cancer Res., 2000, 6, 1693–1701 CAS.
- T. Kimura, J. R. McKolanis, L. A. Dzubinski, K. Islam, D. M. Potter, A. M. Salazar, R. E. Schoen and O. J. Finn, Cancer Prev. Res., 2013, 6, 18–26 CrossRef CAS PubMed.
- M. Palmer, J. Parker, S. Modi, C. Butts, M. Smylie, A. Meikle, M. Kehoe, G. MacLean and M. Longenecker, Clin. Lung Cancer, 2001, 3, 49–57 CrossRef CAS PubMed.
- R. M. Perlmutter, D. Hansburg, D. E. Briles, R. A. Nicolotti and J. M. Davie, J. Immunol., 1978, 121, 566–572 CAS.
- C. H. M. J. Van Elssen, H. Clausen, W. T. V Germeraad, E. P. Bennet, P. P. Menheere, G. M. J. Bos and J. Vanderlocht, J. Immunol. Methods, 2011, 365, 87–94 CrossRef CAS PubMed.
- M. Tosolini, A. Kirilovsky, B. Mlecnik, T. Fredriksen, S. Mauger, G. Bindea, A. Berger, P. Bruneval, W.-H. Fridman, F. Pages and J. Galon, Cancer Res., 2011, 71, 1263–1271 CrossRef CAS PubMed.
- K. Palucka and J. Banchereau, Immunity, 2013, 39, 38–48 CrossRef CAS PubMed.
- G. Lizée, G. Basha, J. Tiong, J.-P. Julien, M. Tian, K. E. Biron and W. A. Jefferies, Nat. Immunol., 2003, 4, 1065–1073 CrossRef PubMed.
- P. R. Wolf and H. L. Ploegh, Annu. Rev. Cell Dev. Biol., 1995, 11, 267–306 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00446j |
| ‡ Present address: Program in Cellular and Molecular Medicine, Boston Children's Hospital, Boston, MA 02115, USA. |
| § Present address: Tumor Immunology, Maastricht University, Maastricht 6229ER, The Netherlands. |
| ¶ Present address: Koch Institute for Integrated Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. |
| This journal is © The Royal Society of Chemistry 2017 |