
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular assembly of a phosphole-based moiety into nanostructures dictated by alkynylplatinum(II) terpyridine complexes through non-covalent Pt⋯Pt and π–π stacking interactions: synthesis, characterization, photophysics and self-assembly behaviors†
Sammual Yu-Lut
Leung
a,
Sloane
Evariste
b,
Christophe
Lescop
 b,
Muriel
Hissler
*b and
Vivian Wing-Wah
Yam
*a
b,
Muriel
Hissler
*b and
Vivian Wing-Wah
Yam
*a
aInstitute of Molecular Functional Materials (Areas of Excellence Scheme, University Grants Committee (Hong Kong)) and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: wwyam@hku.hk
bInstitut des Sciences Chimiques de Rennes, UMR 6226 CNRS-Université de Rennes 1, Campus de Beaulieu, Rennes Cedex, France. E-mail: muriel.hissler@univ-rennes1.fr
First published on 19th April 2017
Abstract
A new class of platinum(II) terpyridine complexes with a phosphole-derived bridging alkynyl ligand have been prepared. The X-ray crystal structure of complex 2 has been determined, and reveals a polymeric zig-zag chain structure with the existence of π–π stacking interactions. The photophysical properties have also been studied, with 3MLCT/3LLCT phosphorescence exhibited in degassed CH2Cl2; the energy of which is varied by the π-conjugation of the terpyridine ligands. The solvent-induced assembly of complex 1 has been studied. The incorporation of hydrophobic hydrocarbon chains has been shown to play an important role in assisting the formation of self-assembled nanostructures via Pt⋯Pt, π–π stacking and hydrophobic–hydrophobic interactions. It has been established that an isodesmic growth mechanism operates in polar media to give nanospheres, while fibrous networks originate from the self-assembly of the complexes in non-polar media, predominantly driven by π–π stacking interactions.
Introduction
Studies on metallosupramolecular π-conjugated amphiphiles and their use in the construction of supramolecular architectures have aroused increasing interest recently.1–4 This is not only because of their interesting spectroscopic and luminescence properties, but also their propensity to exhibit the unprecedented complexity of multiple non-covalent interactions such as hydrogen bonding, π–π stacking interactions, metal–metal interactions and hydrophobic–hydrophobic interactions, among others. Particularly, directional Pt⋯Pt interactions have emerged as a key driving force for providing precise control and tuning of the self-assembly of metallosupramolecular π-conjugated amphiphiles into designated molecular architectures with well-defined morphologies.2 The unique chromophore alkynylplatinum(II) terpyridine as well as platinum(II) complexes with other related cyclometalating pincer ligands that exhibit the formation of Pt⋯Pt and π–π stacking interactions have been demonstrated to undergo self-assembly into metallogels2a,b and liquid crystals,2c,d together with a variety of nanosize molecular architectures such as hairpin conformations,2e tubes,2f helical ribbons,2f,g rods,2h rings2i,j and plates2k,letc.2m,n These studies have provided an in-depth understanding into metallosupramolecular π-conjugated amphiphiles, which have been found to play an important role in dictating the well-organized molecular packings that determine nanostructures in supramolecular chemistry.1–4Moreover, there has been a surge of interest in electronic materials derived from organo main group motifs due to the exhibition of unique structural and electronic properties.5 Among these organo main group molecule motifs, phospholes are one of the unique classes of compounds in electronic material applications.6 Indeed, not only have they aroused attention in the development of electronic devices,7,8 but also in the development of self-assembled materials.8 The development of self-assembled phosphole-based species has established interesting self-assembly behaviors via the subtle balance between the intermolecular π–π interactions of the phosphole backbones, electrostatic interactions as well as the thermal disorder in the hydrocarbon chains.8 Distinct from previously reported phosphole compounds, metal-containing phospholes are relatively less explored.8 Although there have been a few examples in the literature of chloro- and alkynyl-gold(I) phosphole systems, which are confined to studies into their application in organic light-emitting diodes (OLEDs) and self-assembly behaviors,9 research into functionalization of the phosphole system with a platinum(II) metal center, which is well-known to exhibit interesting and rich luminescence and self-assembly behaviors, is essentially unexplored. Exploration in this area may lead to the discovery of unique coordination, supramolecular and material properties. Thus, in view of the rich self-assembly behaviors of square-planar d8 platinum(II) polypyridine systems10–12 and their propensity towards the formation of directional Pt⋯Pt interactions,12 it is anticipated that such directional and noncovalent interactions can act as additional driving forces for the self-assembly of metallosupramolecular π-conjugated phosphole derivatives that may demonstrate interesting spectroscopic, luminescence and morphological properties. These may also provide important insight into the supramolecular assembly of the phosphole-based moiety driven by non-covalent Pt⋯Pt and π–π stacking interactions, which is distinct from previous studies on pure organic analogues and gold(I) systems. The utilization of such directional and non-covalent Pt⋯Pt interactions is anticipated to dictate the assembly behavior of the phosphole-based moiety in a controllable manner under various external stimuli.
Herein, we report the design and synthesis of dinuclear platinum(II) terpyridine complexes as well as that of a mononuclear complex containing a π-conjugated phosphole-derived alkynyl ligand. To the best of our knowledge, this study represents the first example of platinum(II)-containing phosphole complexes, which can exhibit non-covalent metal–metal and π–π stacking interactions in the self-assembly and lead to morphological changes under different conditions. Its photophysical properties of the complexes have been studied. It is also found that the dinuclear complexes with long hydrocarbon chains would self-assemble into spheres in DMSO, while they would aggregate into fibrous networks in diethyl ether. Temperature-dependent UV-vis absorption spectroscopic studies have provided an in-depth understanding into the nature of molecular self-assembly and the role of metal–metal interactions in the formation of these nanostructures, with an isodesmic growth mechanism established for their formation.
Results and discussion
Synthesis and characterization
The detailed synthetic route for the phosphole-based bridging alkynyl ligand is given in Scheme S1.† This class of bridging phosphole-containing ligand would steadily polymerize upon deprotection of the triisopropylsilyl (TIPS) groups. Thus, dinuclear alkynylplatinum(II) terpyridine complexes have been prepared by reacting a mixture of the TIPS-protected phosphole-containing bridging alkynyl ligand with the corresponding chloroplatinum(II) terpyridine precursor in degassed dichloromethane containing triethylamine and a catalytic amount of copper(I) iodide. The bridging phosphole-containing alkynyl ligand was deprotected in situ by the dropwise addition of tetra-n-butylammonium fluoride (TBAF) at 273 K. The reaction mixture was then stirred overnight at room temperature. Chromatography on silica gel using a dichloromethane–acetone mixture (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the eluent, followed by the diffusion of diethyl ether vapor into the CH2Cl2 solution, gave complexes 1 and 2 (Fig. 1) as dark red and red solids, respectively. The mononuclear platinum(II) complex with the bridging phosphole alkynyl ligand was isolated as a side-product in the preparation of complex 1. All complexes were found to be air-stable upon storage in the dark. These cationic complexes were found to be very soluble in common organic solvents such as acetonitrile and acetone, and soluble in dichloromethane and chloroform. In particular, complex 1 is barely soluble in diethyl ether, because of the four long hydrocarbon chains (–C18H37). All dinuclear complexes were characterized by 1H NMR and IR spectroscopy, FAB mass spectrometry and elemental analyses. The 31P{1H} NMR signal of 2 is situated at δ = +51.37 ppm, which is typical of the phosphole moiety. The IR spectra of complexes 1 and 2 exhibit characteristic C
:1 v/v) as the eluent, followed by the diffusion of diethyl ether vapor into the CH2Cl2 solution, gave complexes 1 and 2 (Fig. 1) as dark red and red solids, respectively. The mononuclear platinum(II) complex with the bridging phosphole alkynyl ligand was isolated as a side-product in the preparation of complex 1. All complexes were found to be air-stable upon storage in the dark. These cationic complexes were found to be very soluble in common organic solvents such as acetonitrile and acetone, and soluble in dichloromethane and chloroform. In particular, complex 1 is barely soluble in diethyl ether, because of the four long hydrocarbon chains (–C18H37). All dinuclear complexes were characterized by 1H NMR and IR spectroscopy, FAB mass spectrometry and elemental analyses. The 31P{1H} NMR signal of 2 is situated at δ = +51.37 ppm, which is typical of the phosphole moiety. The IR spectra of complexes 1 and 2 exhibit characteristic C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C absorptions at 2081–2086 cm−1, which are in accordance with the presence of the alkynyl ligand in a terminal σ-coordination mode. In addition, the crystal structure of complex 2 has been successfully determined by X-ray crystallography.
C absorptions at 2081–2086 cm−1, which are in accordance with the presence of the alkynyl ligand in a terminal σ-coordination mode. In addition, the crystal structure of complex 2 has been successfully determined by X-ray crystallography.
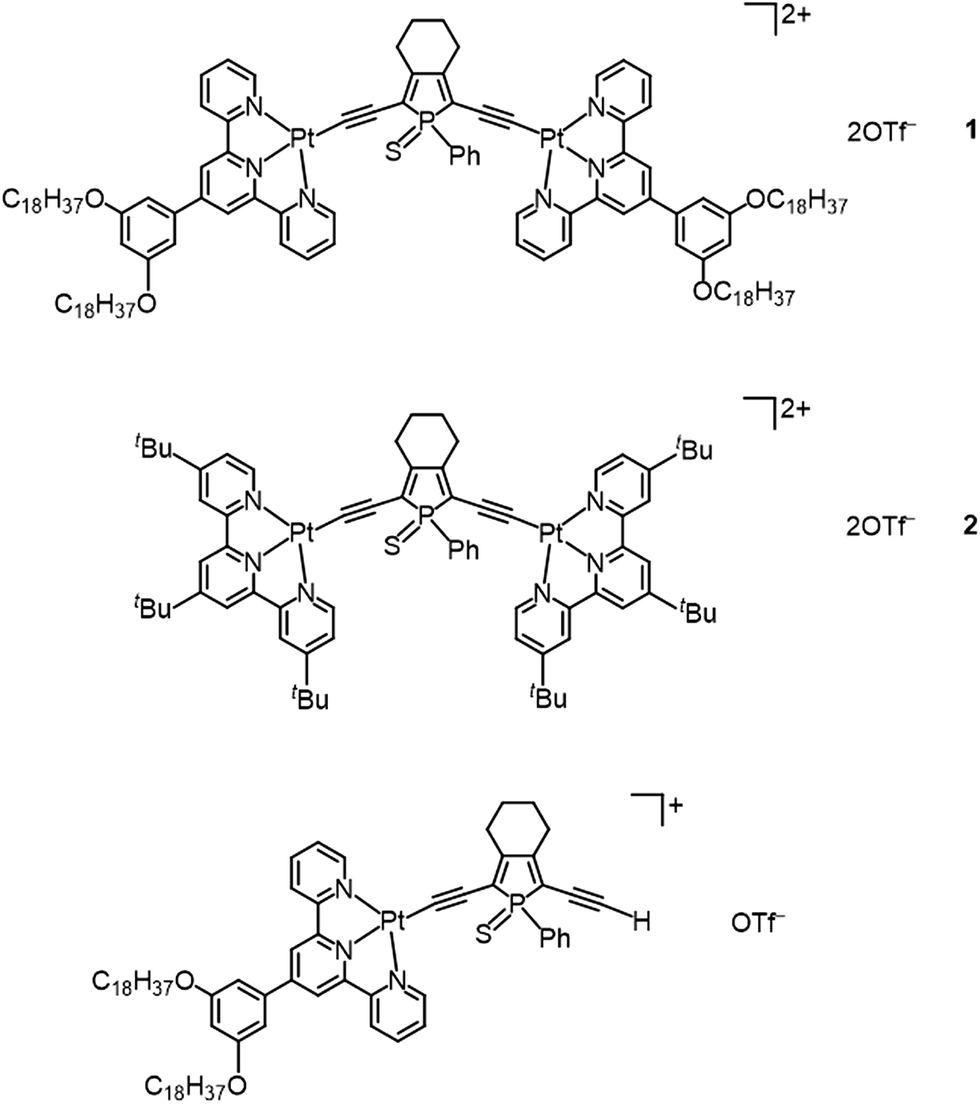 | ||
| Fig. 1 Molecular structures of di- and mononuclear alkynylplatinum(II) complexes with the bridging phosphole-containing alkynyl ligand. | ||
X-ray crystal structure
The coordination geometry at the platinum centre of the alkynylplatinum(II) terpyridine complex is distorted square-planar with the N(1)–Pt(1)–N(3), N(1)–Pt(1)–N(2) and N(2)–Pt(1)–N(3) bond angles being 160.7°, 80.2° and 80.8°, respectively. The steric demand of the tridentate terpyridine ligand distorts the square-planar geometry from the ideal values of 90° and 180°. The square-planar coordination promotes planarization of the terpyridine moieties, which facilitates the formation of supramolecular architectures. A perspective drawing of complex 2 is shown in Fig. 2a. The crystal structure determination data are given in the Experimental section and in Table S1.† The selected bond lengths and angles of complex 2 are summarized in Table 1. The bond lengths of Pt(1)–C(12) and C(11)–C(12) are 1.981 and 1.223 Å, respectively, which are similar to those observed in the previously reported alkynylplatinum(II) terpyridine and bzimpy systems.12a Furthermore, the phosphorus center adopts a distorted tetrahedral geometry with a small endocyclic C(1)–P(1)–C(8) angle in the range of 92.7°. The endocyclic P–C bonds, P(1)–C(1) and P(1)–C(8), are found to have lengths of 1.836 and 1.806 Å, respectively, which are comparable to those observed in the previously reported phosphole systems.7a The crystal packing diagram shows that the dicationic complexes are stacked in a head-to-tail configuration. Interestingly, the crystal packing of complex 2 displays zig-zag chains that are held together mainly through π–π stacking interactions (Fig. 2b). This is because the interplanar distance between the two platinum(II) terpyridine moieties of the dimeric structure is found to be 3.47 Å, which suggests weak to almost insignificant Pt⋯Pt interactions in the polymeric zigzag chain structures.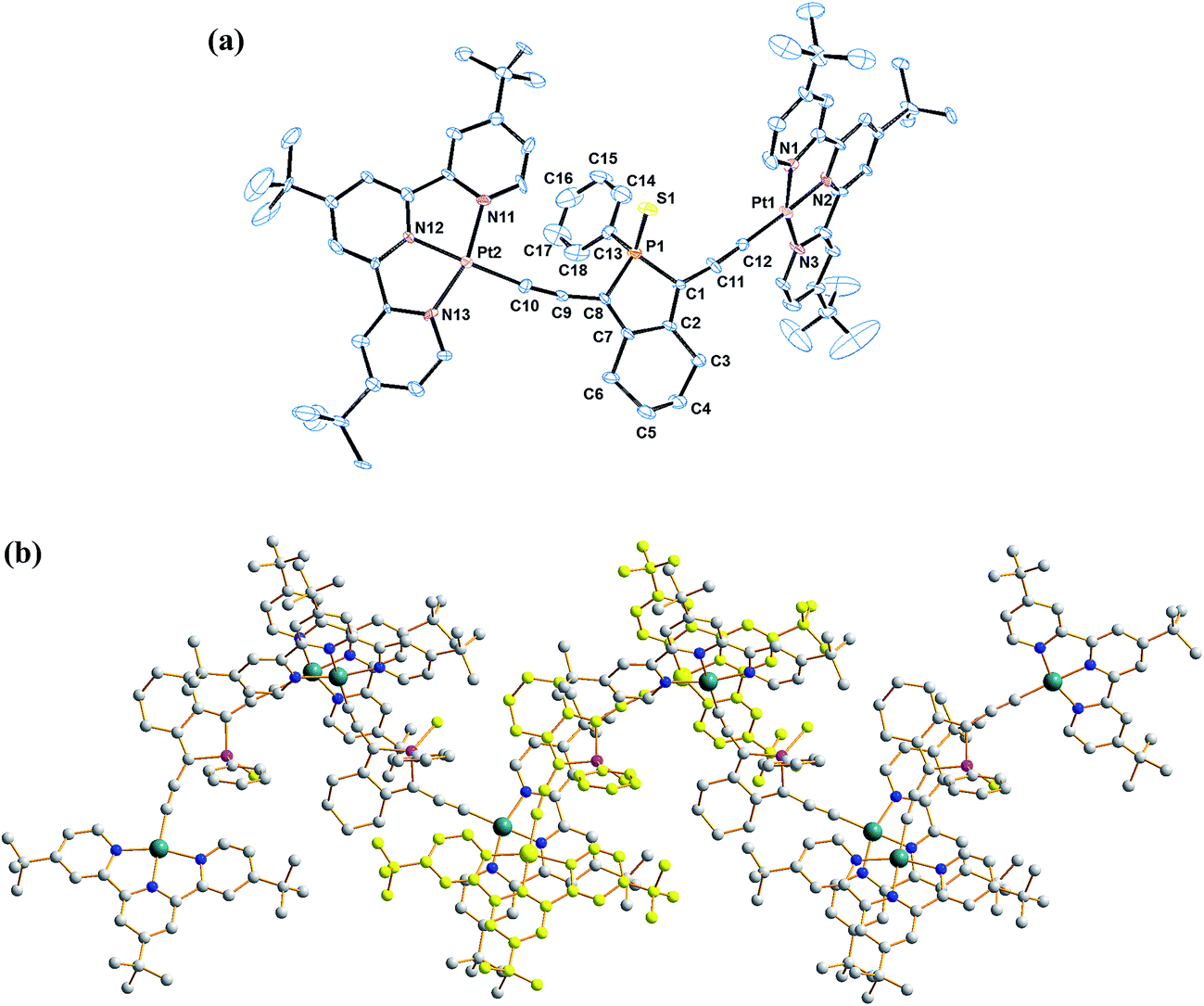 | ||
| Fig. 2 (a) Perspective drawing of the complex cation of 2. Hydrogen atoms, counter-anions and solvent molecules have been omitted for clarity. Thermal ellipsoids are shown at the 30% probability level. (b) Crystal packing diagram of the complex cations of 2. | ||
| Bond angles (deg) | |||
| N1–Pt1–N3 | 160.7(4) | S1–P1–C13 | 112.0(3) |
| N1–Pt1–N2 | 80.2(3) | C1–P1–C8 | 92.7(5) |
| N2–Pt1–N3 | 80.8(4) | C1–P1–C13 | 108.2(5) |
| S1–P1–C1 | 115.8(5) | C8–P1–C13 | 106.1(5) |
| S1–P1–C8 | 120.1(4) | ||
|
|||
| Bond distances (Å) | |||
| Pt1–C12 | 1.981(10) | P1–C1 | 1.181(6) |
| C11–C12 | 1.223(14) | P1–C8 | 1.806(10) |
| P1–S1 | 1.951(5) | P1–C13 | 1.817(8) |
Photophysical studies
The red to orange CH2Cl2 solutions of complexes 1 and 2, together with the mononuclear complex, were examined using UV-vis absorption and emission spectroscopy. Their photophysical data are summarized in Table 2. In the UV-vis absorption spectra, high-energy absorption bands at ca. 313–412 nm, together with low-energy absorption bands at ca. 512–565 nm, are observed. The former are assigned as intense intraligand (IL) [π → π*] transitions of the terpyridine and alkynyl ligands, as supported by the UV-vis absorption spectra of the organic counterparts (Fig. S1†), while the latter are assigned as metal-to-ligand charge-transfer (MLCT) [dπ(Pt) → π*(tpy)] transitions mixed with alkynyl-to-terpyridine ligand-to-ligand charge transfer (LLCT) [π(CCR) → π*(tpy)] character (Fig. 3a).12 In general, the dinuclear complexes exhibit more red-shifted MLCT/LLCT absorption bands (533–565 nm) than the mononuclear complex (512 nm). Notably, complex 1 displays the most red-shifted MLCT/LLCT absorption band due to the more π-conjugated and electron-deficient terpyridine ligand, thus lowering the π*(tpy) orbital energy (LUMO), leading to a lower MLCT/LLCT absorption energy. In the emission spectra, weak luminescence at ca. 596–714 nm in degassed CH2Cl2 was observed upon excitation at λ ≥ 360 nm (Fig. 3b). The structureless emission bands are derived from an excited state which is predominantly 3MLCT [dπ(Pt) → π*(tpy)] in origin, with mixing of 3LLCT [π(CCR) → π*(tpy)] character (vide infra). Concomitant with the findings from the UV-vis absorption spectra, the mononuclear complex exhibits the highest energy 3MLCT/3LLCT emission bands (596 nm) and complex 1 displays the lowest emission energy (714 nm).
| Complex | Absorption | Medium | Emission |
|---|---|---|---|
| λ/nm [ε/dm3 mol−1 cm−1] | (T [K]) | λ [nm] (τ0 [μs]) | |
| a Non-emissive. b Measured in n-butyronitrile. | |||
| 1 | 339 (28990), 419 sh (13350), 565 (11530) |
CH2Cl2 (298) | 714 (<0.1) |
| Solid (298) | —a | ||
| Solid (77) | —a | ||
| Glass (77)b | —a | ||
| 2 | 342 (26340), 414 (11410), 533 (14100) |
CH2Cl2 (298) | 701 (<0.1) |
| Solid (298) | 700 (<0.1) | ||
| Solid (77) | 702 (<0.1) | ||
| Glass (77)b | 633, 698 sh (<0.1) | ||
| Mononuclear complex | 325 (24210), 415 sh (11410), 512 (6720) |
CH2Cl2 (298) | 596 (<0.1) |
| Solid (298) | 582 (<0.1) | ||
| Solid (77) | 583 (<0.1) | ||
| Glass (77)b | 574, 620 sh (<0.1) | ||
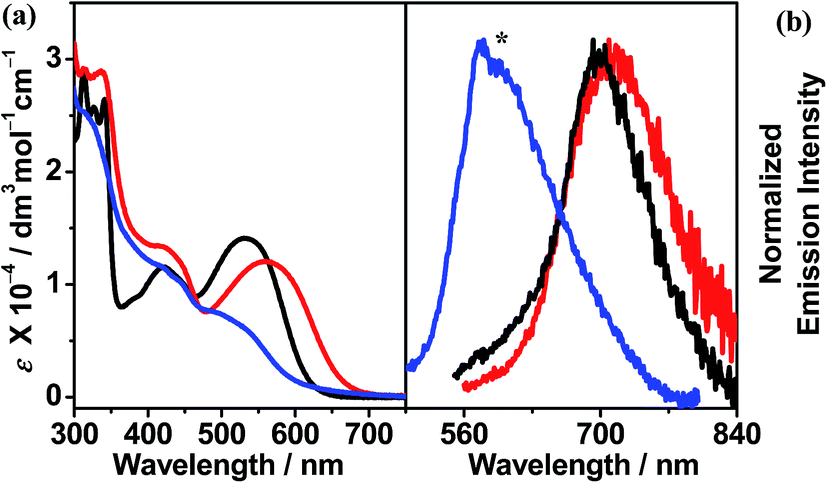 | ||
Fig. 3 (a) UV-vis absorption and (b) emission spectra ([Pt] ≈ 1 × 10−5 M) of ( ) 1, ( ) 1, ( ) 2 and ( ) 2 and ( ) mononuclear complex in CH2Cl2 at 298 K. (The asterisk indicates the instrumental artifact.) ) mononuclear complex in CH2Cl2 at 298 K. (The asterisk indicates the instrumental artifact.) | ||
Complex 1 is found to be non-emissive at room and low temperatures in the solid state as well as in glass matrices at 77 K, while complex 2 and the mononuclear complex exhibit structureless emission bands at 582–700 nm in the solid state at 298 K and 77 K, which can be assigned as being derived from 3MLCT/3LLCT origin. In the glass state, they are found to exhibit vibronically structured emission bands at ca. 574–633 nm with a shoulder at ca. 620–698 nm. The vibrational progressional spacings, ranging from 1292 to 1471 cm−1, are in line with the C![[partial double bond, bottom dashed]](https://www.rsc.org/images/entities/char_e00f.gif) C and CN vibrational modes of the tridentate terpyridine ligands.
C and CN vibrational modes of the tridentate terpyridine ligands.
To provide further insight into the nature of the excited states for this class of complexes, nanosecond transient absorption (TA) measurements were carried out on complex 2 at room temperature in degassed CH2Cl2. The nanosecond TA spectrum of complex 2 recorded immediately after laser flash excitation at 355 nm is shown in Fig. S2.† Upon laser excitation, the TA spectrum displayed a high-energy absorption band at 368 nm and a low-energy absorption band at 610–657 nm, as shown in Fig. S2.† With reference to previous TA studies on the related alkynylplatinum(II) terpyridine system,11f,g the absorption bands were tentatively assigned as the radical anion absorption of the tridentate terpyridine ligand that results from the absorption of the 3MLCT [dπ(Pt) → π*(tpy)]/3LLCT [π(CCR) → π*(tpy)] excited state. The absorption signals for complex 2 were found to exhibit monoexponential decay with a decay time constant of about 20.0 ns. Bleaching was also observed in the spectrum at approximately 650 nm, which was ascribed to the depletion of the ground state upon MLCT/LLCT absorption.
Self-assembly study
The hydrophobic dinuclear complex 1 with long hydrocarbon chains had its self-assembly behavior further examined in solvents with different polarities. In different CH2Cl2 solution concentrations, the plot of the apparent absorbance at 620 nm is found to show good agreement with Beer’s law in the concentration range of 11–180 μM (Fig. S3†). This suggests that there is no significant intermolecular aggregation of the complex molecules in CH2Cl2. Notably, this complex would exhibit strong solvatochromism upon increasing the polarity of the solvent. The solution color of complex 1 is found to change from red to purple in high concentrations of DMSO, which is accompanied by a significant drop in the high-energy absorption band at ca. 350 nm, together with a new absorption shoulder at ca. 620 nm with clear isosbestic points in the UV-vis absorption spectra, as depicted in Fig. 4a. The drop in the absorbance in the high-energy region (ca. 350 nm), which is a result of the hypochromicity effect, indicates that complex 1 is driven to undergo self-assembly through π–π stacking interactions with increasing DMSO concentration. On the other hand, the emergence of the absorption shoulders (ca. 620 nm) in pure DMSO has been further monitored at various concentrations (Fig. S4,† 102–445 μM), which shows a deviation from the linear relationship of Beer’s law. Therefore, the low-energy absorption shoulder is assigned as metal–metal-to-ligand charge-transfer (MMLCT) transitions, resulting from the formation of π–π stacking and metal–metal interactions, which are a result of the intermolecular self-assembly of the hydrophobic alkynylplatinum(II) terpyridine moieties upon increasing the solvent polarity.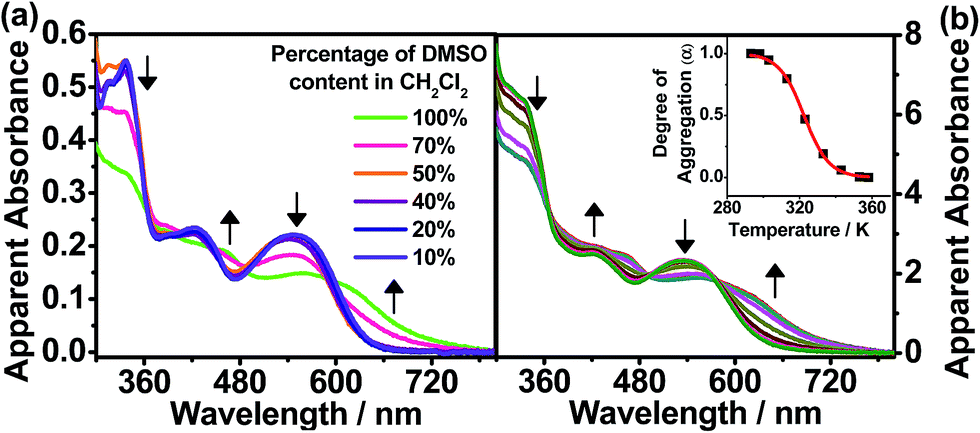 | ||
| Fig. 4 (a) UV-vis absorption spectra of 1 in CH2Cl2 at 298 K upon increasing the DMSO content from 10 to 100% ([Pt] ≈ 1 × 10−5 M). (b) UV-vis absorption spectra of 1 in DMSO upon cooling from 360 to 298 K ([Pt] ≈ 1 × 10−5 M). The apparent absorbance values have been obtained by correcting to a 1 cm path length equivalence. | ||
To further investigate the self-assembly mechanism of complex 1, temperature-dependent UV-vis absorption studies were conducted in pure DMSO. Upon decreasing the temperature from 360 K in DMSO ([Pt] = 218 μM), emergence of the MMLCT absorption band (620 nm) is observed with depletion of the MLCT/LLCT band at ca. 530 nm (Fig. 4b), thus indicating the formation of self-assembled aggregates of complex 1 with the formation of π–π stacking and metal–metal interactions. Interestingly, the cooling curve of complex 1, obtained by plotting the fraction of aggregated species (αagg) against temperature at 620 nm, is clearly sigmoidal (Fig. 4b, inset), which is indicative of an isodesmic self-assembly process. The thermodynamic parameters associated with the self-assembly process of complex 1 in DMSO (Table 3) have been determined by fitting αagg against temperature at 620 nm in accordance with the isodesmic model.13 In particular, the enthalpy and entropy changes are determined to be ΔH = −138 ± 5 kJ mol−1 and ΔS = −428 ± 2 J mol−1 K−1, respectively. The negative enthalpy and entropy values indicate that the self-assembly process is enthalpy driven. Therefore, the origin of the self-assembly behavior can be attributed to the synergistic participation of π–π and Pt⋯Pt interactions in the self-assembly of complex 1 in its aggregated form, thus giving rise to the changes observed in the UV-vis absorption spectra. It is worth noting that the phosphole moiety does play an important role in influencing the self-assembly mechanism. Recently, a series of dinuclear alkynylplatinum(II) terpyridine complexes containing the π-conjugated, planar and linear oligo(para-phenylene ethynylene) (OPE) moiety have been reported to undergo self-assembly through the mechanism of cooperative supramolecular polymerization in DMSO to give nanostructured tubular aggregates.2f In the present system, the mechanism of self-assembly behavior is found to adopt an isodesmic polymerization with the presence of the phosphole moiety. This distinct feature could be attributed to the difference in the molecular design of the bridging alkynyl ligands, in which the OPE unit is π-conjugated and linear while the phosphole unit is bent-shaped with the phosphorus center adopting a tetrahedral geometry. The linear nature of the OPE alkynyl ligands might facilitate lamellar packing to give straight nanotubes via cooperative supramolecular polymerization. In contrast, the tetrahedral geometry at the phosphorus center of the phosphole moiety together with the bent nature of the bridging alkynyl ligands favors the formation of spheres with curved surfaces in the self-assembly process. Complex 1 essentially undergoes the isodesmic polymerization mechanism, gradually undergoing self-assembly into spherical aggregates in the polar DMSO medium. Therefore, the incorporation of the phosphole moiety in the alkynylplatinum(II) terpyridine complexes can provide features that are distinct from those reported previously.2f In addition, the phosphorus centre in phosphole can be readily further functionalized for modification of the bridge. This can allow one to explore the influence of the nature and the geometry of the spacers on the supramolecular assembly of the metal complex systems.
Apart from the self-assembly induced by polar solvents, studies on the addition of non-polar solvents have also been performed. By increasing the diethyl ether content in CH2Cl2, complex 1 exhibits similar changes in the UV-vis absorption spectra, such as a drop in the absorbance in the high-energy region (ca. 350 nm) and the emergence of a MMLCT absorption shoulder (Fig. 5). This indicates that complex 1 undergoes self-assembly, driven by the formation of Pt⋯Pt interactions. However, in comparison with the self-assembly induced by polar solvents, the growth of the red-shifted MMLCT absorption band in the presence of increasing diethyl ether content is less significant. This may possibly be due to the presence of four hydrophobic hydrocarbon chains (–C18H37), which leads to better solvation and a lesser extent of metal–metal interaction formation in non-polar solvents. Nevertheless, the hypochromicity effect in the high-energy region indicates the presence of intermolecular self-assembly of the complexes with the formation of π–π stacking interactions.
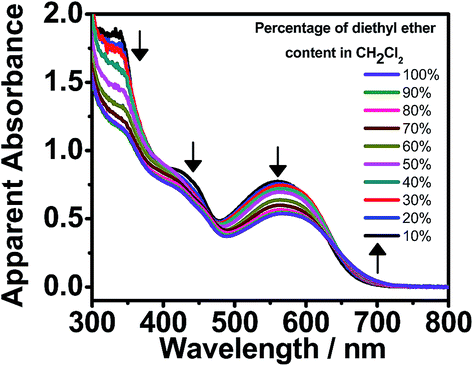 | ||
| Fig. 5 UV-vis absorption spectra of 1 in CH2Cl2 at 298 K upon increasing the diethyl ether content from 10 to 100% ([Pt] ≈ 1 × 10−5 M). | ||
Morphological studies
The distinctive self-assembly behavior of complex 1 in environments of different polarities prompted us to study the self-assembled morphologies using electronic microscopy. In DMSO solution, complex 1 is found to self-assemble into spherical aggregates with diameters of ca. 20–50 nm on carbon-coated copper grids, as demonstrated in the TEM and SEM images depicted in Fig. 6a and b, respectively. Energy-dispersive X-ray (EDX) analysis into the spherical aggregates was conducted and the results are shown in Fig. S5,† which indicates the presence of platinum. Similar spherical architectures on silica wafers can also be observed in the AFM image shown in Fig. 6c, which shows heights of ca. 12 nm. However, the spherical aggregates become flattened on the silica wafers, possibly due to the strong interaction between the soft material and the surface of the substrate. By considering the hydrocarbon chains to be in an all-trans configuration, the molecular length of complex 1 is much smaller than the dimensions of the spherical aggregates. Thus, these supramolecular structures are constructed from multiple layers of the complexes with interdigitated hydrocarbon chains in order to minimize the contact of the polar DMSO media. Together with the observations from the UV-vis absorption spectroscopic study, it is believed that complex 1 undergoes stepwise self-assembly into spherical aggregates with a single equilibrium constant of Ke = 24.2 × 105 M−1, driven by Pt⋯Pt, π–π stacking and hydrophobic–hydrophobic interactions in DMSO media.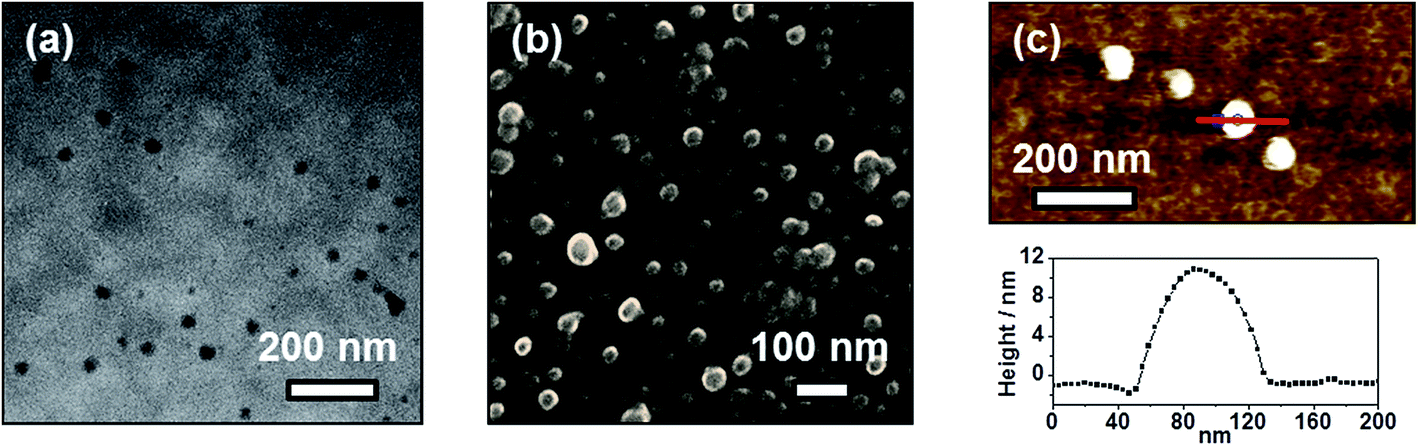 | ||
| Fig. 6 (a) TEM and (b) SEM images prepared from a 300 μM solution of 1 in DMSO on carbon-coated copper grids. (c) Tapping mode AFM image of 1 showing aggregates on silica wafers; bottom: height profile observed along the red line shown. | ||
In contrast to its aggregation behaviour observed in polar solvents, in diethyl ether complex 1 aggregates into a network of irregular fibrous structures with widths in the range of ca. 20–50 nm and lengths of a few hundreds of μm, as shown in the TEM and SEM images (Fig. 7a and b respectively). This discrepancy in size was further evidenced by DLS measurements conducted in two different solvents, in which the hydrodynamic diameter of the fibrous networks is much larger in diethyl ether (Fig. S6†). The four hydrophobic hydrocarbon chains help the doubly-positive charged complexes become solvated in non-polar solvents. It is believed that the fibrous networks originate from the self-assembly of complex 1, predominantly driven by π–π stacking interactions.
 | ||
| Fig. 7 (a) TEM and (b) SEM images prepared from a 300 μM solution of 1 in diethyl ether on carbon-coated copper grids. | ||
Conclusion
To conclude, a new class of platinum(II) terpyridine complexes with a phosphole-based bridging alkynyl ligand have been synthesized through in situ protection of the TIPS groups by TBAF, followed by a catalytic reaction. X-ray crystallography analysis demonstrated that the structure of complex 2 exhibits polymeric zigzag chain structures. The photophysical properties of the complexes have also been studied. They exhibit 3MLCT/3LLCT phosphorescence in degassed CH2Cl2 with emission energies that are varied by the π-conjugation of the terpyridine ligands. Furthermore a solvent-induced aggregation study on complex 1 was also carried out. The incorporation of hydrophobic hydrocarbon chains is thought to play an important role in governing the formation of the nanostructure. The formation of spheres is stabilized by Pt⋯Pt, π–π stacking and hydrophobic–hydrophobic interactions via the isodesmic mechanism in polar media, while fibrous networks originate from the self-assembly of the complexes in non-polar media, predominantly driven by π–π stacking interactions.Experimental section
Materials and reagents
1,7-Diiodooctadiyne was prepared as described in the literature.14 [Pt(R–tpy)Cl](OTf), where R–tpy = 4′-(3,5-bis(octadecyloxy)phenyl)-2,2′:6′,2′′-terpyridine,2f and [(tBu3tpy)PtCl](OTf)12a were synthesized according to modifications of previously reported procedures. Pd(PPh3)2Cl2, ethynyltrimethylsilane, I2, (triisopropylsilyl)acetylene and TBAF (1.0 M) were obtained from Aldrich Chem. Co. Ltd. 1,7-Octadiyne (Alfa Aesar Chem. Co. Ltd), K2PtCl4 (Strem Chemicals), triethylamine (Apollo Scientific Ltd) and CuI (Acros Chem. Co. Ltd) were purchased from the corresponding chemical suppliers. All other reagents and solvents were of analytical grade and were used as received.Synthesis and characterization of the phosphole-based alkynyl ligand
All reactions were carried out under an inert atmosphere of argon using standard Schlenk techniques.CCH2), 1.66 (m, 4H, CCCH2CH2), 1.08 (s, 42H, SiiPr3). 13C NMR (101 MHz, CDCl3, 298 K): δ = 90.1 (s, CC), 80.6 (s, CC), 78.1 (s, CC), 66.5 (s, CC), 27.3 (s, CCCH2), 19.0 (s, CCCH2CH2), 18.7 (s, Si–CH–(CH3)2), 11.4 (s, Si–CH). HR-MS (EI): m/z found 467.3528 [M + H]+; C30H51Si2 calcd 467.35293. Elemental analyses calcd (%) for C30H50Si2: C 77.18, H 10.79; found: C 77.02, H 11.15.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2CH2), 1.72 (m, 4H, CCCH2CH2), 1.06 (m, 42H, SiiPr3). 13C{1H} NMR (101 MHz, CD2Cl2): δ = 157.0 (d, 2J(C,P) = 20.5 Hz, PCC), 132.6 (s, 4J(C,P) = 3.1 Hz, Cpara phenyl), 131.1 (d, 2J(C,P) = 11.2 Hz, Cmeta phenyl), 11.6 (s, C–TIPS), 129.7 (d, 1J(C,P) = 80.0 Hz, Cipso phenyl), 129.0 (d, 3J(C,P) = 12.8 Hz, Cortho phenyl), 120.0 (d, 1J(C,P) = 89.1 Hz, PCC), 107.2 (d, 3J(C,P) = 6.5 Hz, TIPS–CC), 99.6 (d, 2J(C,P) = 13.2 Hz, TIPS–CC), 28.4 (d, 3J(C,P) = 10.8 Hz, CCCH2), 22.5 (s, CCCH2CH2), 18.8 (s, C–TIPS). 31P{1H} NMR (162 MHz, CD2Cl2): δ = +51.0 (s). HR-MS (EI): m/z found 607.3377 [M + H]+; C36H56Si2PS calcd 607.33789. Elemental analysis calcd (%) for C36H55Si2PS: C 71.23, H 9.13, S 5.28; found: C 71.45, H 9.01, S 5.24.
CCH2CH2), 1.72 (m, 4H, CCCH2CH2), 1.06 (m, 42H, SiiPr3). 13C{1H} NMR (101 MHz, CD2Cl2): δ = 157.0 (d, 2J(C,P) = 20.5 Hz, PCC), 132.6 (s, 4J(C,P) = 3.1 Hz, Cpara phenyl), 131.1 (d, 2J(C,P) = 11.2 Hz, Cmeta phenyl), 11.6 (s, C–TIPS), 129.7 (d, 1J(C,P) = 80.0 Hz, Cipso phenyl), 129.0 (d, 3J(C,P) = 12.8 Hz, Cortho phenyl), 120.0 (d, 1J(C,P) = 89.1 Hz, PCC), 107.2 (d, 3J(C,P) = 6.5 Hz, TIPS–CC), 99.6 (d, 2J(C,P) = 13.2 Hz, TIPS–CC), 28.4 (d, 3J(C,P) = 10.8 Hz, CCCH2), 22.5 (s, CCCH2CH2), 18.8 (s, C–TIPS). 31P{1H} NMR (162 MHz, CD2Cl2): δ = +51.0 (s). HR-MS (EI): m/z found 607.3377 [M + H]+; C36H56Si2PS calcd 607.33789. Elemental analysis calcd (%) for C36H55Si2PS: C 71.23, H 9.13, S 5.28; found: C 71.45, H 9.01, S 5.24.
Synthesis and characterization of the di- and mononuclear alkynylplatinum(II) terpyridine complexes with the phosphole-based alkynyl ligand
All reactions were carried out under an inert atmosphere of nitrogen using standard Schlenk techniques.The synthesis of the chloroplatinum(II) terpyridine precursor was carried out according to modification of a literature procedure for [Pt(tpy)Cl](OTf)9a using R–tpy = 4′-(3,5-bis(octadecyloxy)phenyl)-2,2′:6′,2′′-terpyridine. AgOTf (174 mg, 0.66 mmol) was added to a suspension of Pt(PhCN)2Cl2 (283 mg, 0.60 mmol) in acetonitrile solution with further heating under reflux for 15 h. The solution was filtered and the corresponding terpyridine (169 mg, 0.20 mmol) was then added to the clear solution. The reaction mixture was heated under reflux overnight. The solution was then filtered and evaporated under reduced pressure. The crude product was dissolved in chloroform and washed twice with saturated NaCl solution. The organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure. The product was further purified by precipitation of the chloroform solution in hexane twice, and the product was obtained as a yellow powder.
:1 v/v) as the eluent, followed by the diffusion of diethyl ether vapor into a CH2Cl2 solution of the complex to give 1 as red crystals. Yield: 20 mg (32%).
1H NMR (400 MHz, DMSO-D6, 350 K): δ = 9.01 (d, 3J = 7.6 Hz, 4H, terpy), 8.87 (s, 4H, terpy), 8.81 (d, 3J = 7.6 Hz, 4H, terpy), 8.60 (d, 3J = 10.8 Hz, 4H, terpy), 8.43 (t, 3J = 7.6 Hz, 4H, terpy, tpy), 7.89 (m, 1H, Ph), 7.75 (t, 3J = 7.6 Hz, 4H, terpy), 7.62 (dd, 2H, Ph), 7.54 (m, 2H, Ph), 7.19 (s, 2H, terpy), 4.01 (t, 8H, –OCH2–), 2.92 (m, 4H, –CH2–), 2.20 (m, 4H, –CH2–), 2.83 (m, 4H, –C18H37), 1.23 (s, 66H, –C18H37). Positive FAB-MS: ion clusters at m/z 1633.5 [M − OTf]+, 742.4 [M − 2OTf]2+. FTIR: 2081 cm−1ν(CC). Elemental analyses calcd (%) for C134H187F6N6O10PPt2S3 CH2Cl2: C 58.79, H 6.91, N 3.05; found: C 58.88, H 7.07, N 2.89.
C). Elemental analyses calcd (%) for C74H83F6N6O6PPt2S3·2.5(CH2Cl2): C 46.03, H 4.44, N 4.21; found: C 45.87, H 4.47, N 4.17.
Synthesis of mononuclear alkynylplatinum(II) terpyridine complex
1H NMR (400 MHz, CDCl3, 298 K, relative to Me4Si): δ = 9.05 (d, 3J = 7.2 Hz, 2H, terpy), 8.76 (s, 2H, terpy), 8.68 (s, 2H, terpy), 7.99 (t, 3J = 7.7 Hz, 2H, terpy), 7.88 (dd, 3J = 13.7 Hz, 4J = 7.6 Hz, 2H, –Ph), 7.42 (m, 5H, terpy and –Ph), 7.08 (s, 2H, terpy), 6.45 (s, 1H, terpy), 4.08 (m, 4H, –OCH2–), 2.73 (m, 4H, –CH2–), 2.65 (m, 4H, –CH2–), 2.83 (m, 4H, –C18H37), 1.23 (s, 66H, –C18H37). 31P{1H} NMR (161 MHz, CDCl3, 298 K, relative to 85% H3PO4): δ 51.55. Positive ESI-MS: ion clusters at m/z 1334.7 [M − OTf]+.Physical measurements and instrumentation
1H and 31P{1H} NMR spectra were recorded using a Bruker DPX-400 Fourier transform NMR spectrometer at 400 MHz (1H NMR) and 161 MHz (31P NMR) with chemical shifts recorded relative to tetramethylsilane (Me4Si) and 85% phosphoric acid (H3PO4), respectively. High-resolution mass spectra for the ligands were obtained using Varian MAT 311, Waters Q-TOF 2 or ZabSpec TOF Micromass Instruments at the CRMPO, University of Rennes 1. Elemental analyses of the ligands were carried out by the CRMPO. Positive FAB mass spectra of the complexes were recorded using a Thermo Scientific DFS High Resolution Magnetic Sector Mass Spectrometer at The University of Hong Kong. IR spectra of the complexes were recorded using a Perkin Elmer Spectrum Two IR Spectrometer at The University of Hong Kong. Elemental analyses of the metal complexes were carried out using a Carlo Erba 1106 elemental analyzer at the Institute of Chemistry, Chinese Academy of Sciences in Beijing. UV-visible spectra were obtained using a Uvikon 942 spectrophotometer or a Hewlett-Packard 8452A diode array spectrophotometer. The temperatures for the variable-temperature UV-vis measurements were monitored using a Varian Cary Single-Cell Peltier Thermostat. Steady-state excitation and emission spectra in solution and in the solid state at room temperature were recorded using a Spex Fluorolog-3 model FL3-211 fluorescence spectrofluorometer equipped with an R2658P PMT detector. Photophysical measurements of the low-temperature solid and glass states were carried out with the sample solution loaded in a quartz tube inside a quartz-walled Dewar flask. Liquid nitrogen was placed into the Dewar flask for the low temperature (77 K) photophysical measurements. Transient absorption measurements were performed using an LP920-KS Laser Flash Photolysis Spectrometer (Edinburgh Instruments Ltd, Livingston, UK) at ambient temperature. The excitation source was the 355 nm output (third harmonic) of the Spectra-Physics Quanta-Ray Lab-130 pulsed Nd:YAG laser and the probe light source was a Xe900 450 W xenon arc lamp. Transient absorption spectra were obtained using an image-intensified CCD camera (Andor) with a PC plug-in controller, fully operated by the L900 spectrometer software. The absorption kinetics were detected using a Hamamatsu R928 photomultiplier tube and recorded using a Tektronix Model TDS3012B (100 MHz, 1.25 GSs−1) digital oscilloscope and analyzed using the same software for the exponential fit (tail-fit data analysis). TEM and SEM images were taken by the Gatan MultiScan, Model 794 and the Hitachi S4800 FEG, respectively, with both obtained at the Electron Microscope Unit at The University of Hong Kong. AFM topographical images and phase images were obtained using an Asylum MFP3D Atomic Force Microscope with an ARC2 SPM Controller under constant temperature and atmospheric pressure at The University of Hong Kong.X-ray diffraction measurement
Crystals of 2 were obtained, by the slow diffusion of vapors of pentane into dichloromethane solutions, as very thin and small dark crystalline blocks that were in most cases not single crystals. After several attempts, a small single crystal that presented a satisfactory but weak diffraction pattern was obtained and used for the X-ray diffraction studies. Single-crystal data collection was carried out using an APEX II Bruker-AXS instrument (Centre de Diffractométrie, Université de Rennes 1). The reflections were indexed, and the Lorentz polarization corrected and integrated using the DENZO program of the KappaCCD software package. The data-merging process was performed using the SCALEPACK program.15 Structure determinations were performed by direct methods using the SIR97 solving program,16 which revealed all the non-hydrogen atoms. The SHELXL program17 was used to refine the structures using the full-matrix least squares method based on F2. All of the non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were included in idealized positions and refined with isotropic displacement parameters (as well as two non-H atoms that could not be refined with anisotropic displacement parameters). In the studied crystal lattices of 2, dichloromethane solvent molecules were found in addition to the cationic coordination complexes and their counter-anions. These solvent molecules in most cases have a strong tendency to leave the bulk crystal via evaporation once the crystals are removed from their mother liquor, a process that induces rapid degradation of the single-crystal integrity of the crystals investigated. In order to slow down this process, the single crystal was always coated in paratone oil once removed from the mother liquor, and mounted at a low temperature of 150 K as quickly as possible on the diffractometer goniometer, and X-ray data collection was carried out at 150 K. Nevertheless, even by applying such a procedure, the mounted single crystal most probably partially lost its included dichloromethane solvent molecules, which induced partial degradation of the bulk single crystal integrity and resulted in the collection of a weak diffraction data set. The included dichloromethane solvent molecules and triflate counter-anions were found to be highly disordered, and modeling of the disorder of these solvent molecules was not possible, which led to rather high anisotropic displacement parameters for some of their atoms. Therefore a ‘SQUEEZE’ treatment18 was carried out in order to remove the scattering contribution of these molecules that could not be satisfactorily modeled. In addition, some of the carbon atoms of the six tert-butyl groups present a high ADP max/min ratio and the large Hirshfeld difference is a result of the disordered arrangement of these tert-butyl groups in the crystal structure. Nevertheless, correct modeling of this disorder was not possible, which led to unstable refinement cycles together with an unjustified increase in the number of modeled parameters. Consequently, the final R1 and wR2 factors were quite high both before and after the ‘SQUEEZE’ procedure was applied, thus reflecting the unfavorable parameters. The crystal data and structure refinement at 150 K for 2 before and after this ‘SQUEEZE’ treatment are given in Table S1.† Atomic scattering factors for all of the atoms were taken from the International Tables for X-ray Crystallography.19 The CCDC reference number 1506200 contains the supplementary crystallographic data for 2.Acknowledgements
V. W-W. Y. acknowledges the support from The University of Hong Kong and the URC Strategic Research Theme on New Materials. This work has been supported by the French National Research Agency (ANR)-Research Grants Council (RGC) Joint Research Scheme (A-HKU704/12) and the University Grants Committee Areas of Excellence Scheme (AoE/P-03/08). M. H. acknowledges the support from the Ministère de la Recherche et de l’Enseignement Supérieur and the CNRS, Brittany. Dr Alan Kwun-Wa Chan is gratefully acknowledged for his technical assistance in finalizing the manuscript. We also thank Mr Frankie Yu-Fee Chan at the Electron Microscope Unit of The University of Hong Kong for his helpful technical assistance.References
- (a) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed; (b) A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179–183 CrossRef CAS PubMed; (c) W. Zhang, W. Jin, T. Fukushima, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2009, 48, 4747–4750 CrossRef CAS PubMed; (d) G. Golubkov, H. Weissman, E. Shirman, S. G. Wolf, I. Pinkas and B. Rybtchinski, Angew. Chem., Int. Ed., 2009, 48, 926–930 CrossRef CAS PubMed; (e) R. Charvet, Y. Yamamoto, T. Sasaki, J. Kim, K. Kato, M. Takata, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2012, 134, 2524–2527 CrossRef CAS PubMed; (f) H. Yamagishi, T. Fukino, D. Hashizume, T. Mori, Y. Inoue, T. Hikima, M. Takata and T. Aida, J. Am. Chem. Soc., 2015, 137, 7628–7631 CrossRef CAS PubMed.
- (a) A. Y.-Y. Tam, K. M.-C. Wong, G. Wang and V. W.-W. Yam, Chem. Commun., 2007, 20, 2028–2030 RSC; (b) F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J.-P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659–2662 CrossRef CAS PubMed; (c) V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286–6289 CrossRef CAS PubMed; (d) W. Lu, Y. Chen, V. A. L. Roy, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621–7625 CrossRef CAS PubMed; (e) V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and B. W.-K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169–6173 CrossRef CAS PubMed; (f) S. Y.-L. Leung, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845–2850 CrossRef CAS PubMed; (g) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, Chem.–Eur. J., 2009, 15, 4775–4778 CrossRef CAS PubMed; (h) C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136–13143 CrossRef CAS PubMed; (i) W. Lu, S. S.-Y. Chui, K.-M. Ng and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 4568–4572 CrossRef CAS PubMed; (j) H.-L. Au-Yeung, S. Y.-L. Leung, A. Y.-Y. Tam and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17910–17913 CrossRef CAS PubMed; (k) C.-M. Che, C. F. Chow, M. Y. Yuen, V. A. L. Roy, W. Lu, Y. Chen, S. S. Y. Chui and N. Zhu, Chem. Sci., 2011, 2, 216–220 RSC; (l) C. Po, A. Y.-Y. Tam and V. W.-W. Yam, Chem. Sci., 2014, 5, 2688–2695 RSC; (m) C. A. Strassert, C. H. Chien, M. D. G. Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS PubMed; (n) F. C.-M. Leung, S. Y.-L. Leung, C. Y.-S. Chung and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 2989–2992 CrossRef CAS PubMed.
- (a) V. K.-M. Au, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2013, 52, 558–567 CrossRef CAS PubMed; (b) K.-C. Yim, E. S.-H. Lam, K. M.-C. Wong, V. K.-M. Au, C.-C. Ko, W. H. Lam and V. W.-W. Yam, Chem.–Eur. J., 2014, 20, 9930–9939 CrossRef CAS PubMed; (c) X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem.–Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed; (d) A. K.-W. Chan, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2015, 137, 6920–6931 CrossRef CAS PubMed; (e) A. K.-W. Chan, D. Wu, K. M.-C. Wong and V. W.-W. Yam, Inorg. Chem., 2016, 55, 3685–3691 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- (a) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (b) T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed; (c) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- (a) M. Hissler, P. W. Dyer and R. Réau, Coord. Chem. Rev., 2003, 244, 1–44 CrossRef CAS; (b) O. Fadhel, M. Gras, N. Lemaite, V. Deborde, M. Hissler, B. Geffroy and R. Réau, Adv. Mater., 2009, 21, 1261–1265 CrossRef CAS; (c) D. Joly, D. Tondelier, V. Deborde, W. Delaunay, A. Thomas, K. Bhanuprakash, B. Geffroy, M. Hissler and R. Réau, Adv. Funct. Mater., 2012, 22, 567–576 CrossRef CAS; (d) D. Joly, P.-A. Bouit and M. Hissler, J. Mater. Chem. C, 2016, 4, 3686–3698 RSC; (e) M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 5296–5310 RSC.
- (a) J. C.-H. Chan, W. H. Lam, H.-L. Wong, W.-T. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 11504–11508 CrossRef CAS PubMed; (b) P. Gong, K. Ye, J. Sun, P. Chen, P. Xue, H. Yang and R. Lu, RSC Adv., 2015, 5, 94990–94996 RSC.
- (a) Y. Ren, W. H. Kan, M. A. Henderson, P. G. Bomben, C. P. Berlinguette, V. Thangadurai and T. Baumgartner, J. Am. Chem. Soc., 2011, 133, 17014–17026 CrossRef CAS PubMed; (b) Y. Ren, W. H. Kan, V. Thangadurai and T. Baumgartner, Angew. Chem., Int. Ed., 2012, 51, 3964–3968 CrossRef CAS PubMed; (c) P.-A. Bouit, A. Escande, R. Szűcs, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524–6527 CrossRef CAS PubMed; (d) X. He, J.-B. Lin, W. H. Kan, P. Dong, S. Trudel and T. Baumgartner, Adv. Funct. Mater., 2014, 24, 897–906 CrossRef CAS; (e) C. Fave, M. Hissler, K. Sénéchal, I. Ledoux, J. Zyss and R. Réau, Chem. Commun., 2002, 1674–1675 RSC; (f) F. Leca, M. Sauthier, V. Deborde, L. Toupet and R. Réau, Chem.–Eur. J., 2003, 9, 3785–3795 CrossRef CAS PubMed; (g) V. Vreshch, M. El Sayed Moussa, B. Nohra, M. Srebo, N. Vanthuyne, C. Roussel, J. Autschbach, J. Crassous, C. Lescop and R. Réau, Angew. Chem., Int. Ed., 2013, 52, 1968–1972 CrossRef CAS PubMed.
- (a) C. Fave, T.-Y. Cho, M. Hissler, C.-W. Chen, T.-Y. Luh, C.-C. Wu and R. Réau, J. Am. Chem. Soc., 2003, 125, 9254–9255 CrossRef CAS PubMed; (b) H.-C. Su, O. Fadhel, C.-J. Yang, T.-Y. Cho, C. Fave, M. Hissler, C.-C. Wu and R. Réau, J. Am. Chem. Soc., 2006, 128, 983–995 CrossRef CAS PubMed; (c) E. Y.-H. Hong, H.-L. Wong and V. W.-W. Yam, Chem. Commun., 2014, 50, 13272–13274 RSC.
- (a) J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591–4599 CrossRef CAS; (b) M. G. Hill, J. A. Bailey, V. M. Miskowski and H. B. Gray, Inorg. Chem., 1996, 35, 4585–4590 CrossRef CAS; (c) W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913–922 CrossRef CAS; (d) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529–1533 CrossRef CAS; (e) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446–4452 CrossRef CAS; (f) V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145–152 CrossRef CAS; (g) H. Kunkely and A. Vogler, J. Am. Chem. Soc., 1990, 112, 5625–5627 CrossRef CAS; (h) T. K. Aldrige, E. M. Stacy and D. R. McMillin, Inorg. Chem., 1994, 33, 722–727 CrossRef; (i) R. Büchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans, 1999, 5, 711–718 RSC; (j) C.-M. Che, K. T. Wan, L. Y. He, C.-K. Poon and V. W.-W. Yam, J. Chem. Soc., Chem. Commun., 1989, 943–944 RSC; (k) C.-M. Che, L.-Y. He, C.-K. Poon and T. C. W. Mak, Inorg. Chem., 1989, 28, 3081–3083 CrossRef CAS; (l) H. K. Yip, L.-K. Cheng, K. K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans, 1993, 19, 2933–2938 RSC; (m) C.-W. Chan, L.-K. Cheng and C.-M. Che, Coord. Chem. Rev., 1994, 132, 87–97 CrossRef CAS; (n) K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem. Commun., 2006, 32, 3441–3443 RSC; (o) K. M.-C. Wong and V. W.-W. Yam, Coord. Chem. Rev., 2007, 251, 2477–2488 CrossRef CAS; (p) K. M.-C. Wong and V. W.-W. Yam, Chem. Commun., 2011, 47, 11579–11592 RSC.
- (a) K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839–3843 CrossRef CAS PubMed; (b) K. W. Jennette, J. T. Gill, J. A. Sadownick and S. J. Lippard, J. Am. Chem. Soc., 1976, 98, 6159–6168 CrossRef CAS; (c) S. J. Lippard, Acc. Chem. Res., 1978, 11, 211–217 CrossRef CAS; (d) T. J. Wadas, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2003, 42, 3772–3778 CrossRef CAS PubMed; (e) S. Chakraborty, T. J. Wadas, H. Hester, C. Flaschenreim, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6284–6293 CrossRef CAS PubMed; (f) P. Jarosz, K. Lotito, J. Schneider, D. Kumaresan, R. Schmehl and R. Eisenberg, Inorg. Chem., 2009, 48, 2420–2428 CrossRef CAS PubMed; (g) P. Jarosz, P. W. Du, J. Schneider, S. H. Lee, D. McCamant and R. Eisenberg, Inorg. Chem., 2009, 48, 9653–9663 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed; (b) C. Yu, K. M.-C. Wong, K. H.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791–794 CrossRef CAS PubMed; (c) S. Y.-L. Leung, A. Y.-Y. Tam, C.-H. Tao, H. S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2012, 134, 1047–1056 CrossRef CAS PubMed; (d) S. Y.-L. Leung, W. H. Lam and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7986–7991 CrossRef CAS PubMed; (e) S. Y.-L. Leung and V. W.-W. Yam, Chem. Sci., 2013, 4, 4228–4234 RSC; (f) Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Chem. Sci., 2012, 3, 1185–1191 RSC; (g) Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 390–399 CrossRef CAS PubMed; (h) Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 14117–14120 CrossRef CAS PubMed.
- (a) R. B. Martin, Chem. Rev., 1996, 96, 3043–3064 CrossRef CAS PubMed; (b) M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- (a) Y. Matano, M. Nakashima and H. Imahori, Angew. Chem., Int. Ed., 2009, 48, 4002–4005 CrossRef CAS PubMed; (b) N. Lubin-Germain, A. Hallonet, F. Huguenot, S. Palmier, J. Uziel and J. Auge, Org. Lett., 2007, 9, 3679–3682 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor. in Methods in Enzymology, ed. C. W. Carter Jr and R. M. Sweet, Academic Press, New York, 1997, vol. 276, p. 307 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick., SHELX97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 13 CrossRef; (b) P. van der Stuis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- P. J. Brown, A. G. Fox, E. N. Maslen, M. A. O'Keefe and B. T. M. Willis, International Tables for Crystallography, E. Prince, Springer, Dordrech, 2006, vol. C, ch. 6.1, pp. 554–595 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1506200. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00041c |
| This journal is © The Royal Society of Chemistry 2017 |