
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise crystallographic visualization of dynamic guest binding in a nanoporous framework†
Gabriel
Brunet
 a,
Damir A.
Safin
a,
Mohammad Z.
Aghaji
a,
Koen
Robeyns
b,
Ilia
Korobkov
a,
Tom K.
Woo
*a and
Muralee
Murugesu
*a
a,
Damir A.
Safin
a,
Mohammad Z.
Aghaji
a,
Koen
Robeyns
b,
Ilia
Korobkov
a,
Tom K.
Woo
*a and
Muralee
Murugesu
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ontario K1N 6N5, Canada. E-mail: twoo@uottawa.ca; m.murugesu@uottawa.ca
bInstitute of Condensed Matter and Nanosciences, Université Catholique de Louvain, Place L. Pasteur 1, B-1348 Louvain-la-Neuve, Belgium
First published on 13th February 2017
Abstract
Binding sites are at the heart of all host–guest systems, whether biological or chemical. When considering binding sites that form covalent bonds with the guest, we generally envision a single, highly specific binding motif. Through single-crystal X-ray crystallography, the dynamic binding of a guest that displays a variety of covalent binding motifs in a single site of adsorption is directly observed for the first time. The stepwise crystallographic visualization of the incorporation of I2 within a porous MOF is presented, wherein the preferred binding motifs throughout the uptake process are identified. The guest I2 molecules initially bind with terminal iodide atoms of the framework to form [I4]2− units. However, as the adsorption progresses, the I2 molecules are observed to form less energetically favorable I3− groups with the same framework iodide atoms, thereby allowing for more guest molecules to be chemisorbed. At near saturation, even more binding motifs are observed in the same pores, including both physisorbed and chemisorbed guest molecules. Herein, we present the successful identification of a unique set of host–guest interactions which will drive the improvement of high capacity iodine capture materials.
Introduction
Binding sites are ubiquitous in chemical and biological systems, extending from small molecule adsorption in carbon capture materials to complex ligand binding sites in proteins.1 When considering binding sites with high selectivity towards a certain substrate, we inherently envision a single binding motif organized by specific host–guest interactions. The nature of these interactions are generally elucidated through spectroscopic methods, however, the direct observation of binding sites through single crystal X-ray diffraction (SCXRD) provides the most indisputable evidence of the binding interactions at the atomic level.2 The application of SCXRD is particularly challenging for gas adsorption processes, which are highly fluxional. Only in a few cases has the direct crystallographic observation of adsorption sites in metal–organic frameworks (MOFs) and other nanoporous solids been achieved.3 In all cases, each adsorption site only displayed a single binding motif – even when weak physisorption was involved. When the binding is chemisorptive, one would not expect a binding pocket to accommodate a single substrate in more than one way, since covalent interactions tend to be highly specific, directional and strong. To date, the direct observation of a binding site that can accommodate gaseous substrates in a number of different binding modes has yet to be reported. Herein, we present for the first time the dynamic binding of a gaseous guest that displays a variety of covalent binding motifs in a single adsorption site. This remarkable behavior has been directly observed in a stepwise crystallographic fashion, where the chemisorption of I2 within a porous MOF exhibits changing covalent bonding motifs in the same pore to accommodate a larger number of gas molecules.With an increasing interest in the development of gas capture materials, MOFs serve as a new and exciting avenue to explore due to their facile functionalization and high surface areas. The ability to control the framework components has resulted in a steady year-over-year improvement of the uptake capacities and selectivities for various gases of interest.4 In particular, the capture and sequestration of highly mobile volatile gasses produced from nuclear fission, such as 129I and 131I, is becoming an extensive area of research in the field of porous materials.5 Such radionuclides pose a significant risk to human health and the environment, therefore, strategies to efficiently store radioactive iodine in a durable waste form must be developed. The present work seeks to provide new insights into the fascinating binding interactions exhibited by I2, and hence improve upon the current state-of-the-art radioactive iodine capture methods. Our strategy relies on the use of halide binding sites separated through optimal distances, thereby providing favorable halide–halide interactions between the guest and the host.
While studying materials for the selective uptake of I2, we have surveyed porous MOFs with iodide functional groups lining the pores that would act as anchor sites for I2 adsorption. One such material that we have examined is the {[(ZnI2)3(TPT)2]·5.5(C6H5NO2)}n (1) MOF, developed by Biradha et al., that is built from inorganic nodes of ZnI2 linked by the 2,4,6-tris(4-pyridyl)-1,3,5-triazine (TPT) ligand.6 We found that the material displays a near record breaking I2 gravimetric uptake capacity and that there were significant deformations of the framework triggered by the adsorption. Further investigation of the adsorption process by SCXRD allowed for the direct visualization of I2 binding in 1, and the remarkable discovery that the binding motif changes as the adsorption proceeds. Hence, we provide a detailed investigation, through crystallographic and computational methods, of the changes in the binding sites of I2 throughout the uptake process.
Results and discussion
Inclusion procedure
The porous MOF 1 exhibits a doubly interpenetrated structure, leading to the formation of continuous channels with pore apertures of 8 × 5 Å2 (Fig. 1). As-synthesized single crystals of 1 initially contain nitrobenzene as guest molecules, which interact strongly with one another due to π⋯π stacking interactions. A detailed description of the structure and properties of 1 have been presented elsewhere.6 Upon the exposure of 1 to I2 vapors in a closed vessel at room temperature, a pronounced and rapid color change in the single crystals, from colorless to black, could be observed (Fig. 2). We have successfully obtained three crystal structures by varying the time of exposure of 1 to the I2 vapors from 3 to 15 h, providing us with vital information on the mechanism by which I2 becomes incorporated in the MOF. Exposure times beyond 15 hours resulted in weaker diffraction and eventual loss of crystallinity. Nevertheless, the host framework continues the uptake of I2 up to approximately 72 h, after which the MOF becomes saturated (vide infra). The degree of I2 encapsulation will likely depend on the size of the crystals,7 and therefore, efforts were made to select single crystals of approximately equal size for the SCXRD experiments.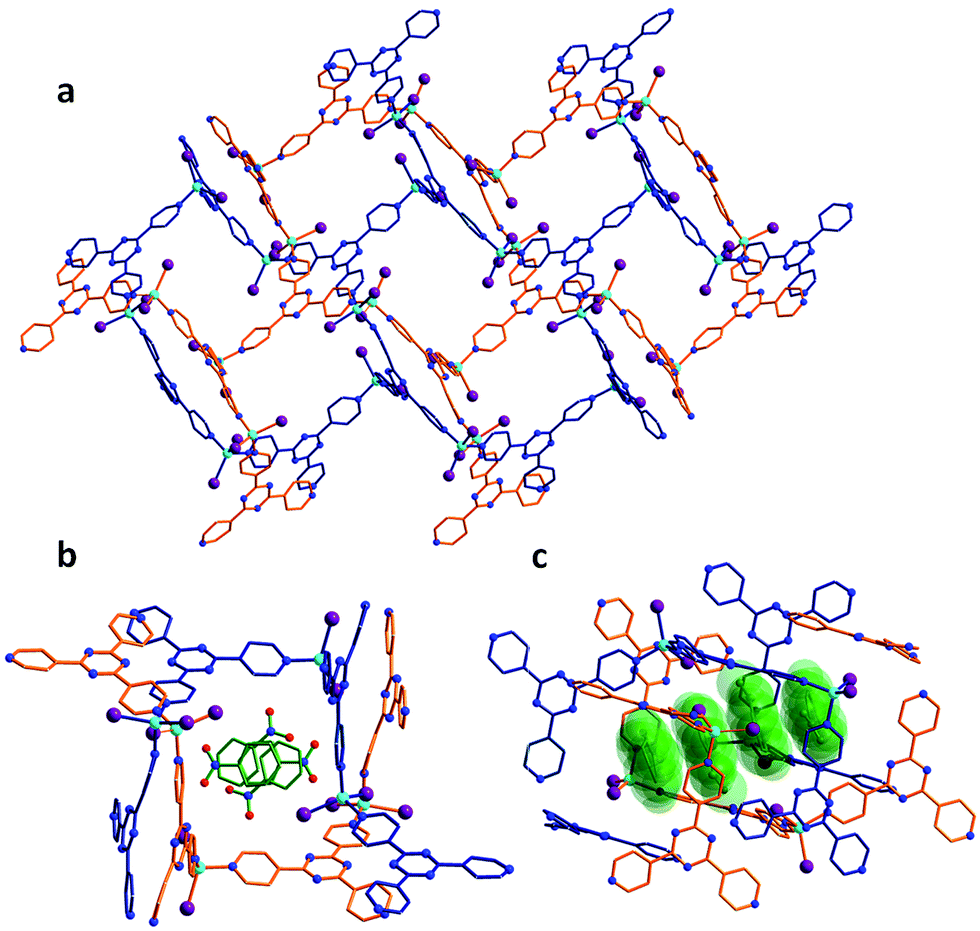 | ||
| Fig. 1 Crystal structure of the as-synthesized MOF 1. (a) Crystal packing of the empty framework 1 along the b-axis, illustrating the doubly-interpenetrated structure. (b) Cross-section of the pores of 1, which are initially filled with nitrobenzene molecules. (c) Side-view of the continuous channels found in 1 and the nitrobenzene guests displayed in space-filling diagrams. | ||
 | ||
| Fig. 2 Time-lapsed photographs demonstrating the color change associated with the exposure of single crystals of 1 to I2 vapors, as viewed under optical microscope. | ||
Formation of [I4]2− bridges
The first SCXRD structure, obtained after 3 h of 1 being immersed in vapors of I2, revealed large peaks of electron density within the pores, which were identified as multiple I2 molecules, giving the first intermediate {[(ZnI2)6(TPT)4]·1.3(I2)·8.65(C6H5NO2)}n (2). It is important to note that the chemical formulas of the iodine-containing MOFs described herein highlight the number of adsorbed I2 molecules, rather than the newly formed iodine species. The crystal system remains in the monoclinic family, however the space group changes from C2/c in 1 to P21/c in 2. Moreover, analysis of the crystal structure indicates that there are approximately 5.2 I2 molecules per unit cell, resulting in an iodine content of ∼7.2 wt% (excluding the framework iodide atoms). With an increase in the iodine content, we conversely observe a decrease in the amount of nitrobenzene molecules from 44 to 34.6 per unit cell. This exchange process, where the nitrobenzene molecules are readily replaced by the encroaching I2, provides evidence that the interactions formed by the I2 species are more favorable than the π⋯π stacking interactions of the nitrobenzene guests (Fig. 1c). Careful analysis of the crystal structure of 2 reveals that the highest occupied I2 guest (I21–I22), and thus the most favourable initial binding site, is positioned between two iodide atoms (I5 and I7) originating from the ZnI2 building units of the host structure (Fig. 3).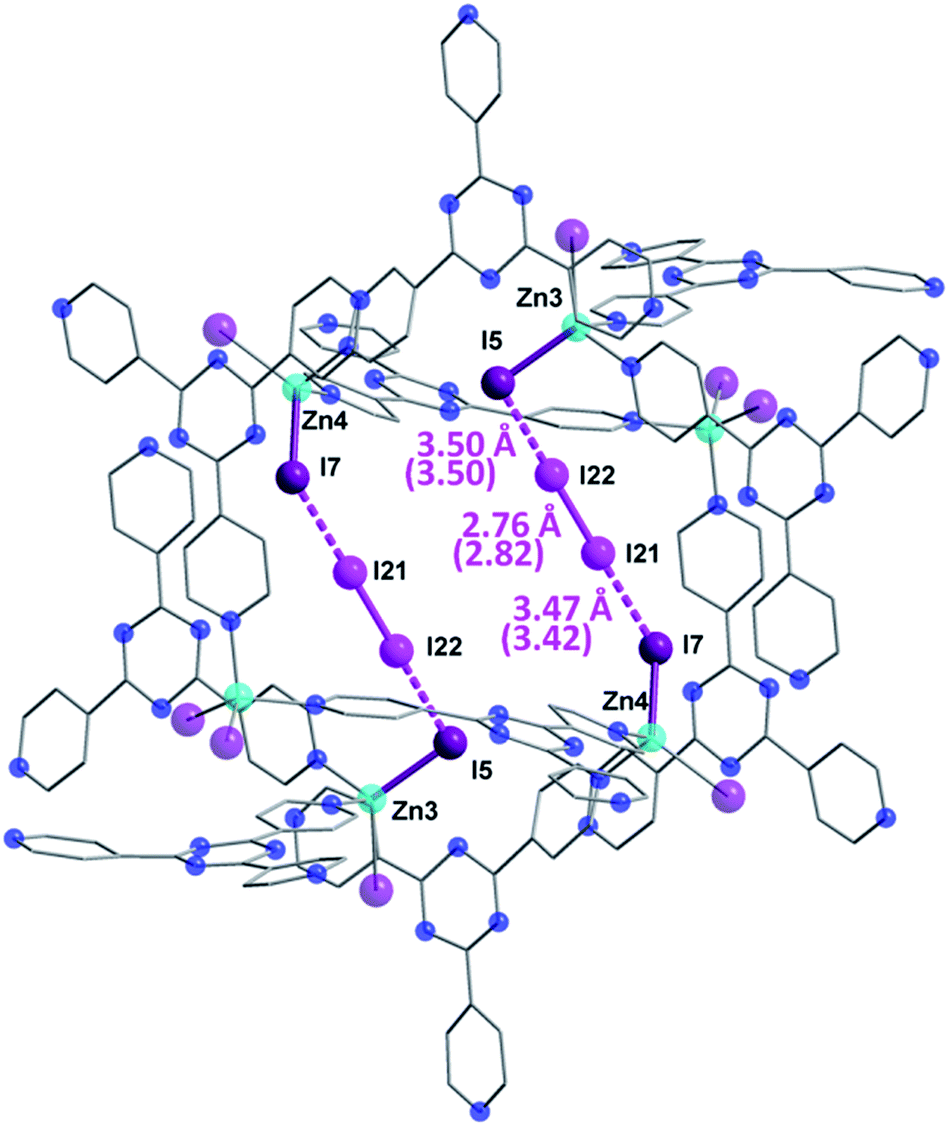 | ||
| Fig. 3 Molecular structure of a single pore of 2, emphasizing the initial preferred binding mode of the I2 guest which forms strong halogen–halogen interactions with the iodide ions of the framework, effectively forming an [I4]2− unit. Selected geometric parameters are shown, with DFT computed values given in parentheses. Guest iodine atoms are shown in a lighter shade of purple than the iodide atoms belonging to the host framework. Hydrogen and disordered atoms are omitted for clarity. | ||
The bond distance between the I2 molecule (I21–I22) and framework iodide atoms are found to be 3.50 and 3.47 Å, while the bond length in the guest I2 elongates from a molecular value of 2.67 Å to 2.76 Å upon formation of 2 (Table S1†). This type of M–I⋯I–I⋯I–M linkage has been previously reported in a handful of discrete compounds and polymeric chains,8 wherein the four iodine atoms are characterized as forming a [I4]2− unit. However, this represents the first MOF structure containing this type of unusual bridging unit. The [I4]2− fragment is approximately linear with the largest deviation coming from the I5⋯I22–I21 angle (∼173.0°). It is also important to note that three other distinct I2 molecules have been identified in 2, with occupancies ranging from 4.6–7.9% (Table S1†). These sparsely occupied guest molecules reveal the subsequent binding sites, which will dominate the uptake behaviour upon further I2 exposure, in the form of I3− anions (vide infra). Additionally, a second [I4]2− unit can also be located in an adjacent pore to the first [I4]2− linkage, however, its occupancy is 15.5% due to the presence of an overlapping nitrobenzene solvent molecule.
In order to provide further insight into the nature of the I2 binding in 2, we have employed dispersion corrected periodic DFT calculations. Geometry optimization of 2 starting from the crystal structure gives a computed structure in excellent agreement with the SCXRD structure (Fig. S1†), with the I–I bond distances deviating no more than 0.06 Å (parenthetic values in Fig. 3). To investigate the degree of covalency in the I−⋯I2 interactions, Wiberg bond orders were calculated (Table S1†). The dual end-on bridging interaction of the I2 guests with the terminal iodides of the framework gave bond orders of 0.20 and 0.24, for I5⋯I22 and I7⋯I21, respectively. By comparison, the I21–I22 pair has a bond order of 0.78 in 2. These results, combined with the calculated I2 bonding energy of 26.2 kcal mol−1, indicate that the I2 adsorption in 2 is chemisorptive in nature.
Increased I2 uptake and replacement of [I4]2− bridges
The same iodine vapor diffusion strategy was employed to obtain the second intermediate structure {[(ZnI2)6(TPT)4]·2.51(I2)·3(C6H5NO2)}n (3), after 6 h of exposure time to the I2 vapors. The dark brown crystals remained in the P21/c space group (Table S2†), yet yielded more electron density within the channels of the MOF, assigned to I2 molecules. The host framework maintains the structural architecture and topology of 1, however, we can observe a further decrease in the unit cell volume going from 2 to 3 (∼0.8%). The encapsulation of additional I2 guests when going from 2 to 3 is accompanied by a further reduction in nitrobenzene solvent molecules, down from 34.6 to 12 molecules per unit cell following 6 h of exposure time to I2 vapors. Accordingly, the iodine guest content increased from 7.2 wt% to 15.3 wt%, yielding ∼10 I2 molecules per unit cell. This drastic increase in merely 3 h of additional exposure time emphasizes the potential of this framework for the rapid capture and sequestration of I2.In comparison to the principal I2 binding motif observed in 2, careful analysis of the crystal structure of 3 reveals eight distinct I2 binding positions with partial occupancies. More interestingly, I2 molecules occupy the same crystallographically equivalent pore in three distinct manners, with occupancies of 69, 20 and 11% (Fig. 4). The high occupancy (69%) binding motif, referred as motif A, is identical to the I2 adsorption observed in 2. The I⋯I distances and computed bond orders (Tables S3 and S4†) are very similar as those in 2 and we can therefore characterize the adsorption as chemisorptive. The lower occupancy binding motif (11%), referred as motif B, not only accommodates an additional I2 molecule compared to motif A, but does so in a completely different manner even though the same atoms of the MOF framework form bonds with the I2 guests. In motif A, two I2 molecules each form [I4]2− moieties with atoms I2 and I10 of the framework. In motif B, each of the two I2B framework atoms instead form an I3− group with the I2 guests (I19B–I20B), and a third I2 molecule forms an [I4]2− moiety with the two I10 atoms (I23). It is noteworthy that the I3− and [I4]2− fragments in motif B are non-interacting with a shortest I⋯I separation of 4.48 Å.
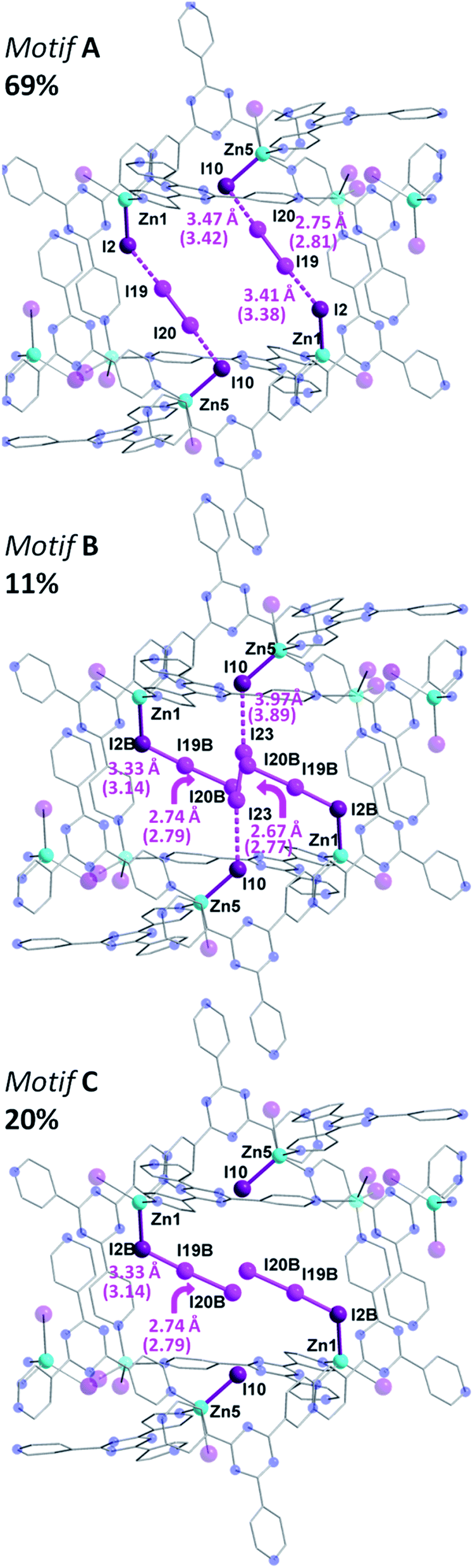 | ||
| Fig. 4 Molecular structure of a single pore of 3, illustrating the disordered and partially occupied I2 guests. In motif A (top), there is the same [I4]2− unit as in 2, while in motif B (middle), there are chemisorbed I3− group along with an I2 molecule forming an [I4]2− unit. Motif C is also shown (bottom). Selected geometric parameters are displayed with DFT computed values given in parenthesis. Guest iodine atoms are shown in a lighter shade of purple than the iodide atoms belonging to the host framework. Hydrogen and additional disordered atoms are omitted for clarity. | ||
The formation of the triiodide group in motif B suggests that the I2 molecule is chemisorbed. The bond distance between the framework I2B atom (which participates in I3− formation, whereas I2 forms the [I4]2− unit) and the I19B atom of the adsorbed I2 is 3.33 Å, consistent with the parameters of an I3− moiety.9 The DFT calculated I2B–I19B bond order is 0.45, while the binding energy was determined to be 21.6 kcal mol−1. These metrics are all consistent with chemisorption. The SCXRD data suggests that the terminal I19B–I20B bond length of the I3− moiety in motif B is 2.74 Å, and is in strong agreement with the DFT optimized geometry of motif B, which gives a I19B–I20B bond length of 2.79 Å.
In addition to the two I2 molecules that adsorb to form triiodide groups, motif B accommodates a third I2 molecule that forms a [I4]2− unit which bisects the pore. DFT calculations also point towards the bonding being chemisorptive. For example, the calculated binding energy was determined to be 20.2 kcal mol−1, which is slightly lower than the 21.6 kcal mol−1 value for the triiodide formation. Furthermore, using the DFT optimized structure, the I10–I23 bond order was found to be 0.15 (Table S3†). It is interesting to note that four additional I2 binding positions are observed in 3, but are located in a different pore (Fig. S2†). This pore accommodates three I2 guest molecules. A more detailed discussion of the bonding in this second pore is given in the ESI.† While motif A and motif B account for the I2 guests in 69 and 11% occupancies, respectively, the remaining 20% is accounted by I19B–I20B, which, as previously stated, forms an I3− unit with I2B and yields motif C (Fig. 4). This third binding motif is identical to motif B, without the bisecting I23–I23 molecule that forms the [I4]2− linkage. As such, the total occupancy of I19B–I20B is 31%, through the combination of motif B and motif C. Thus, motif B, which encapsulates I23, can only be present when motif A is absent, clearly reinforcing the notion of varying bonding motifs with an increase in I2 uptake.
This structural intermediate provides an excellent model to deduce the mechanistic pathway of I2 inclusion. Indeed, each terminal I− ion of the framework serves as a binding site for the I2 guests and display a variety of binding modes. The driving factor towards observing this unprecedented behavior is the energetic stability gained by the incorporation of increasing amounts of I2 molecules within the MOF. Bonding motif A, wherein the I2 molecules bind to form [I4]2− moieties, has the strongest calculated binding energy of 26.6 kcal mol−1 per I2 molecule. However, this motif only accommodates two I2 molecules. In order to accommodate a third I2 molecule, two [I4]2− units can rearrange to form less energetically favorable I3− groups. Evaluation of the bonding energy of isolated I3− groups, without the third I2 molecule, yields a bonding energy of 21.2 kcal mol−1. Thus, while the I3− groups form stronger covalent interactions with the terminal I− ions of the framework, compared to the [I4]2− units, their energetic configuration is less favorable. Overall, however, the energy of motif B, with three I2 molecules adsorbed, is 9.0 kcal mol−1 more stable than the energy of motif A, containing two adsorbed molecules, plus the energy of a free I2 molecule.
Continued sequestration of I2
Following multiple attempts to measure the SCXRD data of 1 saturated with I2 it was determined that 15 h of exposure time to I2 vapors was the limit for retaining adequate crystallinity. Hence, we present the single crystal structure of {[(ZnI2)6(TPT)4]·7.34(I2)}n (4), with the best of our refinement results. Intermediate 4 was refined in the monoclinic P2/n space group and further continues the trend of decreasing unit cell volumes with I2 uptake, reflecting the flexible nature of the MOF. It is important to note that while the agreement factors of the refinement are higher than in 2 and 3, due to the increase in partially occupied and positionally disordered guest molecules, the crystal structure of 4 provides invaluable structural insights regarding the encapsulation of I2. Notably, we can observe the absence of nitrobenzene solvent molecules and their exclusive replacement by I2. The unit cell now comprises approximately ∼29.4 localized I2 guest molecules, a radical increase of 198% going from 3 to 4. Thus, the iodine guest content is ∼37.2 wt%. In other words, 1 g of the empty framework of 1 can uptake around 0.59 g of I2 after 15 h which at this stage outperforms state-of-the-art zeolites, and is comparable to other porous MOFs.10 These results are in good agreement with the TGA measurements performed on single crystals of 4, giving an iodine guest content of 39.8% and an uptake of I2 of 0.66 g g−1 (Fig. S3†). This drastic increase in I2 inclusion from 9 additional hours of I2 vapor exposure results in a significant distortion of the MOF framework. The changes in the pores from the as-synthesized MOF 1, to the structure of 2 and 3 after 3 and 6 h of exposure time to I2 vapors, respectively, followed by the 15 h structure 4 are illustrated in Fig. 5. We can surmise that these distortions, which allow for a greater intake of I2, are a contributing factor in the loss of crystallinity of the single crystals. The connectivity of the framework remains the same, however the pyridine moieties, as well as the central triazine rings of the TPT ligand, are less planar and adopt a more twisted conformation as evidenced by the large thermal parameters. We can hypothesize that these groups exhibit some freedom of rotation, therefore permitting the entry and accommodation of a larger number of I2 guests within the same pore or channel. Structural analysis of 4 reveals that the initial [I4]2− unit found in 2 and 3 has been replaced with several partially occupied physisorbed and chemisorbed I2 molecules, also supporting the previous assessment of reorienting the [I4]2− unit to accommodate a larger number of I2 guests. While 3 displayed a significant increase in electron density assigned to I2, their occupancies remained relatively low, whereas 4 sees the incorporation of an even larger amount of I2 combined with higher occupancies (Table S5†). Chemisorption remains the preferred method of adsorption, with 13 individually refined I2 molecules covalently bound to I− ions of the framework. Furthermore, several [I4]2− units can also be identified in a different pore and in different emplacements than in the structures of 2 and 3 (Fig. S4†). It is worthwhile to mention that several I2 guest molecules are positionally disordered, such as I23–I24, which forms an I3− moiety with I10, undergoes a precession movement leading to a disordered I24 atom. The remaining I3− groups give distances and angles within a reasonable range to those found in the literature.11 The I2 guests involved in the formation of [I4]2− units, are weakly halogen bonded to terminal iodide ions of the framework, as displayed by elongated I−⋯I2 distances (Table S5†). The orientation of the guest along the two terminal I− ions strongly suggests the formation of the Zn–I⋯I–I⋯I–Zn linkage, in similar fashion to the [I4]2− unit described in 2. To complete the analysis of the guests, two physisorbed I2 molecules can also be identified. It is remarkable that even with the inclusion of all these I2 molecules, 4 is still able to encapsulate additional I2 molecules. This was confirmed by the TGA curve of a sample of 1 exposed to I2 vapors over a period of 72 h, giving a weight loss of ∼63.4% (Fig. S3†). Hence, one gram of 1 has the potential of loading an excess of 1.73 g of I2 at room temperature, making it an exceptional material for the capture of iodine. This value is comparable with the highest I2 uptake capacity reported for a MOF, namely Cu-BTC (1.75 g g−1), which was measured at 75 °C.12 This enormous uptake of I2 in 1 can be attributed to a number of factors, including favorable guest–host interactions, framework flexibility, and high porosity (∼60%). More specifically, 1 offers strong sites of adsorption for I2 through the terminal iodide ions of the framework as well as favorable π–halogen interactions with the TPT ligand.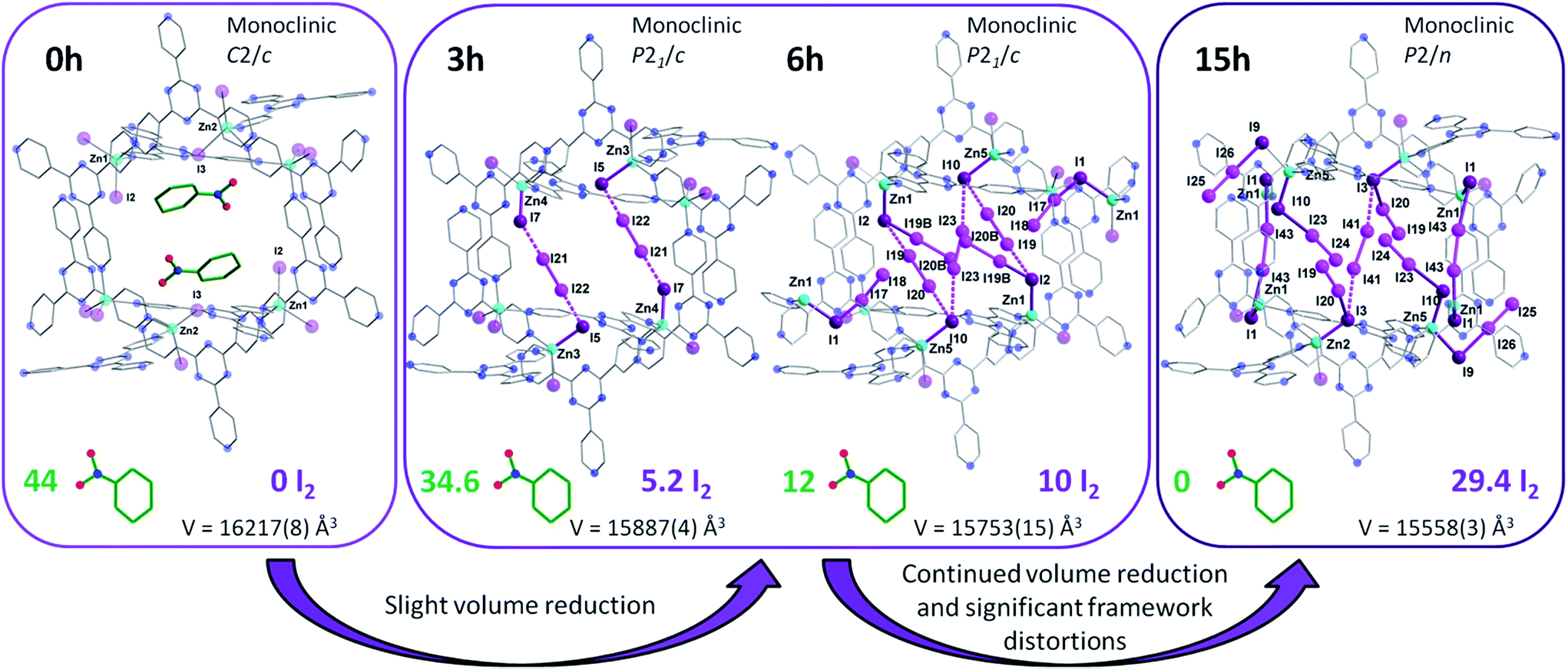 | ||
| Fig. 5 X-ray crystal structure of a single pore of 1 viewed along its channel direction. Evolution of the guests, where nitrobenzene solvent molecules are sequentially exchanged for I2 molecules, are illustrated for the same pore following 3, 6 and 15 h of exposure time to I2 vapors. The amount of localized guest molecules contained per unit cell are listed, along with changes in the space group and unit cell volumes for compounds 1–4. Guest iodine atoms are shown in a lighter shade of purple than the iodine atoms belonging to the host framework. Hydrogen and additional disordered atoms are omitted for clarity. | ||
Stability and release kinetics
In order to probe the release kinetics of iodine, we performed time-dependent diffuse reflectance spectroscopy (DRS) measurements on compound 1 once it was completely saturated by I2. The Kubelka–Munk spectra of TPT, ZnI2, I2 and 1 were first collected to examine the origin of the bands (Fig. S5†). Afterwards, single crystals of 1, saturated with I2, were measured through DRS over a period of 120 h at ambient conditions (Fig. 6). The time-lapsed Kubelka–Munk spectra reveal that stabilization of the compound occurs after approximately four days. This stabilized compound exhibits similar electronic transitions as 1 saturated with I2, providing strong evidence that iodine remains a major component of the MOF. In principle, we can envision that the more weakly encapsulated I2 guests (i.e. physisorbed) are more susceptible of being evacuated, while the chemisorbed iodide molecules would remain part of the framework and require harsher conditions for their removal. A detailed examination of the time-dependent DRS measurements shows a decrease in the intensity of the bands in the UV region, combined with an increase in the intensity and a blue-shift of the shoulders in the visible range. This may be explained by the repulsive interactions of the I2 molecules. The stabilized compound (after 96 h), is weakly diffracting, and therefore, cannot be examined by conventional crystallographic methods. This compound was, however, investigated by FTIR, revealing intense and broad bands at approximately 670, 1640 and 3350 cm−1, which is characteristic of the vibrations for H2O (Fig. S6†). Thus, if 1 is saturated by I2 and subsequently stored at ambient conditions, we can expect evaporation of the physisorbed I2 followed by incorporation of water molecules. Nevertheless, the DRS results demonstrate the potential of 1 for the irreversible capture of radioactive I2, through a number of strong chemisorption sites.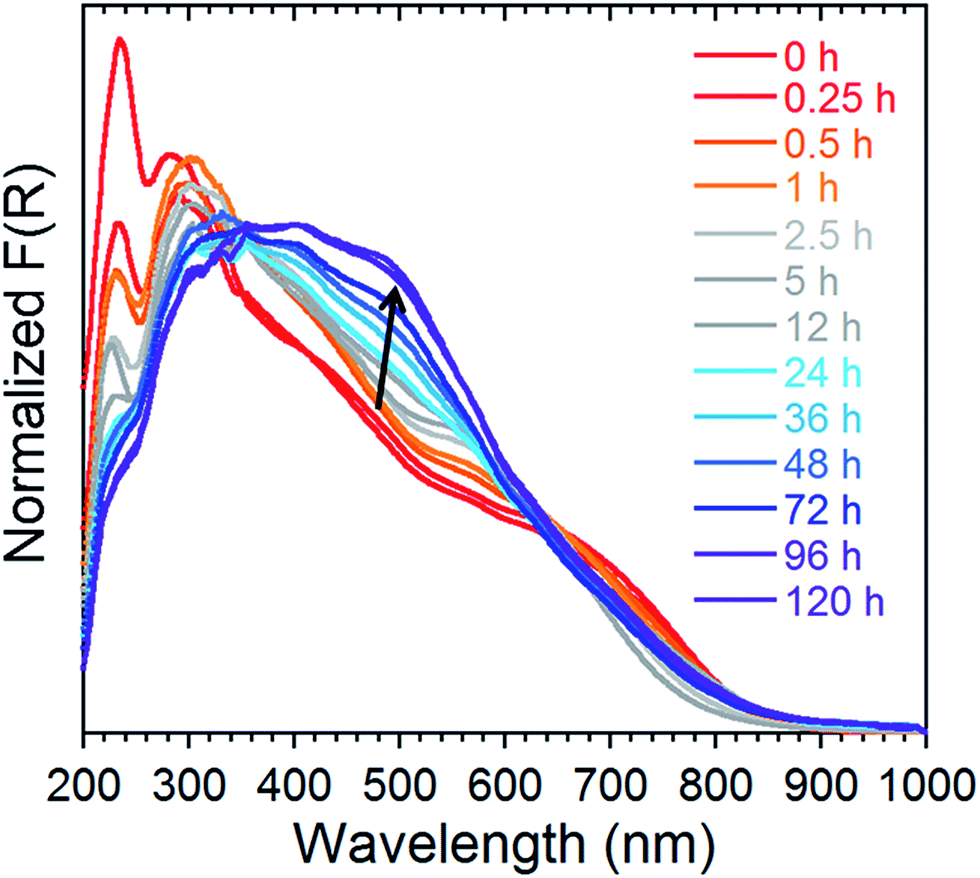 | ||
| Fig. 6 Normalized Kubelka–Munk spectra of 1 saturated with I2 followed over time at ambient conditions for a period of 120 h. | ||
Concluding remarks
The direct single crystal observation of gaseous substrate binding is exceedingly rare, due to the difficulty in retaining adequate crystallinity following guest encapsulation and the lability of gases. In this work, we have successfully elucidated the process by which gaseous I2 is systematically incorporated in the cavities of a highly porous MOF, through the use of stepwise crystallographic experiments and computation. Remarkably, the guest I2 molecules are found to adopt multiple covalent bonding motifs in the same adsorption site, even with the same framework atoms, depending on the percentage of uptake. This is unique in that covalent bonding tends to be highly directional, specific and strong, thereby commanding a single binding motif organized by these interactions. The participation of both physisorption and chemisorption in the uptake of gaseous guests is another fascinating feature of the MOF, since chemisorption is generally associated with frameworks exhibiting open metal sites that can bind guest molecules.13 This illustrates the optimization potential of MOFs, where, notably, chemisorption can be obtained without open metal sites, leading to higher chemical stabilities. Hence, we envision that this study will aid in the rational design of MOFs for the capture of gaseous fission products, through a fundamental understanding of the dynamics and site selection of gaseous substrates. The design strategy involving anchor sites based on halide–halide interactions is highly promising for the enhancement of radioactive iodine capture materials.Acknowledgements
The work was financially supported by the University of Ottawa, the Canadian Foundation for Innovation (CFI), and the Natural Sciences and Engineering Council of Canada (NSERC).Notes and references
-
(a) M. Lim, T. A. Jackson and P. A. Anfinrud, Science, 1995, 269, 962 CrossRef CAS PubMed
; (b) X. Nan, H.-H. Ng, C. A. Johnson, C. D. Laherty, B. M. Turner, R. N. Eisenman and A. Bird, Nature, 1998, 393, 386 CrossRef CAS PubMed
-
(a) H.-L. Zhou, R. B. Lin, C.-T. He, Y.-B. Zhang, N. Feng, Q. Wang, F. Deng, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2013, 4, 2534 Search PubMed
-
(a) J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard and O. M. Yaghi, Science, 2005, 309, 1350 CrossRef CAS PubMed
-
(a) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed
-
(a) S. U. Nandanwar, K. Coldsnow, M. Green, U. Utgikar, P. Sabharwall and D. E. Aston, Chem. Eng. J., 2016, 287, 593 CrossRef CAS
- K. Biradha and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3542 CrossRef
-
(a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai and M. Fujita, Nat. Protoc., 2014, 9, 246 CrossRef CAS PubMed
-
(a) L. Hewison, S. H. Crook, B. E. Mann, A. J. H. M. Meijer, H. Adams, P. Sawle and R. A. Motterlini, Organometallics, 2012, 31, 5823 CrossRef CAS
- C. M. Aragoni, M. Arca, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis and A. Mancini, Bioinorg. Chem. Appl., 2007, 2007, 17416 Search PubMed
-
(a) Q.-K. Liu, J.-P. Ma and Y.-B. Dong, Chem. Commun., 2011, 47, 7185 RSC
-
(a) S.-B. Zhao, R.-Y. Wang and S. Wang, Inorg. Chem., 2006, 45, 5830 CrossRef CAS PubMed
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. M. Nenoff, Chem. Mater., 2013, 25, 2591 CrossRef CAS
-
(a) A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials and instrumentations, synthesis and characterization, experimental details, X-ray refinement details, TGA, DRS and FTIR. CCDC 1475220 (2), 1475221 (3), 1475222 (4). Full crystallographic data for the solved structures have been deposited in the Cambridge Crystallographic Data Center. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00267j |
| This journal is © The Royal Society of Chemistry 2017 |