
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of aristolactam alkaloids via synergistic C–H bond activation and dehydro-Diels–Alder reactions†
Mallu Chenna
Reddy
 a and
Masilamani
Jeganmohan
*ab
a and
Masilamani
Jeganmohan
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research, Pune 411021, India. E-mail: mjeganmohan@iiserpune.ac.in
bDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, Tamil Nadu, India. E-mail: mjeganmohan@iitm.ac.in
First published on 23rd March 2017
Abstract
A concise total synthesis of aristolactam alkaloids by a synergistic combination of C–H bond activation and dehydro-Diels–Alder reactions is described. To achieve the synthesis two new synthetic methodologies, namely the oxidative cyclization of benzamides with vinyl sulfone leading to 3-methyleneisoindolin-1-ones via a ruthenium-catalyzed C–H bond activation, and a dehydro-Diels–Alder reaction followed by the fluoride ion mediated desulfonylation of 3-methyleneisoindolin-1-ones with benzynes, were developed. The method presented allows the opportunity for the construction of all the rings of aristolactams from easily available starting materials.
Introduction
Aristolactams are naturally occurring phenanthrene lactam alkaloids. These alkaloids are isolated from Aristolochiaceae, Annonaceae, Piper Piperaceae, and Saururaceae plant species.1–3 Aristolactams are frequently used as folk medicines in several countries.2d–f Meanwhile, these molecules show an interesting array of biological properties such as anti-inflammatory, anti-platelet, anti-mycobacterial, neuroprotective and anti-cancer activities.2,3 Due to their unique structural features and potential biological activities, a considerable amount of effort has been devoted to synthesizing these molecules by several research groups.4 After surveying all these elegant contributions, we understood that a general and easily approachable method for synthesizing these alkaloids with a minimum number of steps from easily affordable starting materials is needed. Meanwhile, the new method should be general for the preparation of numerous aristolactam derivatives in order to explore the utility of these molecules in various areas. Particularly, the utility of these alkaloids in various biological applications has been extensively increased in the past two decades.Herein, we wish to report an efficient two step synthesis of aristolactam alkaloids from easily available and affordable starting materials such as aromatic acids, alkyl amines and alkenes. To execute the synthesis two new synthetic methodologies, namely the preparation of 3-methyleneisoindolin-1-ones via a ruthenium-catalyzed oxidative cyclization of aromatic amides with vinyl sulfone, and a dehydro-Diels–Alder reaction followed by SO2Ph cleavage of 3-methyleneisoindolin-1-ones with benzynes, were developed. The present method is compatible for the preparation of various aristolactam derivatives including sensitive I, Br, Cl, F and CF3 functional groups. The combination of C–H bond activation and dehydro-Diels–Alder reactions allows a short and efficient synthesis of several aristolactam alkaloids in good yields.
 | (1) |
 | (2) |
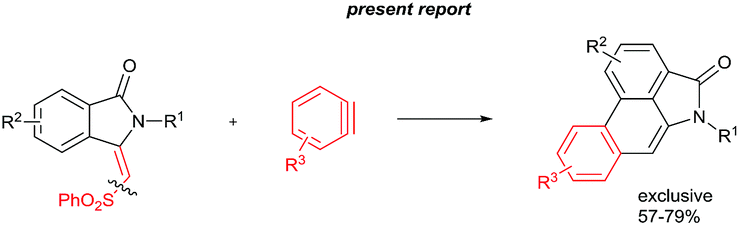
The goal of this work is to construct aristolactam cyclic rings A–D in a simple manner from easily affordable starting materials (Scheme 1). Rings A and B having 3-methyleneisoindolone can be constructed via a metal-catalyzed C–H/N–H annulation of substituted benzamides with alkenes in one pot.5–7 Substituted benzamides can be easily prepared from benzoic acids and amines. Rings C and D can be constructed in one pot via the dehydro-Diels–Alder reaction of 3-methyleneisoindolin-1-ones with benzynes.8,9 However, this type of cycloaddition reaction is not very effective, because it provides competing side products along with the expected product (eqn (1)).4i To overcome this problem, we engineered a molecule that has a cleavable SO2Ph group at the β-carbon of alkene of 3-methyleneisoindolin-1-one. After the cycloaddition reaction, the sulfonyl group can be easily cleaved by a fluoride source in the same step (eqn (2)). Thus, the cycloaddition reaction can be done in a highly selective manner.
 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Results and discussion
Our continuous interest in ruthenium-catalyzed C–H bond activation reaction prompted us to explore the possibility of developing a new synthetic route for the synthesis of key intermediates 3-methyleneisoindolin-1-ones via the ruthenium-catalyzed oxidative cyclization of benzamides with vinyl phenyl sulfone.6c,d,7l The oxidative cyclization of N-methyl benzamide 1a with phenyl vinyl sulfone (2a) in the presence of [{RuCl2(p-cymene)}2] (5 mol%), AgSbF6 (20 mol%) and Cu(OAc)2·H2O (0.5 equiv.) under oxygen at 120 °C for 36 h provided 3-methyleneisoindolin-1-one 3a in 78% yield in an 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 E/Z ratio (Scheme 2).
:5 E/Z ratio (Scheme 2).
 | ||
| Scheme 2 Cyclization of N-substituted benzamides. | ||
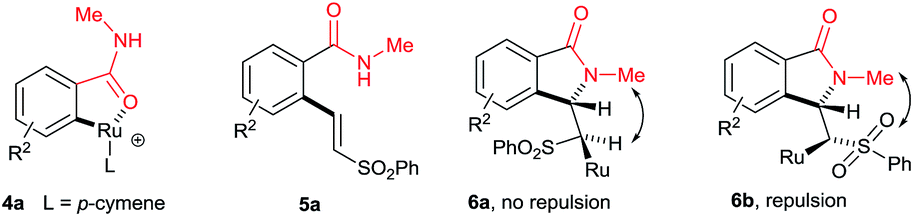
Initially, the cyclization reaction was examined with various solvents such as 1,2-dichloroethane, THF, 1,4-dioxane, DMF, toluene, CF3COOH and CH3COOH (Table 1). Among them, acetic acid was effective yielding product 3a in 78% yield (entry 7). Other solvents such as toluene and THF were less effective, affording product 3a in 20% and 15% yields, respectively (entries 2 and 5). The remaining solvents were not effective. The cyclization reaction was further examined with additives AgSbF6, AgBF4, AgOTf and KPF6. Among them, AgSbF6 was effective, providing product 3a in 78% yield (entry 7). The remaining additives were less effective for the cyclization reaction (entries 8–10). The cyclization reaction did not proceed without AgSbF6 (entry 11). AgSbF6 is used to generate a cationic ruthenium species for activating weak amide group assisted C–H bonds.5c,d Cu(OAc)2·H2O has been widely used as an oxidant for weak chelating group assisted C–H bond activation.5c Usually, a 2.0 equiv. amount of copper source is needed for this type of reaction. However, in the present reaction, a 0.5 equiv. amount of copper source was used and the remaining amount of the copper source was regenerated under oxygen. The cyclization reaction was examined with various substrates such as methyl, propyl, butyl, isopropyl, cyclohexyl and benzyl substituted benzamides 1b–f (Scheme 2). These reactions worked very well, providing the expected cyclization products 3b–f in 69%, 67%, 58%, 56% and 71% yields, respectively, in 96:4 to 99:1 E/Z ratios.
|
|
|||
|---|---|---|---|
| Entry | Solvent | Additive | Yield of 3ab (%) |
| a All reactions were carried out under the following conditions: 1a (75 mg), 2a (1.5 equiv.), [{RuCl2(p-cymene)}2] (5 mol%), additive (20 mol%) and Cu(OAc)2·H2O (50 mol%) in solvent at 120 °C for 36 h under an oxygen atmosphere. b Isolated yield. | |||
| 1 | ClCH2CH2Cl | AgSbF6 | — |
| 2 | THF | AgSbF6 | 15 |
| 3 | 1,4-Dioxane | AgSbF6 | — |
| 4 | DMF | AgSbF6 | — |
| 5 | Toluene | AgSbF6 | 20 |
| 6 | CF3COOH | AgSbF6 | — |
| 7 | CH3COOH | AgSbF6 | 78 |
| 8 | CH3COOH | AgBF4 | 42 |
| 9 | CH3COOH | AgOTf | 46 |
| 10 | CH3COOH | KPF6 | 15 |
| 11 | CH3COOH | — | NR |

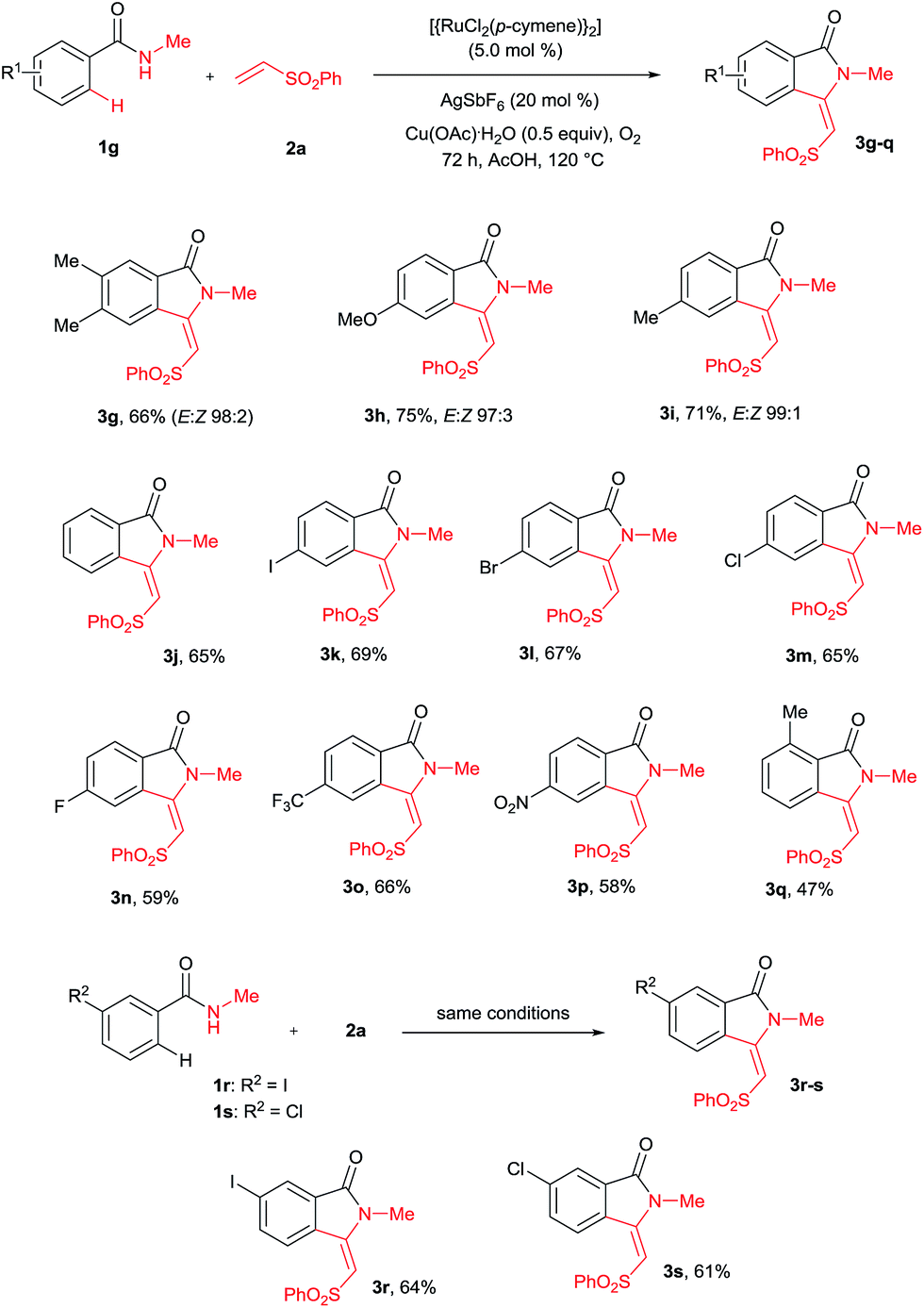
A variety of substituted benzamides 1g–s were compatible for the cyclization reaction (Scheme 3). Electron-releasing (OMe and Me) and halogen (I, Br, Cl and F) substituted benzamides 1g–n efficiently reacted with 2a affording isoindolin-1-ones 3g–n in good yields. The less reactive electron withdrawing (CF3 and NO2) substituted benzamides 1o–p also efficiently reacted with 2a providing products 3o and 3p in good yields. Similarly, ortho and meta substituted benzamides 1q–s also efficiently participated in the reaction, giving products 3q–s in 47%, 64% and 61% yields, respectively. Particularly, in the meta substituted benzamides 1r–s, C–H bond activation takes place at a less hindered C6–H.
 | (3) |
 | ||
| Scheme 3 Scope of substituted benzamides. | ||
The cyclization reaction proceeds via a cationic ruthenium(II) catalyzed ortho alkenylation of benzamide 1a with alkene 2avia intermediate 4a, providing ortho alkenylated benzamide 5a.7i Intramolecular addition of the amide N–H bond of 5a into an alkene moiety affords product 3 (eqn (1), for the detailed mechanism see the ESI†). It is important to note that a minor amount of Z stereoisomer was observed in the cyclization of electron-rich OMe and Me substituted benzamides. Intermediate 6b accounts for the formation of the Z stereoisomer. Presently, the exact reason for the observation of a minor amount of the Z stereoisomer is unclear. However, in halogen and electron-withdrawing substituted benzamides, the E stereoisomer was observed exclusively. The observation of high E stereoselectivity for product 3 is mainly due to the formation of intermediate 6a in which the sulfonyl moiety of the alkene and the cyclic tertiary C–N–Me bond are anti to each other (eqn (3)). Syn coplanarity is avoided due to the steric hindrance of the methyl and SO2Ph groups of intermediate 6b.7i
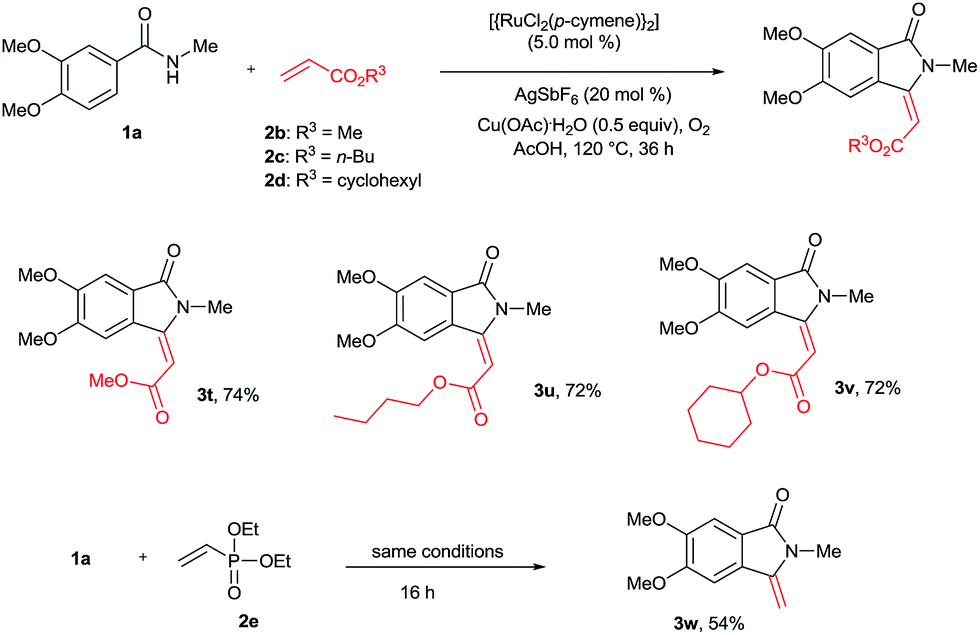
The cyclization reaction was further examined with various alkenes (Scheme 4). Methyl, n-butyl and cyclohexyl acrylates 2b–d efficiently reacted with 1a yielding cyclization products 3t–v in good yields. In these reactions, only E stereoselectivity was observed. Diethyl vinylphosphonate (2e) was also efficiently involved in the reaction, giving product 3w in 54% yield with a free exo double bond. In the product 3w, phosphonate (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(OEt)2) was cleaved under the present reaction conditions. The cyclization reaction was not compatible with acrylonitrile, methyl vinyl ketone and styrene.
O(OEt)2) was cleaved under the present reaction conditions. The cyclization reaction was not compatible with acrylonitrile, methyl vinyl ketone and styrene.
 | ||
| Scheme 4 Scope of alkenes. | ||
To explore the possibility of the preparation of aristolactam derivatives, the dehydro-Diels–Alder reaction of 3 with benzyne was examined (Scheme 5). The cycloaddition of 3g with benzyne precursor 7a in the presence of CsF in CH3CN at 30 °C for 24 h gave aristolactam derivative 9a in 66% yield. It is believed that after cycloaddition reaction, intermediate 8a is formed in which SO2Ph is cleaved by a fluoride ion. The formation of intermediate 8a was confirmed by MALDI-TOF experiment (for the detailed mechanism, see the ESI†).8 However, in the cycloaddition reaction of 3w with 7a, no product was observed. In the cycloaddition of 3t in which an ester substituent is present at the β-carbon of the alkene with 7a, a mixture of heterocyclic molecules 9b and 9b′ was observed in a 42% combined yield in a 4:1 diastereoselective ratio. In the reaction, the CO2Me group did not eliminate like SO2Ph. This result clearly reveals that the SO2Ph group is crucial in order to obtain aristolactams in greater yield with high selectivity.
 | ||
| Scheme 5 Dehydro-Diels–Alder reaction with benzynes. | ||
The cycloaddition reaction was examined with various N-substituted indolin-1-one derivatives 3b–f (Scheme 6). N-propyl, butyl, iso-propyl, cyclohexyl and benzyl substituted isoindolin-1-ones 3b–f underwent cycloaddition with 7a providing aristolactam derivatives 9c–g in good yields. Meanwhile, OMe, Me, I, Br, Cl, F and CF3 substituted isoindolin-1-ones 3h–s also efficiently participated in the reaction yielding products 9h–q in good yields.
 | ||
| Scheme 6 Scope of substituted isoindolin-1-ones. | ||
The scope of the cycloaddition reaction was further examined with substituted benzynes 7b–g (Scheme 7). Symmetrical benzynes such as 3,4-dimethoxy benzyne, 3,4-dimethyl benzyne, indene derivative and 1,3-benzodioxale reacted with 3j, providing cyclization products 9r–u in good yields. When unsymmetrical benzyne 7f was used, regioisomeric products 9v and 9v′ were observed in 66% yield in a 9:1 ratio. Interestingly, the unsymmetrical benzyne precursor 7g provided aristolactam 9w in 69% yield in a highly regioselective manner. The structure of compound 9w was supported by single crystal X-ray diffraction analysis. It is important to note that by using benzyne precursor 7g, several natural products can be prepared by changing the substituent on the benzamides.
 | ||
| Scheme 7 Scope of substituted benzynes. | ||
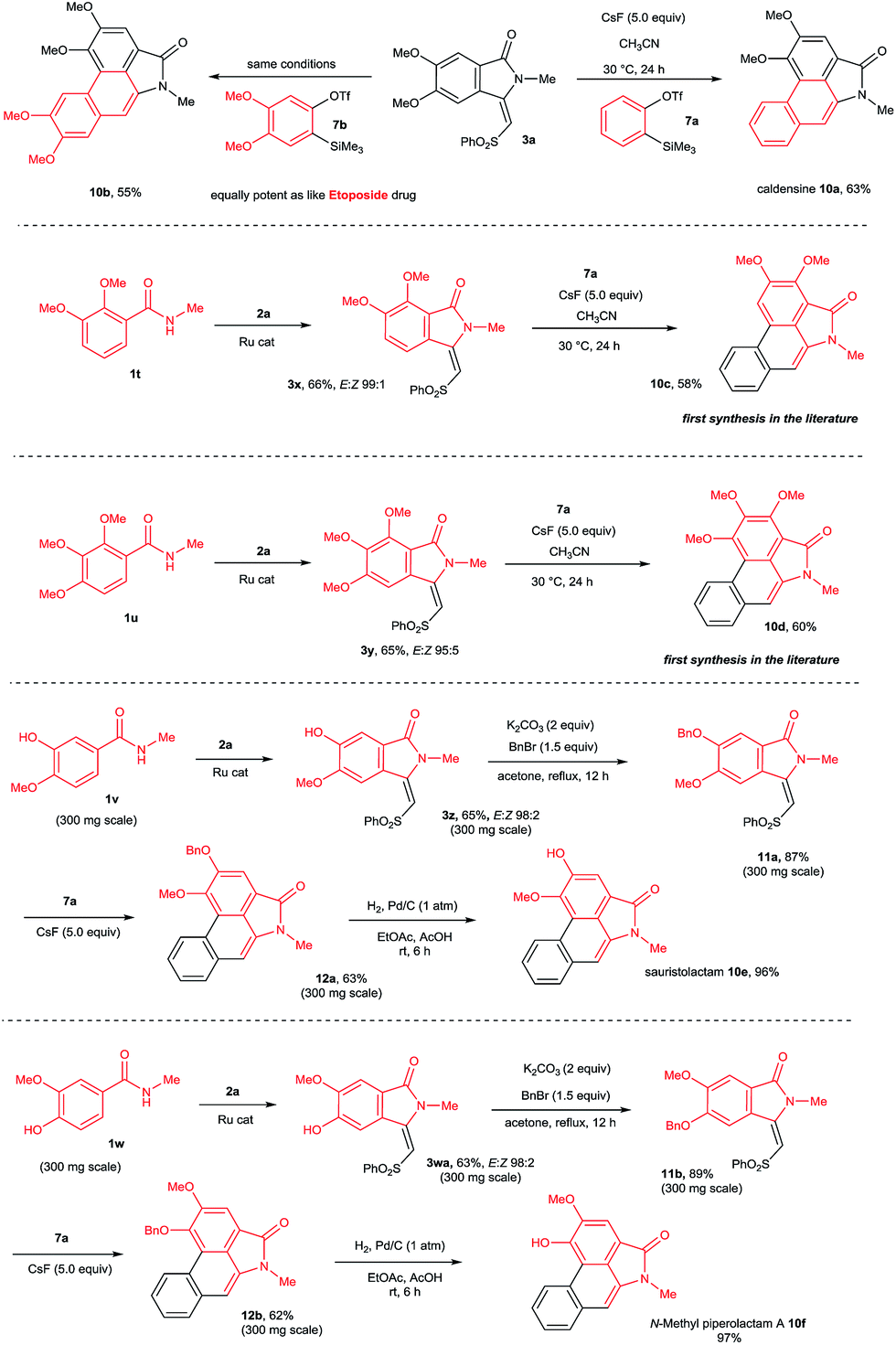
This result prompted us to explore the possibility of preparing N-methyl aristolactam alkaloids (Scheme 8). Treatment of compound 3a with benzyne precursors 7a or 7b in the presence of CsF in CH3CN at 30 °C for 24 h gave caldensine 10a and 10b in 63% and 55% yields, respectively. Caldensine exhibited an IC50 value of 25 mM against chloroquine-sensitive and also showed antiplasmodial activity.3a Compound 10b is equally potent towards multidrug-resistant cell lines compared with the commercially available drug etoposide.2a In a similar fashion, other alkaloids such as 2,3-dimethoxy-N-methyl-aristolactam 10c and 2,3,4-trimethoxy-N-methyl-aristolactam 10d were prepared in good yields. It is important to note that the alkaloids 10c–d were prepared for the first time in the literature. A highly useful sauristolactam (10e) and N-methyl piperolactam A (10f) were prepared in three steps. The reaction of 3-hydroxy-4-methoxy (1v) and 3-methoxy-4-hydroxy (1w) benzamides with 2a provided products 3z and 3wa in good yields. Later, a free hydroxy group of 3z and 3wa was protected with benzyl bromide followed by a cycloaddition reaction with 7a affording products 12a–b. Later, the benzyl group was deprotected by a palladium-catalyzed hydrogenation reaction, yielding alkaloids 10e–f in excellent yields. Sauristolactam (10e) and N-methyl piperolactam A (10f) have shown cytotoxic activity against several cancer cell lines1c,2a and neuroprotective activity.3b
 | ||
| Scheme 8 Synthesis of N-methyl aristolactam alkaloids. | ||
By employing the present protocol, N-H aristolactams were also prepared by using N-PMB substituted benzamides (Scheme 9). The reaction of 1x with 2a at 120 °C for 16 h under similar reaction conditions provided product 3xa in 63% yield. Later, 3xa was treated with benzyne precursors 7a or 7b in the presence of CsF in CH3CN at 30 °C for 24 h followed by PMB cleavage yielding cepharanone B (10g) and norcepharanone (10h) in good yields. In a similar fashion, piperolactam C alkaloid (10i) was prepared by the cyclization of 1y with 2a in the presence of a ruthenium catalyst followed by cycloaddition with 7a and subsequent PMB cleavage. Meanwhile, by using cepharanone B (10g), aristolactam FI (10j) can be prepared easily using a known procedure.4j Cepharanone B (10g) showed antimalarial activity with IC50 values of 7.51–11.01 μg mL−1 (ref. 3c) and also exhibited significant cytotoxic activity against human CNS carcinoma cells.3d Piperolactam C showed cytotoxicity against P-388 cells with an IC50 value of 78 μM.3e It is important to note that the E/Z ratio of indolin-1-one does not affect the yield of the benzyne cycloaddition reaction.
 | ||
| Scheme 9 Synthesis of N-H aristolactam alkaloids. | ||
Conclusions
In conclusion, we have demonstrated an efficient route to synthesize aristolactam alkaloids in good yields using a synergistic combination of C–H bond activation, dehydro-Diels–Alder and desulfonylation reactions. To prepare the target molecules two new synthetic methodologies namely, a ruthenium-catalyzed oxidative cyclization and dehydro-Diels–Alder reaction, were developed. A library of aristolactam derivatives that have substituents on all rings was prepared from easily available starting materials.Acknowledgements
We thank the DST-SERB (EMR/2014/000978), India for the support of this research. M. C. R. thanks the CSIR for a fellowship.Notes and references
- (a) V. Kumar, Poonam, A. K. Prasad and V. S. Parmar, Nat. Prod. Rep., 2003, 20, 565 RSC; (b) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444 RSC; (c) J. Michle, M. J. Ingrouille, M. S. J. Simmonds and M. Heinrich, Nat. Prod. Rep., 2014, 31, 676 RSC.
- (a) Y. L. Choi, J. K. Kim, S.-U. Choi, Y.-K. Min, M.-A. Bae, B. T. Kim and J.-N. Heo, Bioorg. Med. Chem. Lett., 2009, 19, 3036 CrossRef CAS PubMed; (b) S. J. Desai, B. R. Prabhu and N. B. Mulchandani, Phytochemistry, 1988, 27, 1511 CrossRef CAS; (c) Y.-N. Zhang, X.-G. Zhong, Z.-P. Zheng, X.-D. Hu, J.-P. Zuo and L. H. Hu, Bioorg. Med. Chem., 2007, 15, 988 CrossRef CAS PubMed; (d) P. J. Houghton and M. Ogutveren, Phytochemistry, 1991, 30, 253 CrossRef CAS; (e) B. S. Vishwanath and T. V. Gowda, Toxicon, 1987, 25, 929 CrossRef CAS PubMed; (f) H. A. Priestap, Phytochemistry, 1987, 26, 519 CrossRef CAS.
- (a) F. Martin, V. Choomuenwai and R. A. Davis, Phytochemistry, 2013, 86, 121 CrossRef PubMed; (b) S. R. Kim, S. H. Sung, S. Y. Kang, K. A. Koo, S. H. King, C. J. Ma, H.-S. Lee, M. J. Park and Y. C. Kim, Planta Med., 2004, 70, 391 CrossRef CAS PubMed; (c) K. Likhitwitayawuid, L. Wiasathien, V. Jongboonprasert, J. Krungkrai, N. Aimi, H. Takayama and M. Kitajima, Pharm. Pharmacol. Lett., 1997, 7, 99 CAS; (d) S. K. Kim, S. Ryu, Y. J. No, S. U. Choi and Y. S. Kim, Arch. Pharmacal Res., 2001, 24, 518 CrossRef CAS; (e) I. L. Tsai, F. P. Lee, C. C. Wu, C. Y. Duh, T. Ishikawa, J. J. Chen, Y. C. Chen, H. Seki and I. S. Chen, Planta Med., 2005, 71, 535 CrossRef CAS PubMed.
- (a) J. C. Estevez, M. C. Villaverde, R. J. Estevez and L. Castedo, Tetrahedron, 1995, 51, 4075 CrossRef CAS; (b) J. C. Estevez, R. J. Estevez and L. Castedo, Tetrahedron, 1995, 51, 10801 CrossRef CAS; (c) B. Gomez, E. Guitian and L. Castedo, Synlett, 1992, 903 CrossRef CAS; (d) V. Rys, A. Couture, E. Deniau and P. Grandclaudon, Eur. J. Org. Chem., 2003, 1231 CrossRef CAS; (e) C. Hoarau, A. Couture, H. Cornet, E. Deniau and P. Grandclaudon, J. Org. Chem., 2001, 66, 8064 CrossRef CAS PubMed; (f) A. Couture, E. Deniau, P. Grandclaudon and C. Hoarau, J. Org. Chem., 1998, 63, 3128 CrossRef CAS; (g) A. Couture, E. Deniau, P. Grandclaudon and S. Lebrun, Synlett, 1997, 1475 CrossRef CAS; (h) J. I. Levin and S. M. Weinreb, J. Am. Chem. Soc., 1983, 105, 1397 CrossRef CAS; (i) N. Atanes, L. Castedo, E. Guitian, C. Saa, J. M. Saa and R. Suau, J. Org. Chem., 1991, 56, 2984 CrossRef CAS; (j) J. K. Kim, Y. H. Kim, H. T. Nam, B. T. Kim and J.-N. Heo, Org. Lett., 2008, 10, 3543 CrossRef CAS PubMed; (k) J.-N. Heo, T. J. Kim and J. K. Kim, Bull. Korean Chem. Soc., 2011, 32, 4431 CrossRef CAS; (l) K. Sakthivel and K. Srinivasan, Eur. J. Org. Chem., 2013, 3386 CrossRef CAS; (m) L. Benesch, P. Bury, D. Guillaneux, S. Houldsworth, X. Wang and V. Snieckus, Tetrahedron Lett., 1998, 39, 961 CrossRef CAS; (n) T. Yao and R. C. Larock, J. Org. Chem., 2005, 70, 1432 CrossRef CAS PubMed; (o) J. C. Estevez, R. J. Estevez, E. Guitihn, M. C. Villaverde and L. Castedo, Tetrahedron Lett., 1989, 30, 5786 CrossRef.
- C–H activation: (a) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bermann and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) C. Zhu, R. Wang and J. R. Flack, Chem.–Asian J., 2012, 7, 1502 CrossRef CAS PubMed; (d) P. B. Arokiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef PubMed; (e) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (g) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (h) J. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef PubMed; (i) P. Gandeepan and C.-H. Cheng, Chem. Asian. J., 2015, 10, 824 CrossRef CAS PubMed; (j) R. Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed.
- Ruthenium papers: (a) E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161 CrossRef CAS PubMed; (b) T. Ueyama, S. Mochida, T. Fukutani, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 706 CrossRef CAS PubMed; (c) K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144 CrossRef CAS PubMed; (d) R. Manikandan, P. Madasamy and M. Jeganmohan, ACS Catal., 2016, 6, 230 CrossRef CAS; (e) K. Muralirajan, K. Parthasarathy and C.-H. Cheng, Org. Lett., 2012, 14, 4262 CrossRef CAS PubMed; (f) A. Bechtoldt, C. Tirler, K. Raghuvanshi, S. Warratz, C. Kornhaab and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 264 CrossRef CAS PubMed.
- Isoindolinone synthesis: (a) J. W. Wrigglesworth, B. Cox, G. C. Lloyd-Jones and K. I. Booker-Milburn, Org. Lett., 2011, 13, 5326 CrossRef CAS PubMed; (b) D.-D. Li, T.-T. Yuan and G.-W. Wang, Chem. Commun., 2011, 47, 12789 RSC; (c) F. W. Patureau, T. Besset and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1064 CrossRef CAS PubMed; (d) X. Wei, F. Wang, G. Song, Z. Dub and X. Li, Org. Biomol. Chem., 2012, 10, 5521 RSC; (e) A. M. Martínez, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem. Commun., 2014, 50, 6105 RSC; (f) C. Shangjun, C. Chao, S. Peng and X. Chanjuan, Org. Lett., 2014, 16, 3142 CrossRef PubMed; (g) B. Zhou, W. Hou, Y. Yang and Y. Li, Chem.–Eur. J., 2013, 19, 4701 CrossRef CAS PubMed; (h) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed; (i) J. Li, M. John and L. Ackermann, Chem.–Eur. J., 2014, 20, 5403 CrossRef CAS PubMed; (j) J. Zhou, B. Li, Z.-C. Qian and B.-F. Shi, Adv. Synth. Catal., 2014, 356, 1038 CrossRef CAS; (k) R. Manoharan and M. Jeganmohan, Chem. Commun., 2015, 51, 2929 RSC; (l) M. C. Reddy and M. Jeganmohan, Org. Lett., 2014, 16, 4866 CrossRef CAS PubMed , references therein.
- SO2R cleavage: (a) L. Li, Y. Liu, Y. Peng, L. Yu, X. Wu and H. Yan, Angew. Chem., Int. Ed., 2016, 55, 331 CrossRef CAS PubMed; (b) V. Sikervar, J. C. Fleet and P. L. Fuchs, Chem. Commun., 2012, 48, 9077 RSC.
- Selected benzyne reviews and papers: (a) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (b) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140 RSC; (c) K. M. Allan and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 17270 CrossRef CAS PubMed; (d) J. Li, N. Wang, C. Li and X. Jia, Org. Lett., 2012, 14, 4994 CrossRef CAS PubMed; (e) S. S. Bhojgude, A. Bhunia, R. G. Gonnade and A. T. Biju, Org. Lett., 2014, 16, 676 CrossRef CAS PubMed; (f) H. X. Siyang, X. R. Wu, H. L. Liu, X. Y. Wu and P. N. Liu, J. Org. Chem., 2014, 79, 1505 CrossRef CAS PubMed; (g) A. F. C. Rossini, A. C. A. Muraca, G. A. Casagrande and C. Raminelli, J. Org. Chem., 2015, 80, 10033 CrossRef CAS PubMed; (h) G. P. Perecim, A. Rodrigues and C. Raminelli, Tetrahedron Lett., 2015, 56, 6848 CrossRef CAS; (i) J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 15798 CrossRef CAS PubMed; (j) S.-E. Suh, S. A. Barros and D. M. Chenoweth, Chem. Sci., 2015, 6, 5128 RSC; (k) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208 CrossRef CAS PubMed; (l) A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54 CrossRef CAS PubMed; (m) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766 CrossRef CAS PubMed; (n) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658 CrossRef CAS PubMed; (o) S. S. Bhojgude, A. Bhunia, R. G. Gonnade and A. T. Biju, Org. Lett., 2014, 16, 676 CrossRef CAS PubMed; (p) H. X. Siyang, X. R. Wu, H. L. Liu, X. Y. Wu and P. N. Liu, J. Org. Chem., 2014, 79, 1505 CrossRef CAS PubMed; (q) J.-C. Castillo, J. Quiroga, R. Abonia, J. Rodriguez and Y. Coquerel, Org. Lett., 2015, 17, 3374 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data. CCDC 1526825. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00161d |
| This journal is © The Royal Society of Chemistry 2017 |