
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of electrostatic interactions to reveal a reaction network unifying the aggregation behaviour of the Aβ42 peptide and its variants†
Georg
Meisl
 a,
Xiaoting
Yang
b,
Christopher M.
Dobson
*a,
Sara
Linse
*b and
Tuomas P. J.
Knowles
*a
a,
Xiaoting
Yang
b,
Christopher M.
Dobson
*a,
Sara
Linse
*b and
Tuomas P. J.
Knowles
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: tpjk2@cam.ac.uk; cmd44@cam.ac.uk
bChemistry Department and Molecular Protein Science, Lund University, P. O. Box 124, SE221 00 Lund, Sweden. E-mail: sara.linse@biochemistry.lu.se
First published on 26th April 2017
Abstract
The aggregation of the amyloid β peptide (Aβ42), which is linked to Alzheimer's disease, can be altered significantly by modulations of the peptide's intermolecular electrostatic interactions. Variations in sequence and solution conditions have been found to lead to highly variable aggregation behaviour. Here we modulate systematically the electrostatic interactions governing the aggregation kinetics by varying the ionic strength of the solution. We find that changes in the solution ionic strength induce a switch in the reaction pathway, altering the dominant mechanisms of aggregate multiplication. This strategy thereby allows us to continuously sample a large space of different reaction mechanisms and develop a minimal reaction network that unifies the experimental kinetics under a wide range of different conditions. More generally, this universal reaction network connects previously separate systems, such as charge mutants of the Aβ42 peptide, on a continuous mechanistic landscape, providing a unified picture of the aggregation mechanism of Aβ42.
Introduction
Most functional proteins have a net charge under normal physiological conditions, which helps confer solubility,1–3 and is governed by the protein sequence and structure, as well as the solution conditions such as pH, salt concentration and the concentration of other charged species.4–7 The interactions involving charged and polar groups modulate properties such as solubility, stability and reaction rates.2,8–12 In addition to their importance in the functional interactions of proteins, electrostatic interactions play a key role in the formation of aberrant protein aggregates.13–16 In particular, charged proteins with embedded hydrophobic segments can be highly aggregation-prone and their assembly into amyloid fibrils is associated with Alzheimer's disease (the Aβ peptide), Parkinson's disease (the protein α-synuclein) and a range of other debilitating human diseases. The aggregation kinetics of these proteins are strongly influenced by electrostatic interactions and therefore depend on solution conditions and the presence of species able to shield charges.17,18Recent years have seen a significant advance in the mechanistic understanding of the aggregation of disease-associated proteins under controlled conditions in vitro.19–21 The mechanistic effects of variations in solution conditions, however, have often not been characterised in detail and therefore only the part of the overall reaction network relevant under a given set of conditions has been investigated. The individual systems under different conditions are not linked together into a continuous mechanistic picture. A more complete reaction network will be particularly important in vivo where aggregation-prone proteins are found in the presence of a large number of other molecules, which modulate their interactions.
Here, we present a method of sampling a large region of the reaction network of an aggregating system by modulating electrostatic interactions. This approach provides a means of altering the relative importance of different processes contributing to the overall reaction network and thereby allows the sampling of a broad range of macroscopic behaviour that can be explained by a single reaction network. In the present work we investigate the aggregation kinetics of the 42-residue amyloid β peptide, Aβ42, at different peptide and salt concentrations under quiescent conditions. We develop a model that quantitatively accounts for the observed lag times, kinetic profiles and peptide concentration dependences over the range of ionic strengths studied and rationalizes the interplay of the individual microscopic rates and their dependence on the magnitude of the electrostatic screening.
Results and discussion
Monomeric Aβ42 has a net charge of between −3 and −4 at pH 8.0 where the C-terminus and Asp, Glu, Lys and Arg side chains are mostly ionized and the His side chains and the N-terminus may be partly protonated (Fig. 1a).22,23 Due to interactions of the charged groups, the specific value of the net charge is likely to change upon the alteration of conformation and close packing associated with assembly of the peptides into aggregates.24,25 The number of hydrophobic residues in the C-terminal segment of Aβ42 (Fig. 1a) gives this peptide a high aggregation propensity, despite the strong electrostatic repulsion between individual monomers.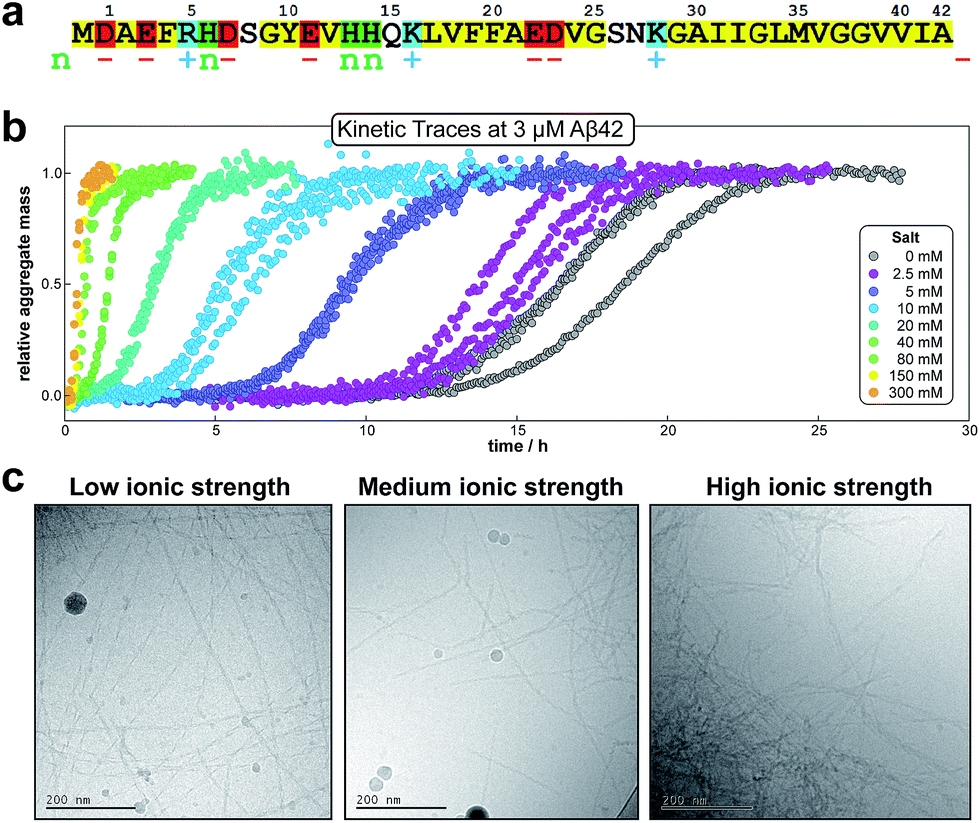 | ||
| Fig. 1 Amino acid sequence and aggregation data for Aβ42. (a) Hydrophobic residues are shown in yellow, groups that are positively charged, negatively charged or having pK value close to neutral pH are shown in blue (+), red (−) and green (n), respectively. (b) The kinetic data for the aggregation of 3 μM Aβ42 in the presence of varying concentrations of salt, measured by ThT fluorescence; three replicates at each salt concentration are shown. (c) Cryo TEM images at a low (29 mM), intermediate (57 mM) and high (329 mM) ionic strength. The fibrils were obtained by aggregation of solutions with a monomer concentration of 10 μM (note: the large circular objects are particles of ice). | ||
In the present study the aggregation kinetics of Aβ42 at monomer concentrations ranging form 0.55 to 7 μM, and concentrations of up to 300 mM of added NaCl, in 4 mM phosphate buffer (giving an ionic strength of ≈12 mM in the absence of added NaCl), were recorded in triplicate repeats by monitoring thioflavin T (ThT) fluorescence (Fig. 1b), which is proportional to the total fibril mass (ESI Section 4.2†). Fibrillar structures of similar morphology were found to be formed at all monomer and salt concentrations as monitored by cryo electron microscopy (TEM) (Fig. 1c), however, fibrils are packed more densely at higher salt concentrations. The entire set of experiments was repeated with a fresh batch of purified peptide, at salt concentrations in the same range, yielding similar results, which are shown in the ESI, Section 1.3.† In order to analyse such a large body of kinetic data with a complex underlying mechanism we first set out to obtain general constraints on possible mechanisms by considering the qualitative features of the data.
Half times and scaling
The half time of the aggregation process is defined as the time by which half the final aggregate concentration has formed. In the first instance, the value of the half time is a guide to the aggregation propensity of a given system. For a charged peptide such as Aβ42, the aggregation propensity is expected to increase as the electrostatic repulsion between peptides becomes screened with increasing ionic strength. Indeed this accelerating effect of salt on the overall reaction rate was observed at all peptide concentrations examined in the present study, as is evident both from the kinetic curves in Fig. 1b and from the monotonic decrease in half times with increasing ionic strength, Fig. 2b. Similar results have been previously reported also for Aβ40 studied at a single peptide concentration.22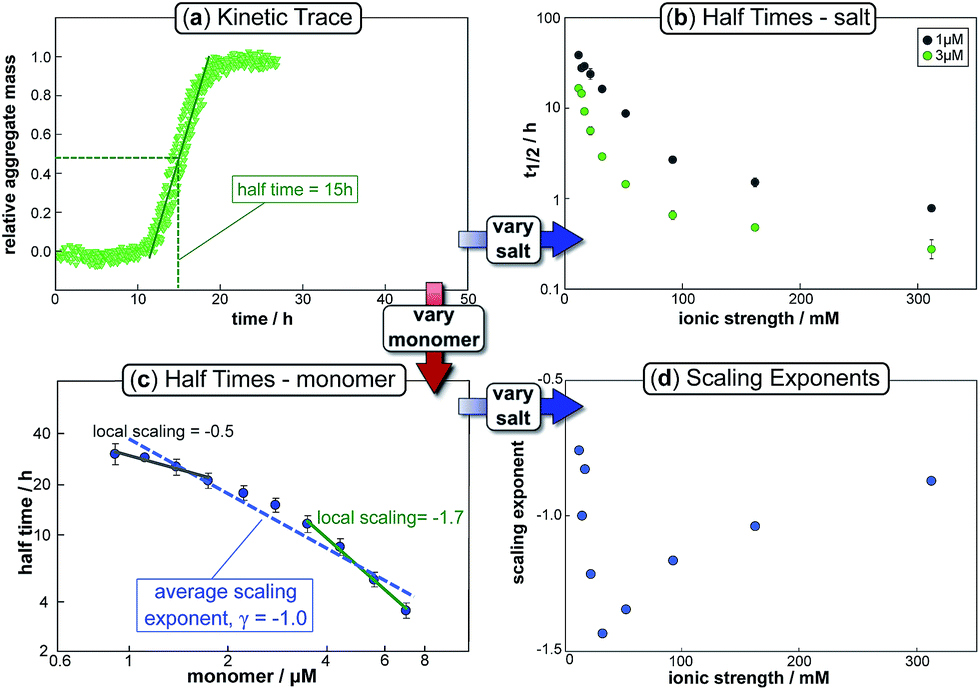 | ||
| Fig. 2 Half time dependence on monomer and salt concentration. The top left panel (a) shows a typical kinetic trace, in this case at a free monomer concentration of 3 μM and an ionic strength of 14.5 mM. The half time of aggregation can easily be extracted from such traces and plots of its variation with varying salt or monomer concentration are shown in the top right (b) and bottom left panels (c) respectively. The average slope of the double logarithmic plot of half time against monomer concentration gives the scaling exponent, γ, whose variation with ionic strength is plotted in the bottom right panel (d). Note the curvature in (c), indicative of a scaling exponent that depends on the monomer concentration. | ||
To go beyond this very qualitative result and obtain mechanistic information, we consider the dependence of the half time t1/2 on the monomer concentration, described by the scaling exponent γ where t1/2 ≈ m0γ and m0 is the monomer concentration at the beginning of an aggregation reaction. As outlined in Fig. 2a, the half time was determined for each kinetic curve at each monomer and salt concentration. The variation of the half time with varying salt concentration, at the same monomer concentration, shows the increase in aggregation propensity with increasing ionic strength, Fig. 2b. By contrast, from the variation of the half time with monomer concentration at the same salt concentration, the scaling exponent20 can be extracted, Fig. 2c, giving one scaling exponent at each salt concentration. We observed that the average scaling exponent has a biphasic dependence on the ionic strength, Fig. 2d; at low ionic strengths the half time scales with γ = −0.7. As the ionic strength increases, the average scaling exponent reaches a minimum of −1.4 at an ionic strength of around 32 mM and then increases again to −0.9 at an ionic strength of 312 mM, the highest value used here. Therefore, as the ionic strength is increased, the half times decrease monotonically for all monomer concentrations, but the dependence of the aggregation rate on the monomer concentration, given by the magnitude of the scaling, is largest at intermediate ionic strengths of circa 30 mM.
In addition we observe that at some salt concentrations there are variations in the scaling exponent as the monomer concentration changes, resulting in curvature of the double logarithmic plots of half time versus monomer concentration: at low ionic strengths the scaling exponent increases significantly in magnitude (i.e. the monomer dependence of the reaction increases) as the monomer concentration is increased, for example from γ = −0.5 to γ = −1.7 at an ionic strength of 14.5 mM, Fig. 2c. The opposite curvature is evident at some higher ionic strengths where the scaling exponent decreases in magnitude (i.e. the monomer dependence of the reaction decreases, Fig. 5) as the monomer concentration increases.
In summary three distinct features emerge from this half time analysis: (1) the half times decrease with increasing ionic strength (as expected due to shielding of charge repulsion between peptides). (2) The half time scaling with monomer concentration is strongest at an ionic strength of approximately 30 mM and weaker at both lower and higher ionic strengths. (3) At some ionic strengths, the scaling exponent depends on the monomer concentration, i.e. there is curvature in double logarithmic plots of half time versus monomer concentration.
Development of an aggregation model
We next set out to develop a unifying model that reproduces these half time features and fits the time evolution of aggregate mass at all monomer and salt concentrations. Such a model will allow a determination of the underlying reaction network of the aggregation reaction and show how the dominant pathways through this network are shifted by changes in ionic strength. In order to derive a quantitative description of the kinetics of an aggregating system,26,27 we have used a master equation approach19–21 that aims to classify all the processes that contribute significantly to the aggregation reaction by their mathematical dependence on the monomer concentration, the overall fibril number and the fibril mass concentrations.The elongation of the ends of growth competent fibril by the addition of monomers dominates the formation of fibril mass. In studies of the aggregation of other proteins, this elongation step has been found to saturate,28 in our system, however, no saturation effects are observed (see ESI Fig. S9†) hence elongation is modelled as a single step reaction.
Whereas the production of fibril mass is dominated by a single process, three classes of processes are responsible for the formation of new aggregates and thereby new fibril ends (see Fig. 3): (1) processes that depend only on the monomer concentration, m(t), (e.g. homogeneous nucleation of monomers in solution) which form new aggregates with rate constant kn and are of reaction order nc in monomer concentration (referred to as primary nucleation). (2) Processes that depend on the monomer concentration and the fibril mass (e.g. heterogeneous nucleation on the fibril surface) produce new aggregates with rate constant k2 and are of order n2 in monomer (referred to as secondary nucleation). (3) Processes that depend only on the fibril mass (e.g. breakage of fibrils) and create new free ends with rate constant k− (referred to as fragmentation). The latter two types of event are referred to as secondary processes as they involve the catalysis of the formation of new aggregates by existing aggregates.
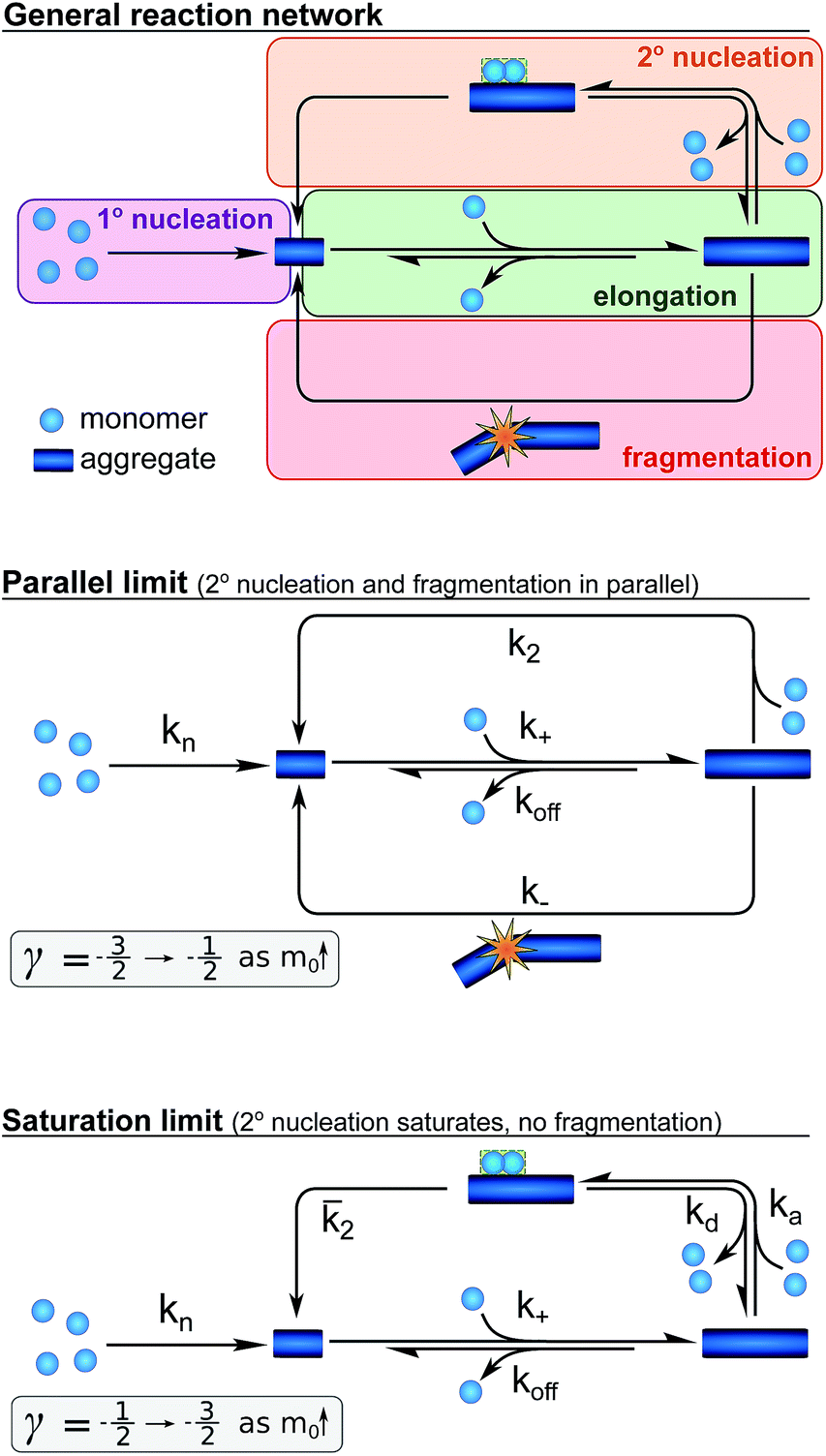 | ||
Fig. 3 A schematic depiction of the reaction network for the aggregation of Aβ42. The general network (top), the special case when both fragmentation and unsaturated secondary nucleation are important (centre), resulting in a parallel network, and the special case where saturation of secondary nucleation is important (bottom) but fragmentation is negligible. The ranges of approximate scaling exponents for each of these cases as the monomer concentration increases are given below each scheme. The rate constants given denote primary nucleation (kn), elongation (k+), depolymerisation (koff), fragmentation (k−), single-step secondary nucleation (k2), and the rates associated with multi-step secondary nucleation: the monomer attachment (ka) and detachment rate constants (kd), and the nucleus conversion/detachment rate constant (![[k with combining macron]](https://www.rsc.org/images/entities/i_char_006b_0304.gif) 2). 2). | ||
Whilst primary nucleation and fragmentation may be assumed to be single step processes, secondary nucleation consists of multiple steps in series, as we have established previously.29,30 The first step of secondary nucleation is a monomer-dependent attachment step in which free monomers interact with the fibril surface and the second step is a monomer-independent detachment of a newly formed nucleus. The overall process can be dominated by either step, depending on the conditions, and the concentration at which the second step becomes rate limiting, i.e. the concentration at which all binding sites on the fibril are occupied, is determined by the constant KM. By combining all these processes the full reaction network of aggregation is obtained, as displayed at the top of Fig. 3.
Experiments in which the aggregation was monitored following the addition of monomeric Aβ42 to preformed fibrils confirmed that the production of new aggregates in the present system is dominated by secondary processes, as shown in detail in the ESI, Section 3.1, Fig. S8.† Under these conditions fragmentation and secondary nucleation produce significantly more new aggregates than primary nucleation even at very low aggregate concentrations.31 In order to simplify the fitting and analysis we have considered two limits of this general reaction network, differing in their mechanism of the secondary process: in the first case both fragmentation and secondary nucleation, which act in parallel, may be significant, but we assume that there is no significant saturation of secondary nucleation (i.e. KM ≫ m(t)n2). We will refer to this case as the parallel limit and the corresponding scheme is shown in Fig. 3. The second case allows for saturation of secondary nucleation, but assumes that there is no significant contribution from fragmentation (i.e. k2m(t)n2 ≫ k−). We will refer to this case as the saturation limit and the corresponding scheme is shown in Fig. 3c. The detailed solutions to the kinetics of these two limiting cases (see Methods) can be used to estimate the expected range of scaling exponents which are shown below each scheme in Fig. 3.
Kinetic analysis
We fitted the equations for both limits to the experimental data, obtained as described above, simultaneously for all monomer concentrations at each ionic strength (all results see ESI Section 1.3†). The parallel limit fits the data well at low and intermediate ionic strengths (see e.g.Fig. 4a), but becomes invalid at high ionic strengths (see ESI, Fig. S5†). The saturation limit fits the data at intermediate and high ionic strengths (Fig. 4c), but fails to reproduce the data at low ionic strengths (see Fig. 4b).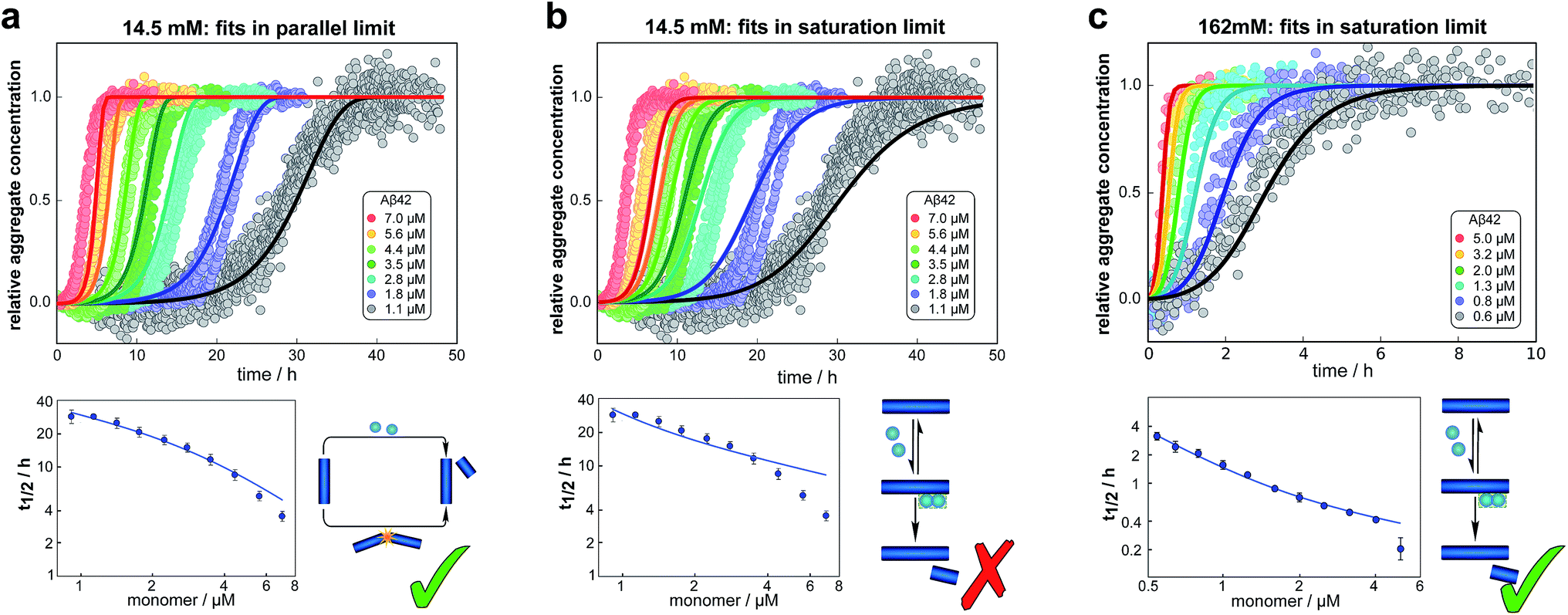 | ||
| Fig. 4 Global fits of the aggregation curves. (a) The top panel shows a global fit of the parallel limit, eqn (1), with three free parameters, to the all kinetic traces at 14.5 mM ionic strength. Shown below is a fit of just the half time behaviour for the same dataset. (b) The same data set as in (a) is fitted by the saturation limit, eqn (3). The fit of the network in the parallel limit (a) is significantly better than that of the saturation limit (b), the mean squared error in the parallel limit being half of that in the saturation limit. Note in particular how in the saturation limit we fail to reproduce the correct half times, as this limit cannot produce a decrease in scaling exponent with increasing monomer concentration. The saturation mechanism hence cannot explain the data at low ionic strengths. (c) By contrast to (b), a fit of the network in the saturation limit, eqn (3) with three free fitting parameters, to the data at 162 mM ionic strength shows good agreement with the experimental data. For clarity not all sampled concentrations are displayed in the plots of the full time-courses; the fits were, however, performed on the entire dataset (see the ESI, Sections 1.1 and 1.2,† for plots of the full dataset and an explanation of the deviation of the parallel limit fits at high monomer concentrations). | ||
These results paint a clear picture of how the system goes through four distinct regimes, each defined by which mechanism dominates the production of new fibrils as the ionic strength increases (Fig. 5):
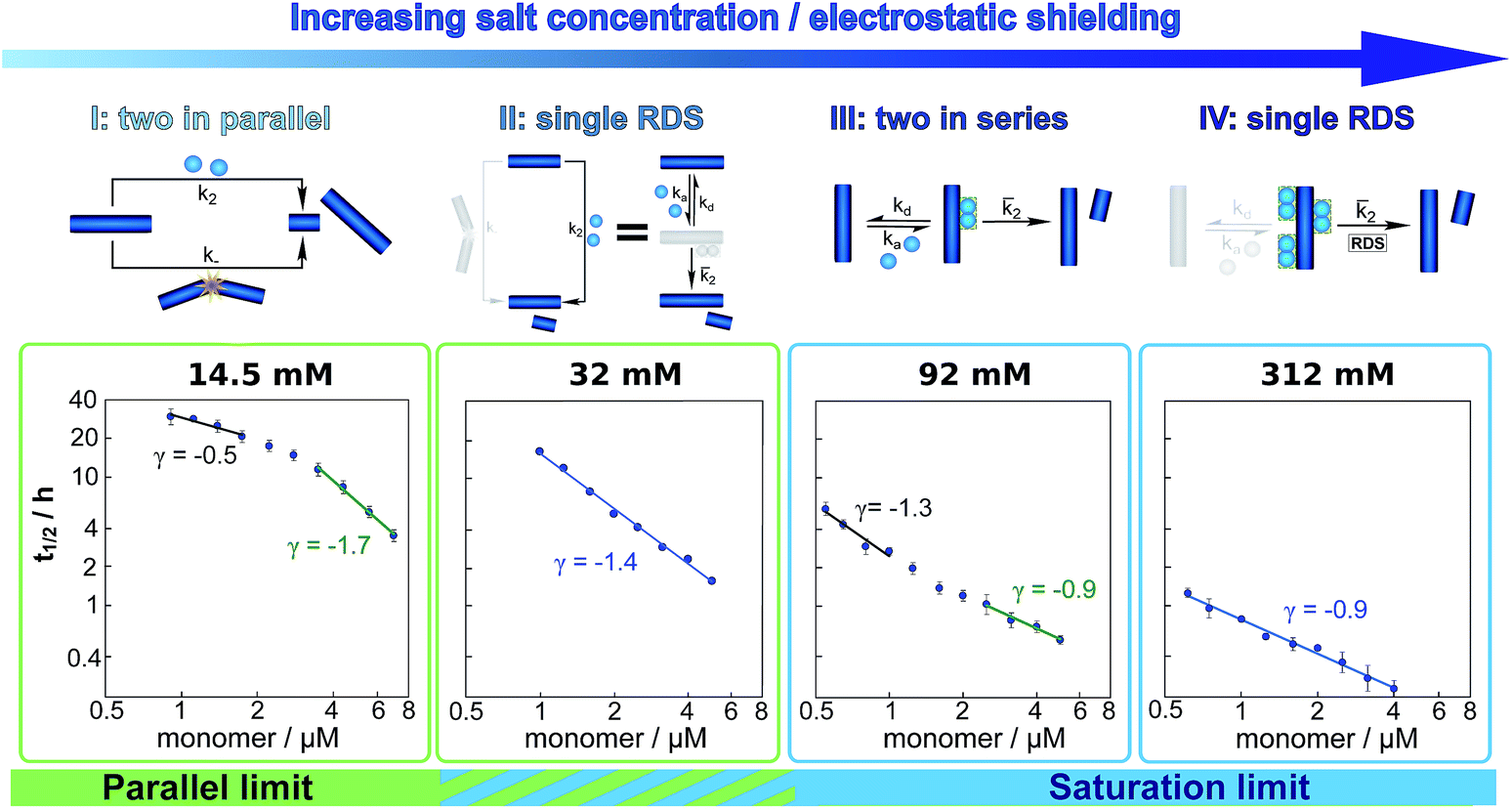 | ||
| Fig. 5 Dependence of the mechanism on ionic strength. Double logarithmic plots of half time versus initial monomer concentration for representative salt concentrations, one from each of the four regimes detailed in the main text. Above each plot a schematic of the secondary process dominating the aggregation network is displayed. The differing slopes and curvature can be explained by considering which secondary mechanism dominates the generation of new fibrils. From left to right: at an ionic strength of 14.5 mM, a combination of fragmentation and secondary nucleation, with fragmentation dominating at low monomer concentrations and secondary nucleation at high monomer concentrations results in negative curvature. At an ionic strength of 32 mM, non-saturated secondary nucleation alone, i.e. a single rate determining step (RDS), describes the data over the entire monomer range. At an ionic strength of 92 mM, secondary nucleation saturates as the monomer concentration increases, giving positive curvature. At an ionic strength of 312 mM, secondary nucleation is fully saturated over the entire monomer concentration range, again requiring only a single RDS. | ||
(I) At low ionic strengths (12–20 mM) the number of new growth competent ends produced by secondary nucleation and fragmentation is comparable (Fig. 5, 14.5 mM ionic strength): at low monomer concentrations fragmentation produces the largest number of new aggregates, resulting in a shallow dependence of the half time on monomer concentration, i.e. a scaling exponent of between −0.5 and −1. At high monomer concentrations secondary nucleation becomes the main source of new aggregates, resulting in a steeper dependence of the half time on monomer concentration with a scaling exponent of around −1.5. The average scaling exponent is circa −1.0.
(II) At intermediate ionic strengths (22–62 mM) fragmentation is negligible and secondary nucleation is not yet saturated: the monomer dependence of the half times is the same at all monomer concentrations, and the points in the double logarithmic plots of half time versus peptide concentration in Fig. 5 at an ionic strength of 32 mM fall on a straight line. In this region the data follows a simple, single step secondary nucleation mechanism and therefore both the parallel and the saturation limit are equally valid. The average scaling exponent is close to its minimum value of −1.44.
(III) At ionic strengths beyond values where the average scaling exponent is at a minimum (circa 92 mM) secondary nucleation starts to saturate: at low monomer concentrations it is barely saturated, giving a scaling exponent of circa −1.5, whereas at high monomer concentrations saturation starts to become significant, giving a scaling exponent of circa −1.0. Some curvature is evident in the half time plots (92 mM ionic strength Fig. 5).
(IV) At high ionic strengths (circa 200 mM and above) secondary nucleation is fully saturated at all monomer concentrations investigated here. Repulsion between Aβ42 monomers and between monomers and fibrils is effectively screened. Detachment of newly formed nuclei becomes rate limiting during secondary nucleation. The monomer dependence of the half times is constant with monomer concentration and the half times in Fig. 5, at an ionic strength of 312 mM, can be seen to lie on a straight line.
Effect of ionic strength on individual microscopic processes
The global fitting of the aggregation curves above provides an explanation of the observed changes in half times and scaling exponents: the differential effect of a change in ionic strength on the rates of the individual processes in the aggregation reaction leads to shifts in the dominant mechanism of aggregate multiplication. In order to rationalize this difference in susceptibility to electrostatic shielding we consider the variation of each rate constant with ionic strength.In the plots in Fig. 6a and b we show the logarithm of the various rate constant versus the square root of ionic strength (this latter value includes the contribution from the added salt as well as the buffer; in the absence of added salt the buffer alone (4 mM sodium phosphate, 40 uM EDTA, pH 8.0) results in an ionic strength of approximately 12 mM). In a simple Debye–Hückel (DH)16,32–34 model of the effect of ionic strength, these points would be expected to lie on a straight line. However, DH is accurate only at low ionic strengths, which are not accessible experimentally due to the need for a buffer during monomer isolation by size exclusion chromatography and to control the pH during the reaction and due to issues of irreproducibility which emerge in experiments with very low aggregation rates and long lag times as observed at low ionic strengths. Straight line fits up to an ionic strength of 100 mM are shown in Fig. 6, and although there is significant deviation from linearity, we can use the value of the slope to obtain on order of magnitude estimate of the charge of the species involved, as a consistency check. The slope corresponds approximately to the product of the valency of the reacting ions. In case of the elongation rate, Fig. 6a, the slope is approximately 6, which is determined by the charge of a monomer and that of a free fibril end, consistent with a charge of −2 to −3 for each of these species. In the case of the combined elongation–nucleation rate constant, Fig. 6b, the slope is approximately 13. If we assume the contribution from elongation is again approximately 6 (this is additive), the contribution from nucleation to the slope is then 7, which again is consistent with a charge of −2 to −3 for the monomers reacting during primary nucleation (details see ESI Section 2.5†). Hence, although DH theory is inadequate to describe fully the behaviour of charged macromolecules at the high ionic strengths studied here, the estimates it yields for the charges of the reacting species are in good agreement with the peptide sequence (Fig. 1a) and hence consistent with an electrostatic effect.
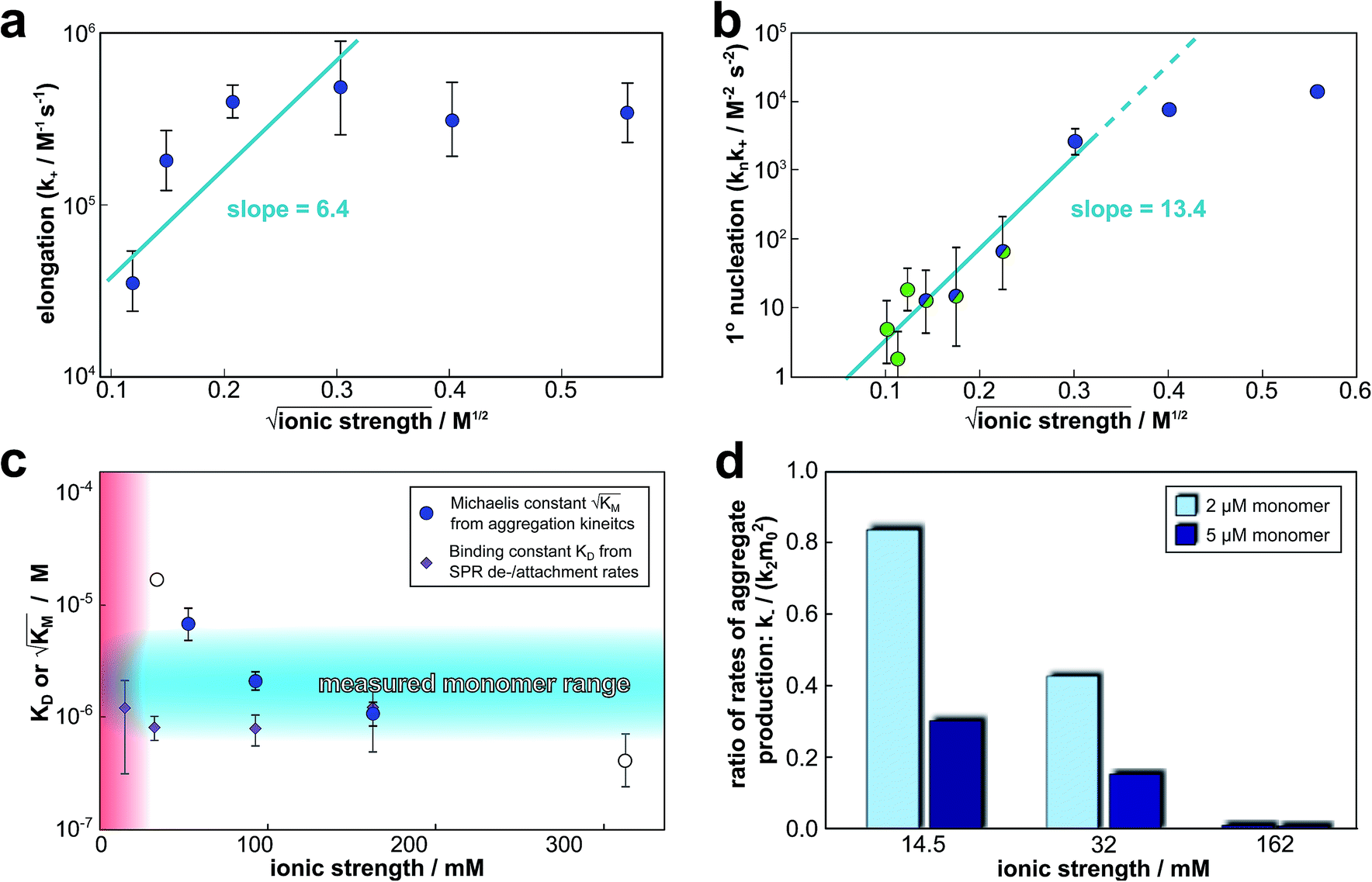 | ||
Fig. 6 Effect of electrostatic screening on the microscopic rates. (a) The elongation rate constant as measured in strongly seeded experiments (blue dots, experimental details in ESI Section 3.2†). (b) The product of the elongation rate constant and the primary nucleation rate constant at different ionic strengths, obtained from global fits of the specific equation valid at each ionic strength: green dots correspond to the parallel limit (three lowest ionic strengths), blue dots to the saturation limit (three highest ionic strengths); the green-blue dots are an average of the fits of the two limits in the region of intermediate ionic strength where both limits are equally valid (three intermediate ionic strengths). The blue lines are straight line fits up to 100 mM ionic strength. (c) The Michaelis constant, KM (blue dots), as obtained from global fits in the saturation limit at high ionic strengths (the region where the saturation limit is no longer valid is marked in red). As 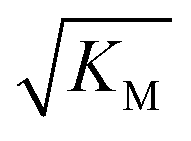 gives the monomer concentration at which saturation effects become important, the region of monomer concentrations used in this study is marked in blue. Values of KM outside the sampled region are likely to be less accurate, as the variation of KM in this region will have very little effect on the aggregation kinetics. The corresponding points are shown as empty circles. The binding constant of monomers to fibrils was measured independently by SPR, and computed from the rates of adsorption and desorption (purple squares). (d) Ratio of the rates of production of new free ends from secondary nucleation and fragmentation at ionic strengths of 14.5 mM, 32 mM and 162 mM, at monomer concentrations of 2 μM (light blue) and 5 μM (dark blue). The ratio of fragmentation to secondary nucleation decreases both with increasing monomer concentration and increasing ionic strength. At ionic strengths of 162 mM and above, fragmentation is too slow to be measured. Error bars in all cases are obtained from fitting subsets of the data (see ESI Section 4.3†). gives the monomer concentration at which saturation effects become important, the region of monomer concentrations used in this study is marked in blue. Values of KM outside the sampled region are likely to be less accurate, as the variation of KM in this region will have very little effect on the aggregation kinetics. The corresponding points are shown as empty circles. The binding constant of monomers to fibrils was measured independently by SPR, and computed from the rates of adsorption and desorption (purple squares). (d) Ratio of the rates of production of new free ends from secondary nucleation and fragmentation at ionic strengths of 14.5 mM, 32 mM and 162 mM, at monomer concentrations of 2 μM (light blue) and 5 μM (dark blue). The ratio of fragmentation to secondary nucleation decreases both with increasing monomer concentration and increasing ionic strength. At ionic strengths of 162 mM and above, fragmentation is too slow to be measured. Error bars in all cases are obtained from fitting subsets of the data (see ESI Section 4.3†). | ||
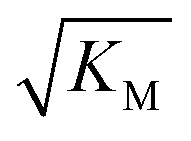 gives the monomer concentration at which saturation effects become important: if
gives the monomer concentration at which saturation effects become important: if 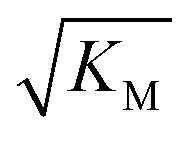 is above the region of monomer concentrations that are sampled in the experiment we expect the system to be unsaturated, if
is above the region of monomer concentrations that are sampled in the experiment we expect the system to be unsaturated, if 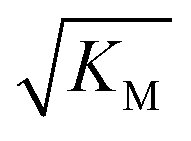 is within the region of monomer concentrations that are sampled we expect the system to start displaying saturation as the monomer concentration increases, and finally if the region of monomer concentrations that are sampled is above
is within the region of monomer concentrations that are sampled we expect the system to start displaying saturation as the monomer concentration increases, and finally if the region of monomer concentrations that are sampled is above 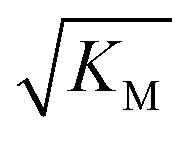 we expect the system to be fully saturated at all sampled monomer concentrations. The results show that
we expect the system to be fully saturated at all sampled monomer concentrations. The results show that 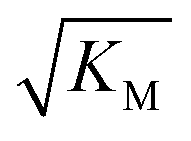 decreases with increasing ionic strength, i.e. the system will begin to saturate at lower monomer concentrations the higher the ionic strength (Fig. 6c, blue dots). This observation can be rationalised by considering the fact that the monomer concentration dependent step in secondary nucleation involves an association reaction between negatively charged monomers and negatively charged fibrils, which will be slower at lower ionic strengths. By contrast the monomer-independent detachment step is dominated by short range interactions and rearrangements, which is therefore likely to be less affected by the screening of electrostatic interactions. At high ionic strengths the monomer-dependent step becomes very fast, whereas the monomer-independent step proceeds at a rate similar to that at low ionic strength, and therefore it becomes rate limiting, leading to the observed saturation of secondary nucleation. To further investigate the origin of this observed behaviour we performed surface plasmon resonance (SPR) measurements to determine the equilibrium dissociation constant of monomers to fibrils, KD. In these experiments the dissociation constant was calculated from the association and dissociation rate constants which were determined by fitting (see ESI Section 2.4, Fig. S7†) rate equations to the increase and the decay of the SPR signal (Fig. 6c, purple squares). The dissociation constant obtained is approximately 1 μM and, given the larger errors on the SPR measurements, shows no significant variation with ionic strength. By comparison, previous studies of dissociation constants of charged macromolecules from SPR have measured ca. one order of magnitude decrease in KD upon an increase from low to physiological salt concentrations.35
decreases with increasing ionic strength, i.e. the system will begin to saturate at lower monomer concentrations the higher the ionic strength (Fig. 6c, blue dots). This observation can be rationalised by considering the fact that the monomer concentration dependent step in secondary nucleation involves an association reaction between negatively charged monomers and negatively charged fibrils, which will be slower at lower ionic strengths. By contrast the monomer-independent detachment step is dominated by short range interactions and rearrangements, which is therefore likely to be less affected by the screening of electrostatic interactions. At high ionic strengths the monomer-dependent step becomes very fast, whereas the monomer-independent step proceeds at a rate similar to that at low ionic strength, and therefore it becomes rate limiting, leading to the observed saturation of secondary nucleation. To further investigate the origin of this observed behaviour we performed surface plasmon resonance (SPR) measurements to determine the equilibrium dissociation constant of monomers to fibrils, KD. In these experiments the dissociation constant was calculated from the association and dissociation rate constants which were determined by fitting (see ESI Section 2.4, Fig. S7†) rate equations to the increase and the decay of the SPR signal (Fig. 6c, purple squares). The dissociation constant obtained is approximately 1 μM and, given the larger errors on the SPR measurements, shows no significant variation with ionic strength. By comparison, previous studies of dissociation constants of charged macromolecules from SPR have measured ca. one order of magnitude decrease in KD upon an increase from low to physiological salt concentrations.35
Note that the saturation concentration 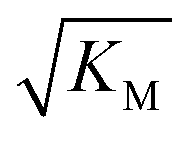 , in addition to being dependent on the strength of the adsorption to the fibrils, also depends on the interaction between the monomers forming the nucleus. Therefore it is not directly comparable in magnitude to the equilibrium dissociation constant, KD, obtained from SPR. The fact that the variation of
, in addition to being dependent on the strength of the adsorption to the fibrils, also depends on the interaction between the monomers forming the nucleus. Therefore it is not directly comparable in magnitude to the equilibrium dissociation constant, KD, obtained from SPR. The fact that the variation of 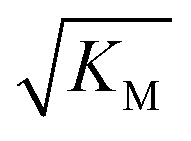 is not reflected in the values of the binding constant from SPR would suggest that the ionic strength dependence largely originates in the contributions of monomer–monomer interactions to
is not reflected in the values of the binding constant from SPR would suggest that the ionic strength dependence largely originates in the contributions of monomer–monomer interactions to 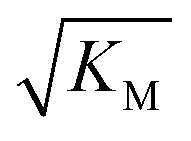 . However, due to the differences of the two systems, (the fibrils are surface bound in SPR experiments, rather than in solution as in the aggregation experiments and effects from partial charges of the dextran may perturb SPR data at low ionic strength) and the large errors associated with the SPR measurements, further experiments will be required to verify this finding.
. However, due to the differences of the two systems, (the fibrils are surface bound in SPR experiments, rather than in solution as in the aggregation experiments and effects from partial charges of the dextran may perturb SPR data at low ionic strength) and the large errors associated with the SPR measurements, further experiments will be required to verify this finding.
Although the aggregation behaviour of Aβ42 is evidently complex over the entire salt and monomer concentration ranges, it can be rationalised completely by considering the effect of ionic strength in terms of an increase of the association rates of the various microscopic processes. The more charged species associate in a given process, the more it is affected by a change in ionic strength. The fact that this simple explanation is sufficient to account for such a large range of complex behaviours strongly supports the minimal mechanistic description of the aggregation process developed here.
Parallels to other Aβ variants
By modulating the electrostatic shielding we have explored large parts of the reaction network of aggregation, yielding a variety of macroscopic behaviours. A similar variety of behaviours is observed in the different variants of Aβ, therefore the question arises if the reaction network obtained here offers a way to connect these different variants in a single mechanistic picture. Indeed many variants of Aβ under a range of conditions aggregate via a pathway that is part of this general reaction network, summarised in Table 1. Generally we find that effects that increase the fibril coverage during secondary nucleation or increase the rate of attachment of monomers to fibrils relative to the detachment of newly formed nuclei lead to an aggregation mechanism that is dominated by a saturating, or saturated, secondary nucleation mechanism, a process often associated with toxicity.39,40 This is the case for the aggregation of Aβ42 at a slightly lower pH of 7.4,37 as well as the aggregation of the mutants A2V37 and E22G.38 The reasons for increased fibril coverage in these cases are believed to be a decreased electrostatic repulsion as more residues become protonated at lower pH, a higher hydrophobicity and hence stronger binding of monomers to fibrils for A2V, and again a lowering of the electrostatic repulsion due to the loss of a charged sidegroup for E22G. This behaviour is analogous to the increased fibril affinity of monomers we observed here at high ionic strengths. We previously found that the other major variant of the Aβ peptide, Aβ40, aggregates via a saturating secondary nucleation mechanism, however the overall rates are lower than in the case of Aβ42, possibly due to a decreased nucleus formation/detachment step, rather than an increased binding, during secondary nucleation. By contrast, N-terminally extended variants of Aβ42 aggregate via an unsaturated secondary nucleation mechanism, which is likely to be due to the reduced number of reactive encounters of fibrils and monomers and therefore a saturation of the fibril surface is not reached at the studied monomer concentrations.36 Finally our extended reaction network also connects the aggregation behaviour of Aβ42 under strong agitation,31 with the behaviour under quiescent conditions: the shear forces induced by shaking significantly increase the rate of fragmentation and the mechanism shifts to one dominated by fragmentation. An analogous effect can be produced by significantly lowering the rate of the other nucleation processes, as we have observed here at low salt concentrations, allowing fragmentation to become kinetically visible.| System | Scaling | Path through reaction network | Analogous to Aβ42 (pH 8) at | Proposed rationalisation (behaviour relative to Aβ42 (ref. 31)) |
|---|---|---|---|---|
| Aβ40 (ref. 29) | −1.2 to −0.5 | Saturating 2° nuc | Medium to high ionic strength | Decreased detachment rate of 2° nuclei |
| Aβ42 (shaking)31 | −0.6 | Fragmentation | Low ionic strength, low monomer | Increased fragmentation from shear forces |
| Aβ42-A2V37 | −1.5 to −0.5 | Saturating 2° nuc | Medium to high ionic strength | Increased 2° nucleation, (more hydrophobic) |
| Aβ42 (pH 7.4)37 | −1.5 to −0.5 | Saturating 2° nuc | Medium to high ionic strength | Increased 2° nucleation, (less charged) |
| Aβ42-ext 15 (ref. 36) | Approx −1.3 | Non-saturated 2° nuc | Medium ionic strength | Unchanged from Aβ42 |
| Aβ42-E22G38 | −0.5 | Fully saturated 2° nuc | High ionic strength | Increased 2° nucleation, (less charged) |
Conclusion
In this work we have shown that electrostatic screening can be used to modulate the relative importance of different microscopic processes and thereby to alter the specific pathway that dominates the conversion of soluble peptides to their amyloid forms. This approach has allowed us to tackle a complex reaction network and to establish a mechanistic framework that accounts fully for the aggregation behaviour of Aβ42 over a wide range of solution conditions and monomer concentrations. As the individual rate constants are altered by the increasing electrostatic shielding, modifying the interplay and competition of the different microscopic processes, four distinct types of macroscopic behaviour emerge. The rate of fragmentation is found to be largely unaffected by variations in ionic strength and only contributes notably at low ionic strengths and monomer concentrations. By contrast, the rate constants for primary and secondary nucleation each increase by approximately two orders of magnitude and the rate constant of elongation increases by circa one order of magnitude, upon increasing the ionic strength from 12 mM to 312 mM.These findings show that the modulation of electrostatic interactions has profound mechanistic effects beyond a simple overall increase in aggregation propensity, and due to its differential effect on the individual rates it represents a means to sample an extended reaction network. The general reaction network we obtained through this sampling of a large proportion of the space of possible aggregation mechanisms serves as a unified framework for describing the aggregation behaviour of the variants of the Aβ peptide. The analysis detailed here, therefore, serves as a basis for interpreting how changes in solution conditions or peptide sequence shift the dominant pathways in the reaction network. It provides a continuum of mechanisms connecting the varied behaviour of different systems. In particular it allows the determination of the extent to which any alterations of the peptide sequence or the solution conditions correlate with those produced by altered electrostatic screening, thereby providing key insights into the physical origin of any observed mechanistic differences.
Materials and methods
Aβ42 expression and purification
Aβ(M1-42), of sequence shown in Fig. 1, here referred to as Aβ42, was expressed in E. coli from a synthetic gene and purified in batch format using ion exchange (see ESI, Fig. S10†) and size exclusion steps as described in Walsh et al.41 This results in highly pure monomeric peptide, which was divided in identical aliquots and stored as lyophilized peptide powder.Samples for aggregation kinetics
Monomeric Aβ42 was isolated twice by gel filtration of aliquots of purified peptides just prior to preparing each of the experiments, to remove any traces of aggregates formed during freezing and thawing and to exchange the buffer. The peptide powder was dissolved in 6 M GuHCl at pH 8.0 and the monomer peak was collected in low-bind Eppendorf tubes (Axygene) on ice and the concentration was determined by absorbance at 280 nm using an extinction coefficient of 1440 l mol−1 cm−1. Gel filtration was performed in 20 mM sodium phosphate, 0.2 mM EDTA, pH 8 at high enough concentrations that the isolated monomeric peptide could be diluted 5-fold with water or NaCl and still contain monomer concentrations of up to 5–7 μM. The final solutions used in the aggregation assays contained 4 mM sodium phosphate buffer, pH 8.0, with 40 μM EDTA, 6 μM ThT and NaCl in the range between 0 to 300 mM. The monomer concentration was varied between 0.5 and 7 μM and the ThT concentration (6 μM) was chosen to be in the range that has been shown to provide a response that is linearly dependent on the total aggregate mass.31 Here, this linearity was found to hold at all salt concentrations. At 0–10 mM NaCl the fluorescence intensity is higher the lower the salt concentration, but the response still varies linearly with monomer concentration for each salt concentration. All solutions used in gel filtration and in the preparation of sample series were extensively degassed. The aggregation kinetics were studied at 37 °C under quiescent condition by recording the ThT fluorescence intensity as a function of time using a plate reader (FluoStar Omega or Optima, BMG Labtech, Offenberg, Germany). The fluorescence was recorded in half-area 96-well PEG-coated black polystyrene plates with clear bottoms (Corning 3881, Massachusetts, USA) measuring from below with a 440 nm excitation filter and a 480 nm emission filter.Surface plasmon resonance (SPR) assays
Aβ42 monomer samples were prepared as described above for aggregation kinetics. Monomeric samples were loaded in a 96-well PEG-coated polystyrene plate (Corning 3881, USA) and 6 μM ThT was added to one of the wells as a fluorescence control. Fibrils were formed at 37 °C in 20 mM sodium phosphate buffer with 0.2 mM EDTA at pH 8. Fluorescence intensity was monitored as described above for aggregation kinetics and the reaction was terminated when the ThT signal of the control well reached the plateau. To immobilize the Aβ42 fibrils covalently on the surface of a CM3 sensor chip (GE Healthcare Life Sciences, USA) through amine coupling, 100 μl of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 fresh mixture of 0.05 M NHS and 0.2 M EDC was injected and flown through the channels to active the chip surface. The Aβ42 fibril sample was then flown through three channels and one channel was left as a negative control. Ethanolamine was used afterwards to block the remaining active site on the chip. Aβ42 monomer samples were diluted 5-fold with water and added to a highly concentrated NaCl solution to reach final concentrations of 7 μM and 3 μM Aβ42, 4 mM sodium phosphate, 40 μM EDTA, and 2.5 mM, 12 mM and 150 mM NaCl, at pH 8.0. The Aβ42 binding test was performed at two monomer concentrations, 7 μM and 3 μM at three ionic strengths, 14.5 mM, 24 mM and 162 mM. The fibril covered surface was equilibrated in buffers of the same ionic strength accordingly before the measurement. The binding test was performed at 37 °C, on Biacore 3000 instrument (GE Healthcare Life Sciences, USA). 40 μl sample was injected at each measurement and the dissociation process was monitored at a flow rate of 30 μl min−1. By fitting the dissociation curves to a sum of a linear and exponential function, the maximum coverage was determined (see ESI Section 2.4†).
:1 fresh mixture of 0.05 M NHS and 0.2 M EDC was injected and flown through the channels to active the chip surface. The Aβ42 fibril sample was then flown through three channels and one channel was left as a negative control. Ethanolamine was used afterwards to block the remaining active site on the chip. Aβ42 monomer samples were diluted 5-fold with water and added to a highly concentrated NaCl solution to reach final concentrations of 7 μM and 3 μM Aβ42, 4 mM sodium phosphate, 40 μM EDTA, and 2.5 mM, 12 mM and 150 mM NaCl, at pH 8.0. The Aβ42 binding test was performed at two monomer concentrations, 7 μM and 3 μM at three ionic strengths, 14.5 mM, 24 mM and 162 mM. The fibril covered surface was equilibrated in buffers of the same ionic strength accordingly before the measurement. The binding test was performed at 37 °C, on Biacore 3000 instrument (GE Healthcare Life Sciences, USA). 40 μl sample was injected at each measurement and the dissociation process was monitored at a flow rate of 30 μl min−1. By fitting the dissociation curves to a sum of a linear and exponential function, the maximum coverage was determined (see ESI Section 2.4†).
Cryogenic transmission electron microscopy (TEM)
Aβ42 monomers were purified the as described above for aggregation kinetics. The sample was diluted with water and highly concentrated NaCl solution was added to give samples with 10 μM Aβ42 monomer in sodium phosphate buffer with ionic strengths of 29 mM, 57 mM and 329 mM. The samples were loaded in a 96-well PEG-coated polystyrene plate (Corning 3881, USA) and ThT was added to one well at each ionic strength as the fluorescence control. Fibrils were formed in the same way as described as described above for SPR and 5 μl sample at each ionic strength was loaded as a liquid film on a lacey carbon filmed cooper grid. A layer of sample less than 300 nm thick was produced on the grid by blotting the extra liquid away at the back of the grid using a filter paper, followed by flash freezing the grid in liquid ethane and stored in liquid nitrogen. The grid preparation was carried out in a controlled environment vitrification system to ensure the stable temperature and humidity in order to maintain the original states of the sample. Images were recorded using a 120 kV electron microscope (Philips CM120 BioTWIN Cryo) with a CCD camera.Theoretical model
The time evolution of aggregate mass, M(t), in the parallel limit is given by: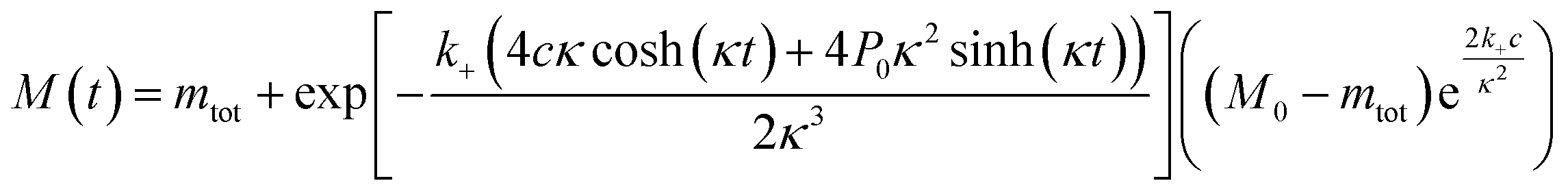 | (1) |
| a = k2m0n2 + k− |
| c = knm0nc + aM0 |
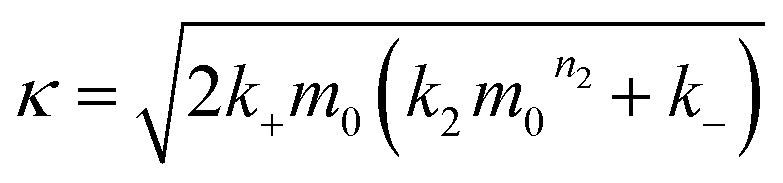 | (2) |
In the saturation limit we obtain:
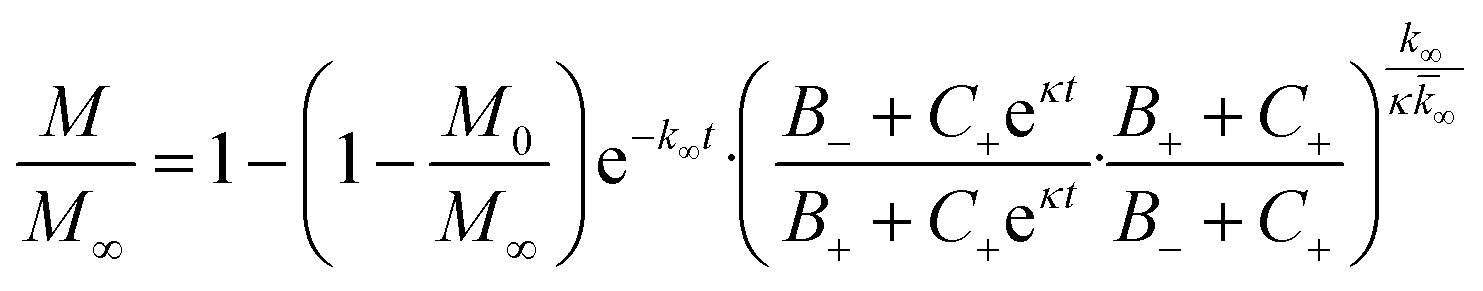 | (3) |
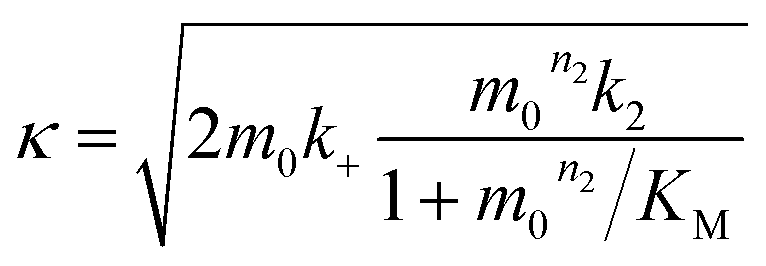 | (4) |
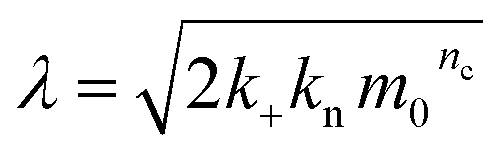 | (5) |
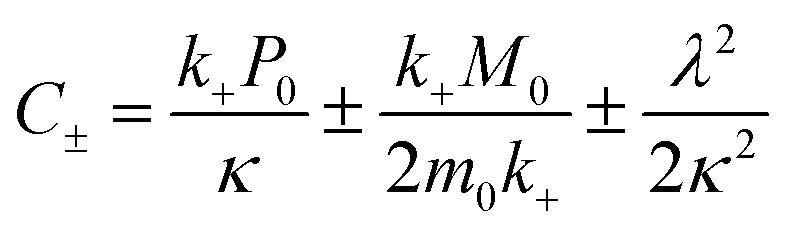 | (6) |
| k∞ = 2k + P∞ | (7) |
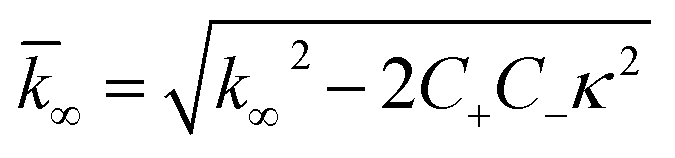 | (8) |
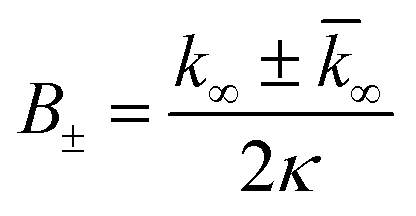 | (9) |
Again m0 is the initial monomer concentration, P0, M0 and P∞, M∞ are the aggregate number and mass concentrations at the beginning of the reaction and at equilibrium, i.e. after completion of the aggregation reaction. The details of the derivation of these equations can be found in the ESI Section 2.1.† A discussion about how these models relate to models explicitly including oligomers can be found in ESI Section 2.3.†
Note that for unseeded experiments (i.e. experiments starting from monomer alone, without preformed fibrils) the two limiting cases only involve three free parameters each (k+k2, k+kn and k+k− in the parallel limit and k+k2, k+kn and KM in the saturation limit), which were found to be sufficient in the present study to produce high quality global fits to all monomer concentrations at any given salt concentration. In the parallel limit, the scaling exponents is given by:
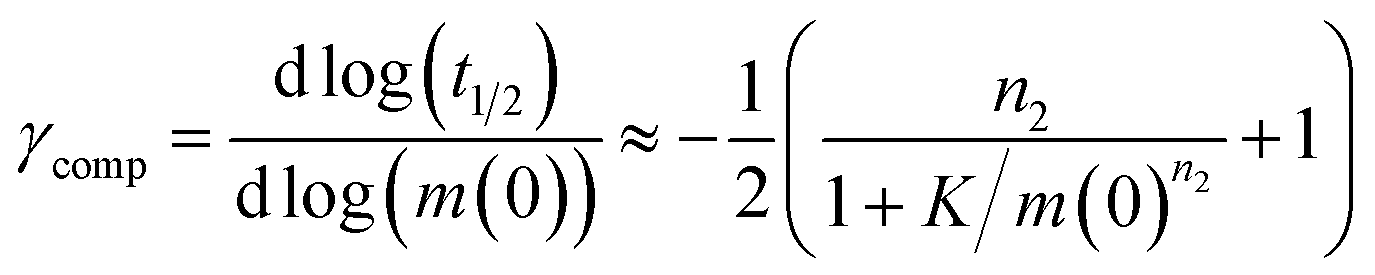 | (10) |
In the saturation limit, the scaling exponent is given by:
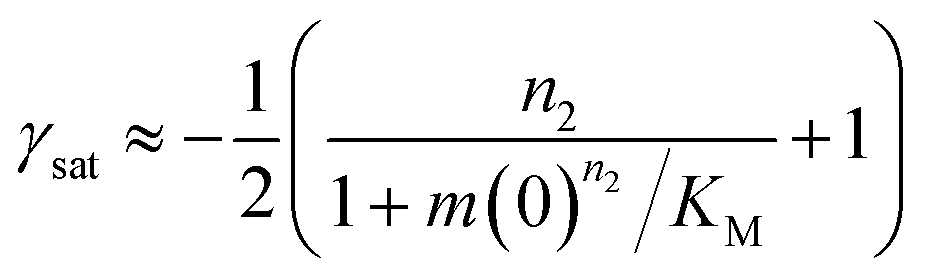 | (11) |
Fitting
The data were normalised to give units of relative fibril mass concentration. The fits were performed using a basin-hopping algorithm29,42 on the AmyloFit fitting platform.21 Please see ESI Section 4.4† and Meisl et al.21 and references therein for details. There were three global fitting parameters (i.e. one value of each parameter for all monomer concentrations) at each salt concentration: knk+, k2k+ and k−k+ in the parallel limit, and knk+, k2k+ and KM for in saturation limit. The reaction orders of primary and secondary nucleation, nc and n2, were both fixed to 2, in line with the values found for Aβ42 in previous work.31 The effect of fitting the reaction orders, which were set to the value previously established at a single salt concentration,31 is discussed in the ESI Section 1.2.†Acknowledgements
We thank Gunnel Karlsson, Lund University, for excellent help with cryo-EM. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) through the ERC grant PhysProt (agreement no 337969) (SL, TPJK), Sidney Sussex College Cambridge (GM), the Frances and Augusta Newman Foundation (TPJK), the Biotechnology and Biological Science Research Council (TPJK), the Swedish research Council (SL), and China Scholarship Council (XY).References
- M. S. Lawrence, K. J. Phillips and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 10110–10112 CrossRef CAS PubMed.
- M. Kurnik, L. Hedberg, J. Danielsson and M. Oliveberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5705–5710 CrossRef CAS PubMed.
- D. N. Marti and H. R. Bosshard, Biochemistry, 2004, 43, 12436–12447 CrossRef CAS PubMed.
- S. Lindman, S. Linse, F. A. A. Mulder and I. Andre, Biochemistry, 2006, 45, 13993–14002 CrossRef CAS PubMed.
- F. da Silva, S. Linse and B. Jönsson, J. Phys. Chem. B, 2005, 109, 2007–2013 CrossRef CAS PubMed.
- T. Kesvatera, B. Jonsson, E. Thulin and S. Linse, J. Mol. Biol., 1996, 259, 828–839 CrossRef CAS PubMed.
- T. Kesvatera, B. Jönsson, E. Thulin and S. Linse, Proteins: Struct., Funct., Bioinf., 1999, 37, 106–115 CrossRef CAS.
- S. Linse, P. Brodin, C. Johansson, E. Thulin, T. Grundstrom and S. Forsen, Nature, 1988, 335, 651–652 CrossRef CAS PubMed.
- H.-X. Zhou, Phys. Biol., 2005, 2, R1 CrossRef CAS PubMed.
- W. M. Matousek, B. Ciani, C. A. Fitch, B. E. Garcia-Moreno, R. A. Kammerer and A. T. Alexandrescu, J. Mol. Biol., 2007, 374, 206–219 CrossRef CAS PubMed.
- S. Lindman, W.-F. Xue, O. Szczepankiewicz, M. C. Bauer, H. Nilsson and S. Linse, Biophys. J., 2006, 90, 2911–2921 CrossRef CAS PubMed.
- W.-F. Xue, A. L. Hellewell, W. S. Gosal, S. W. Homans, E. W. Hewitt and S. E. Radford, J. Biol. Chem., 2009, 284, 34272–34282 CrossRef CAS PubMed.
- M. Vendruscolo and C. M. Dobson, Nature, 2007, 449, 555 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- G. Schreiber, G. Haran and H.-X. Zhou, Chem. Rev., 2009, 109, 839–860 CrossRef CAS PubMed.
- A. K. Buell, P. Hung, X. Salvatella, M. E. Welland, C. M. Dobson and T. P. Knowles, Biophys. J., 2013, 104, 1116–1126 CrossRef CAS PubMed.
- A. Abelein, A. Graslund and J. Danielsson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5407–5412 CrossRef CAS PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. A. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef CAS PubMed.
- S. I. A. Cohen, M. Vendruscolo, M. E. Welland, C. M. Dobson, E. M. Terentjev and T. P. J. Knowles, J. Chem. Phys., 2011, 135, 065105 CrossRef PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. T. C. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. J. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef CAS PubMed.
- K. Klement, K. Wieligmann, J. Meinhardt, P. Hortschansky, W. Richter and M. Fändrich, J. Mol. Biol., 2007, 373, 1321–1333 CrossRef CAS PubMed.
- V. Betts, M. A. Leissring, G. Dolios, R. Wang, D. J. Selkoe and D. M. Walsh, Neurobiol. Dis., 2008, 31, 442–450 CrossRef CAS PubMed.
- M. Lund and B. Jönsson, Biochemistry, 2005, 44, 5722–5727 CrossRef CAS PubMed.
- F. L. B. da Silva, M. Lund, B. Jönsson and T. Akesson, J. Phys. Chem. B, 2006, 110, 4459–4464 CrossRef CAS PubMed.
- F. A. Ferrone, J. Hofrichter and W. A. Eaton, J. Mol. Biol., 1985, 183, 611–631 CrossRef CAS PubMed.
- S. R. Collins, A. Douglass, R. D. Vale and J. S. Weissman, PLoS Biol., 2004, 2, e321 Search PubMed.
- W. P. Esler, E. R. Stimson, J. M. Jennings, H. V. Vinters, J. R. Ghilardi, J. P. Lee, P. W. Mantyh and J. E. Maggio, Biochemistry, 2000, 39, 6288–6295 CrossRef CAS PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. A. Cohen, C. M. Dobson, S. Linse and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef CAS PubMed.
- A. Saric, A. Buell, G. Meisl, T. C. T. Michaels, C. M. Dobson, S. Linse, T. P. J. Knowles and D. Frenkel, Nat. Phys., 2016, 12, 874–880 CrossRef CAS.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- P. Debye and E. Hückel, Phys. Z., 1923, 24, 185 CAS.
- J. N. Bronsted and C. E. Teeter, J. Phys. Chem., 1923, 28, 579–587 CrossRef.
- N. Bjerrum, Z. Phys. Chem., 1924, 108, 82 CAS.
- D. DellâĂŹOrco, W.-F. Xue, E. Thulin and S. Linse, Biophys. J., 2005, 88, 1991–2002 CrossRef PubMed.
- O. Szczepankiewicz, B. Linse, G. Meisl, E. Thulin, B. Frohm, C. S. Frigerio, M. T. Colvin, A. C. Jacavone, R. G. Griffin, T. Knowles, D. M. Walsh and S. Linse, J. Am. Chem. Soc., 2015, 137, 14673–14685 CrossRef CAS PubMed.
- G. Meisl, X. Yang, B. Frohm, T. P. J. Knowles and S. Linse, Sci. Rep., 2016, 6, 18728 CrossRef CAS PubMed.
- B. Bolognesi, S. I. A. Cohen, P. A. Terol, E. K. EsbjÃűrner, S. Giorgetti, M. F. Mossuto, A. Natalello, A.-C. Brorsson, T. P. J. Knowles, C. M. Dobson and L. M. Luheshi, ACS Chem. Biol., 2014, 9, 378–382 CrossRef CAS PubMed.
- D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, Nature, 2002, 416, 535–539 CrossRef CAS PubMed.
- A. Jan, O. Adolfsson, I. Allaman, A.-L. Buccarello, P. J. Magistretti, A. Pfeifer, A. Muhs and H. A. Lashuel, J. Biol. Chem., 2011, 286, 8585–8596 CrossRef CAS PubMed.
- D. M. Walsh, E. Thulin, A. M. Minogue, N. Gustavsson, E. Pang, D. B. Teplow and S. Linse, FEBS J., 2009, 276, 1266–1281 CrossRef CAS PubMed.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00215g |
| This journal is © The Royal Society of Chemistry 2017 |