DOI:
10.1039/C7RA12954H
(Paper)
RSC Adv., 2018,
8, 2818-2823
Maleimidation of dextran and the application in designing a dextran–camptothecin conjugate†
Received
1st December 2017
, Accepted 3rd January 2018
First published on 12th January 2018
Abstract
Camptothecin analogs, as commonly used chemotherapy drugs, usually have poor water solubility which has limited their use in the clinic. In order to improve the water-solubility of camptothecin, a new dextran derivative Dex-Mal was synthesized and used in designing a dextran–camptothecin conjugate which contained a CTB-sensitive linker. This conjugate could efficiently release the therapeutic drug SN-38 in the presence of cathepsin B and the antiproliferative activity of the conjugate was similar to the approved drug Irinotecan hydrochloride. Furthermore, in the presence of dextran, the conjugate could self-assemble into nanoparticles in water, which could improve the targeting ability through the EPR effect. This provides a potential way to formulate a drug delivery system for camptothecin analogs or other drugs which have poor water solubility.
Introduction
Although great efforts have been made in cancer pharmacotherapy in the last few decades, there is still a long way to go to cure cancer. According to the statistics, there will be more than 1.68 million new cancer cases and 0.60 million cancer deaths in the United States in 2017.1 Traditional anticancer drugs such as camptothecin (CPT) and paclitaxel (PTX) analogs are commonly used in clinical cancer therapy due to their high efficiency and wide spectrum.2 However, the poor water solubility of CPT and PTX analogs has limited their clinical application. Moreover, the nonspecific distribution in the body always leads to severe side effects. To overcome these obstacles, various drug delivery systems have been developed, including liposomes,3 micelles4 and polymer–drug conjugates;5 these drug delivery systems can selectively deliver therapeutic agents into tumor tissues via the enhanced permeability and retention (EPR) effect, as well as improving the physicochemical and pharmacokinetic properties.
Being different from liposomes and micelles, polymer–drug conjugates are more stable as the presence of the covalent attachment. These conjugates are usually obtained by coupling the drugs to polymers through cleavable linkers which are sensitive to pH, temperature, reduction, oxidation, enzymes and so on. There are many kinds polymers used for drug conjugation, such as N-(2-hydroxypropyl) methacrylamide (HMPA),6 poly-L-glutamic acid (PG),7 polyethylene glycol (PEG),8 styrene maleic anhydride (SMA)9 and dextran.10
Dextran, which is synthesized from sucrose by certain lactic-acid bacteria like Leuconostoc mesenteroides and Streptococcus mutans, is a natural linear polymer of glucose linked by a 1-6 linked-glucopyranoside.11 The great water solubility and biocompatibility of dextran makes it approved for clinical application by FDA. Unlike PEG, which only has modifiable groups at the terminal, dextran is easy to modify due to the large number of hydroxyl groups which can undergo direct esterification,12 carbonyldiimidazole activation,13 periodate oxidation,14 cyanogens bromide activation and etherification.15 However, these reactions are often involved in severe reaction conditions that are not suitable for many substrates. The thiol–maleimide ‘Click’ reaction is a popular bioorthogonal reaction which is commonly applied in the field of bioconjugation. Meanwhile, the maleimidation is a common method used for modification of polymers and antibodies.16,17 Nevertheless, very few research has been disclosed on the maleimidation of dextran,18 therefore we developed a method to modify dextran by maleimidation.
“Smart linkers” are usually activated by the specificity of tumor tissue such as acidic medium, reduction environment and some specific enzymes.19 Among these linkers, enzyme-sensitive linkers are more attractive because of their high selectivities. Cathepsin B (CTB) is a cysteine protease involved in numerous pathological processes.20 The expression of CTB was found to be relatively higher in tumor tissue than in normal tissue and blood plasma.21 Besides, CTB can selectively recognize several specific dipeptide sequences such valine–citrulline, phenylalanine–arginine and lysine–lysine.22 With these advantages, CTB-sensitive linkers have been successfully applied in designing probes,23 polymer–drug conjugates and antibody–drug conjugates.24–26 In previous studies we have disclosed a CTB-activated nanoprodrug which had great water solubility and in vivo anti-tumor activity.24 Herein we introduce a dextran–camptothecin conjugate based on our earlier report.
Experimental
Materials and methods
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DRX-400 MHz spectrometer (400 and 100 MHz, respectively) using CDCl3, D2O, or DMSO-d6 as solvents with TMS as an internal standard. Chemical shifts were reported as d (ppm) and spin–spin coupling constants as J (Hz) values. The mass spectra (MS) were recorded on a Finnigan MAT-95 mass spectrometer. Melting points were taken on a SGW X-4 melting point apparatus, uncorrected and reported in degrees centigrade. Column chromatography was performed with silica gel (200–300 mesh). UV-vis absorption spectra were recorded on a Varian Cary 100 spectrophotometer.
Maleic anhydride, beta-alanine, acepramin and N-acetyl-L-cysteine were purchased from Energy Chemical Reagent Co. (Shanghai, China). HATU was purchased from Highfine Biotech Co., Ltd (Suzhou, China). Dextran (20 kDa) was purchased from Pharmacosmos. Compound 8 was synthesized before and other chemicals were purchased from Sinopharm Chemical Reagent Co., Ltd. Human colon cancer cell line HCT-116, human hepatoma cell line HepG2 and human cervical cancer cell line HeLa were purchased from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). The cell culture fluid and FBS were purchased from Thermo Fisher Scientific.
Synthetic procedures
Compound 2. Maleic anhydride (10.0 g, 0.102 mol) and β-alanine (9.90 g, 0.112 mol) were dissolved in glacial acetic acid (150 mL) and the solution was heated to reflux for 8 h then acetic acid was removed under vacuum to give the crude product which was recrystallized in diethyl ether to afford a white solid (9.50 g, 55%). Mp 105–107 °C. 1H-NMR (400 MHz, DMSO-d6) δ 12.4 (s, 1H), 7.02 (d, J = 4.4 Hz, 2H), 3.62 (t, J = 7.3 Hz, 2H), 2.53–2.45 (m, 2H).
Compound 4. Compound 4 was synthesized as described in the ref. 28.
Dex-Mal. Compound 4 (calculated for 5.91 mmol) was added to a solution of dextran (T20, 11.8 mmol glucose unit) in anhydrous DMSO. The reaction mixture was heated to 45 °C overnight and dialyzed against deionized water for 24 h in a dialysis tube (MWCO: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Finally, the solution in the dialysis tube was lyophilized and washed with cold ethanol to give Dex-Mal as a white flocculent solid.
000). Finally, the solution in the dialysis tube was lyophilized and washed with cold ethanol to give Dex-Mal as a white flocculent solid.
Compound 6. Compound 6 was synthesized as described in the ref. 27.
Compound 7. To a solution of compound 6 (8.0 g, 19.7 mmol) in anhydrous DMF was added DIEA (6.85 mL, 39.4 mmol) and HATU (7.49 g, 19.7 mmol) in batches. The mixture was stirred at room temperature for 2 h and acepramin (2.58 g, 19.7 mmol) was added and then was stirred overnight. The solution was poured into water and extracted with ethyl acetate. The organic layer was washed with water followed by brine and dried over Na2SO4. The final product was purified by silicagel column to afford a white solid (5.0 g, 49%), mp 156–158 °C. 1H-NMR (400 MHz, DMSO-d6) δ 11.93 (s, 1H), 8.05 (d, J = 8.3 Hz, 1H), 7.89 (s, 1H), 7.41–7.14 (m, 15H), 4.31 (d, J = 7.5 Hz, 1H), 3.11–2.85 (m, 2H), 2.37–2.21 (m, 2H), 2.14 (t, J = 7.4 Hz, 2H), 1.81 (s, 3H), 1.49–1.30 (m, 4H), 1.28–1.15 (m, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 174.3, 169.5, 168.9, 144.3, 129.0, 128.0, 126.7, 65.8, 51.5, 38.3, 34.1, 33.6, 28.5, 25.8, 24.1, 22.4. MS (ESI) m/z = 541.3 [M + Na]+
Compound 8. Compound 8 was synthesized as described in the ref. 24.
Compound 9. Compound 8 (800 mg, 0.74 mmol) was dissolved in dichloromethane and TFA was added. The mixture was stirred at room temperature for 2 h and then the solvent was removed under vacuum to give the crude product directly for the next step. Compound 7 (577 mg, 1.04 mmol) was dissolved in anhydrous DMF and HATU (395.2 mg, 1.04 mmol), DIEA (0.24 mL, 1.4 mmol) was added, then the previous crude product was dissolved in DMF and added to the solution. Subsequently the mixture was stirred at room temperature overnight and the solvent was removed under vacuum to give the residue. Then it was purified by column chromatography to afford the target compound (550 mg, 54%). 1H NMR (400 MHz, DMSO-d6) δ 8.30–8.10 (m, 2H), 8.02–7.84 (m, 2H), 7.62 (dt, J = 26.2, 16.0 Hz, 3H), 7.46–7.26 (m, 15H), 5.58–5.32 (m, 4H), 5.21–5.06 (m, 2H), 4.70–4.54 (m, 2H), 4.36 (d, J = 24.2 Hz, 2H), 4.21 (t, J = 18.7 Hz, 3H), 3.56 (s, 2H), 3.45 (d, J = 9.8 Hz, 2H), 3.27–2.88 (m, 11H), 2.43–2.29 (m, 3H), 2.18 (dt, J = 15.2, 7.8 Hz, 2H), 2.08–1.91 (m, 3H), 1.88 (d, J = 7.3 Hz, 3H), 1.81–1.58 (m, 3H), 1.57–1.20 (m, 12H), 0.92 (dt, J = 16.4, 7.0 Hz, 8H). HR-MS m/z: 1480.7810, (calculated for C80H94N11O15S, 1480.6652).
Dextran–camptothecin conjugate. Compound 9 (100 mg, 0.068 mmol) was suspended in dichloromethane and TFA was added to form a yellow solution. Triethyl silicane (0.05 mL, 0.31 mmol) was added to the solution and it turned light kelly. After stirring for 2 h, NaHCO3 (120 mg, 1.5 mmol) in 10 mL water was added and the mixture turned colourless and Dex-Mal (120 mg, 0.069 mmol maleimide unit) was added. The solution was stirred for another 4 h and then dialyzed against deionized water for 24 h in a dialysis tube (MWCO: 10000). Finally, the solution in the dialysis tube was lyophilized to give the target product as a white solid.
General HPLC method
HPLC analysis was performed at room temperature using a Diamonsil C18 (250 mm × 4.6 mm) and a mobile phase gradient from 10% CH3CN/buffer (0.1% TFA/H2O) to 30% CH3CN/buffer (0.1% TFA/H2O) for 3 min, 30% CH3CN/buffer (0.1% TFA/H2O) to 90% CH3CN/buffer (0.1% TFA/H2O) for 12 min, 90% CH3CN/buffer (0.1% TFA/H2O) for 5 min, a flow rate of 1.0 mL min−1, and plotted at 373 nm. This method was used in stability studies.
Calculation of drug loading
Compound 9 was dissolved in DMSO and diluted to different molar concentrations and the dextran–camptothecin conjugate was diluted to a given mass concentration as well. The UV-vis absorption spectrum was recorded on a Varian Cary 100 spectrophotometer. The calibration curve was obtained and the drug loading was calculated according to the absorption at 365 nm.
Water solubility of the dextran–camptothecin conjugate and Irinotecan hydrochloride
Excess amount of Irinotecan hydrochloride and the dextran–camptothecin conjugate was added into 0.1 mL water respectively and dissolved thoroughly. The undissolved substances were filtered by a 0.22 μm filter, and the filtrate was diluted with DMSO, and the UV-vis absorption spectrum of the dilution was recorded on a Varian Carry 100 Spectrophotometer.
Particle size distribution of the dextran–camptothecin conjugate in water
The Dex-Mal and dextran–camptothecin conjugate was dissolved in water with a suitable concentration and the size distribution was measured on a dynamic light scattering (DLS) (ZetasizerNano ZS, Malvern Instruments, UK).
Enzyme-sensitivity of the dextran–camptothecin conjugate
CTB (10 units) from bovine spleen was dissolved in 1 mL buffer containing 25 mM sodium acetate (pH = 5.0) and 1 mM EDTA at −80 °C before use. The above stock solution (60 μL) was activated with 120 μL buffer containing 30 mM dithiothreitol (DTT) and 15 mM EDTA for 15 min at 37 °C. The activated CTB solution was diluted with 0.82 mL buffer containing 25 mM acetate (pH = 5.0) and 1 mM EDTA. The final concentration of CTB in this solution was 0.6 unit per mL. Then, the dextran–camptothecin conjugate in 10 μL DMSO was added to CTB solution and incubated at 37 °C. The sample (70 μL) was collected and quenched by the addition of 70 μL acetonitrile. The concentration of drug released was determined by HPLC using external standard method.
Cell viability assay
HCT-116, HepG2 and Hela cells were cultured in McCoy's 5A (for HCT-116) or MEM (for HepG2), or DMEM (for Hela) medium, supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin (Thermo Fisher Scientific). All cell lines were maintained at 37 °C with 5% CO2. Cell viability was evaluated by the MTT assay. Cells were seeded in 96-well plates and incubated for 12 h. A range of concentrations of the test compounds were added and the plates were incubated for 72 h. Before the addition of 10 μL 5 mg mL−1 MTT, 100 μL of medium was removed in each well. After 4 h of incubation, 50 μL of lysis solution (10% SDS, 5% isobutanol and 0.012 mol L−1 HCl) was added to each well and incubated for another 12 h. The absorbance was measured using a SpectraMax M5 microplate reader at 570 nm. The concentration of drug inhibiting 50% of cells (IC50) was calculated using the software of Graphpad Prism 5.
Results and discussion
Design of the dextran–camptothecin conjugate
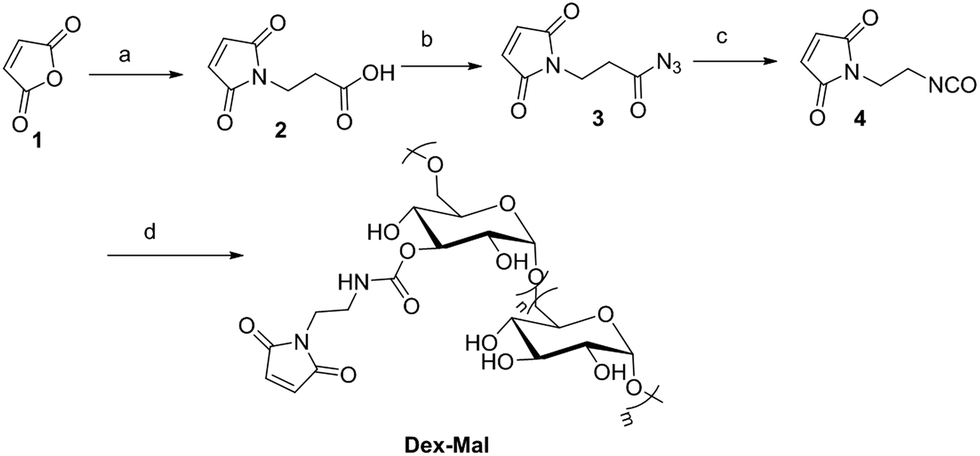
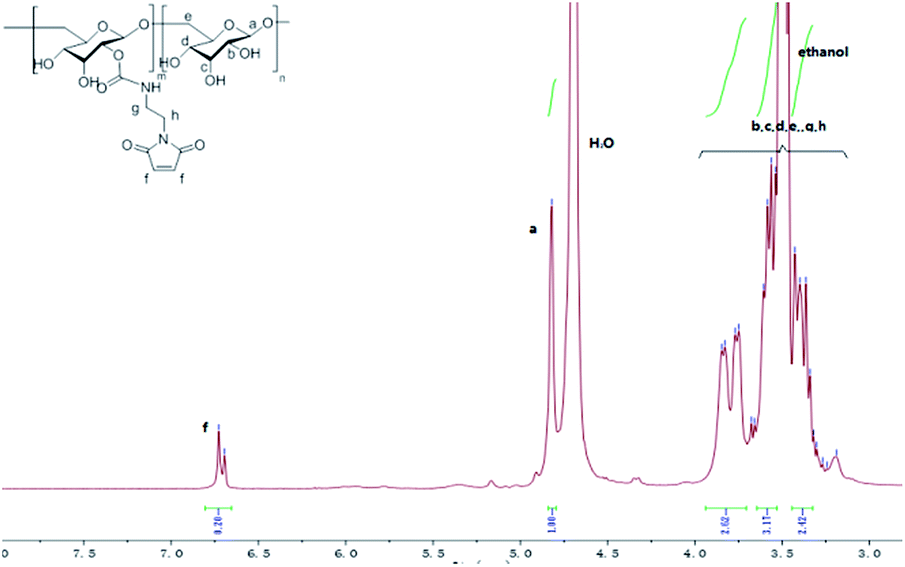
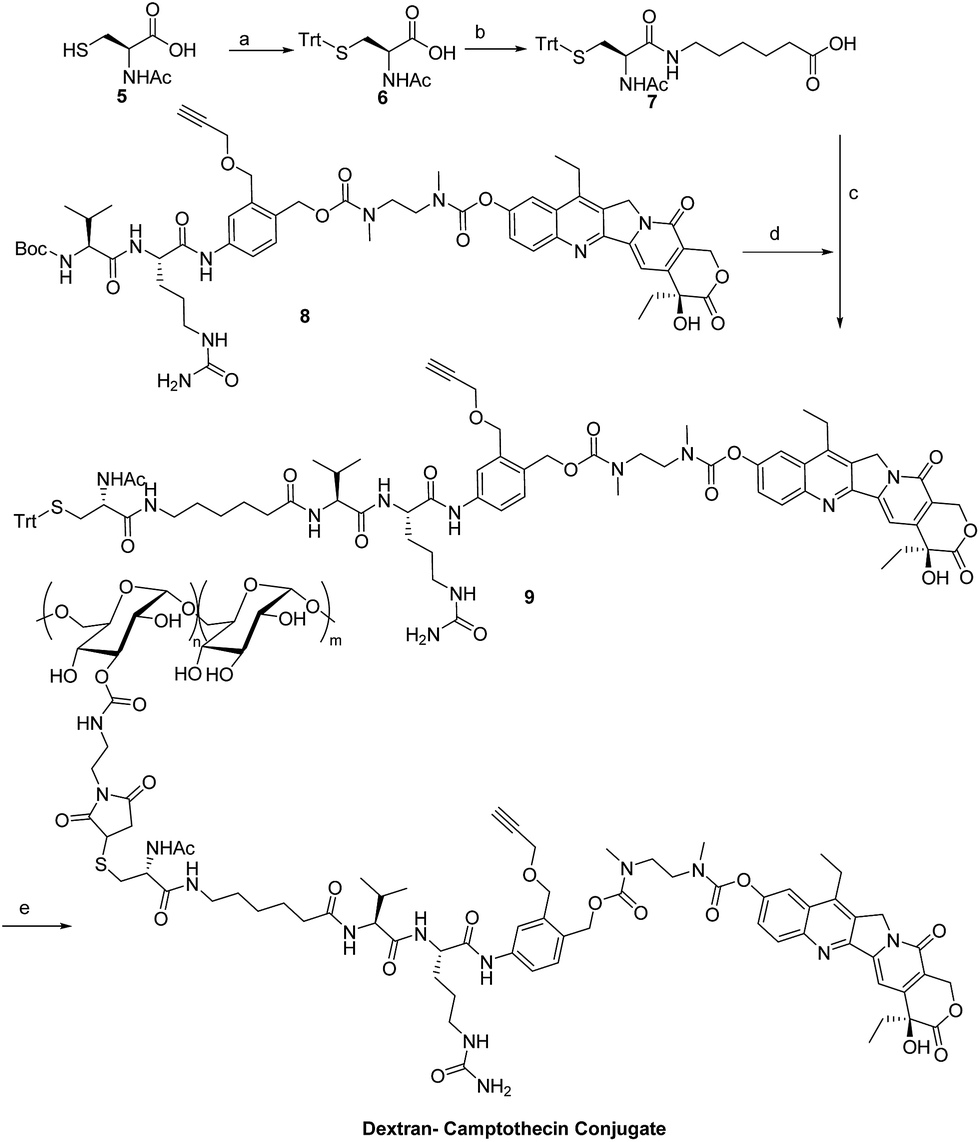
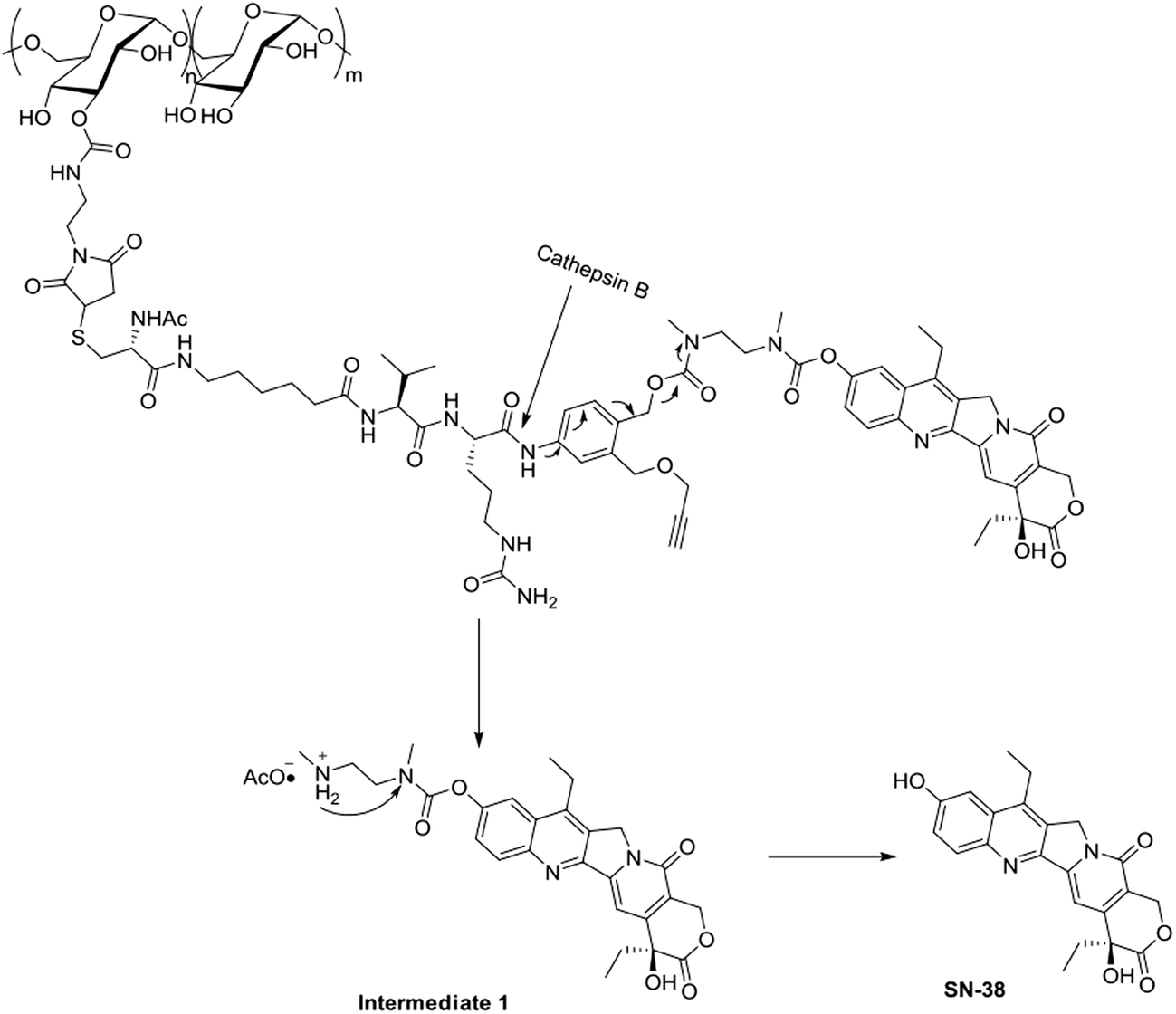
Dex-Mal was synthesized by the reaction of isocyanate and dextran, which is catalyzed by DBTDL (Scheme 1). The graft ratio of maleimide which was defined as the number of maleimide per D-glucose units was calculated by the proton peak areas of maleimide group (f, at about 6.7 ppm) and the proton peak areas of 1-H in the glucose unit (a) of dextran (at about 4.9 ppm) in the 1H-NMR spectrum of Dex-Mal (Fig. 1). From Fig. 1, we were noticed that the graft ratios of maleimide of Dex-Mal were 10%, which means that there was a maleimide group every ten glucose unit in the Dex-Mal. As mentioned in our previous paper,24 compound 8 had been successfully synthesized, a spacer with a sulfydryl terminal was need to attach compound 8 to the polymer. Amidation on the N-terminal of the dipeptide (Val-Cit) is a common strategy and acepramin is a frequently used spacer. Cysteine is an endogenous amino acid, so it was employed as the sulfydryl terminal spacer of compound 7 (Scheme 2). The Boc group of compound 8 was removed successfully but the product was unstable, therefore it was directly treated with the activated intermediate of compound 7 to afford compound 9 which was confirmed by 1H-NMR and HR-MS. Compound 9 was treated with TFA and triethyl silicane to remove the protecting group but it was easily oxidized so it was directly attached to Dex-Mal at the presence of PBS (pH = 7.4, 0.1 N). The final product was purified by dialysis and analyzed by the UV-vis absorption spectrum. Through the UV-vis absorption spectrum, we calculated the graft ratio of SN-38 to the dextran was about 8.3%, and the drug loading rate was about 11.56% in calculation. As all the reaction conditions used in the reaction with dextran were mild, they would not affect the structure of dextran chain, the finial molecular weight of the dextran–camptothecin conjugate is about 35 kDa in calculation. The standard curve was provided in ESI.†
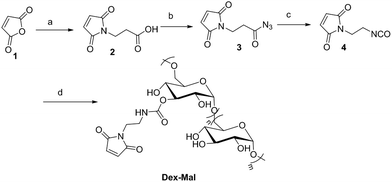 |
| | Scheme 1 Synthesis of Dex-Mal, reagents and conditions: (a) beta-alanine, glacial acetic acid, reflux; (b) anhydrous acetone, TEA, methyl chloroformate, NaN3; (c) toluene, reflux; (d) dextran (T-20), DBTDL, DMSO. | |
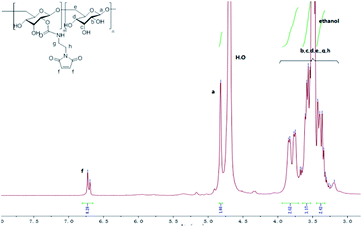 |
| | Fig. 1 The 1H-NMR of the Dex-Mal. | |
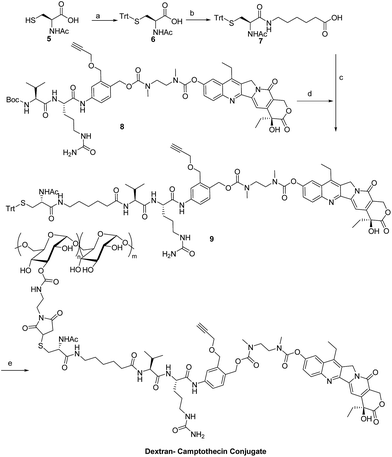 |
| | Scheme 2 Synthesis of the dextran–camptothecin conjugate. Reagents and conditions: (a) TrtCl, DMF; (b) acepramin, HATU, DIEA, DMF; (c) DMF, HATU, DIEA; (d) TFA, DCM; (e) TFA, DCM, NaHCO3, H2O. | |
Water solubility of dextran–camptothecin conjugate
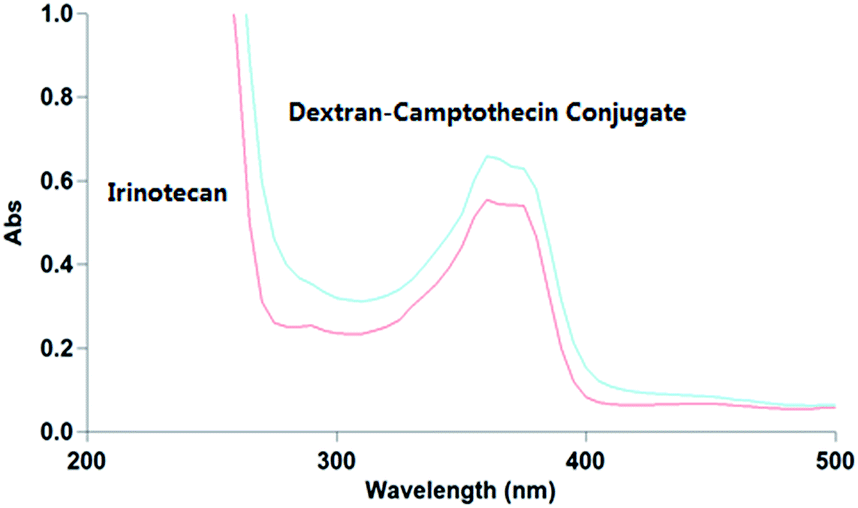
The water solubility of dextran–camptothecin conjugate here was tested with UV-vis absorption spectrum (Fig. 2). As we all know that polymers can be supersaturation when excess amount are dissolved in water. Here we also had the problem, the solution became a gel when excess amount was added to the water, so we increased the amount of conjugate little by little until the solution nearly became a gel. Then we diluted the solution of dextran–camptothecin conjugate and the filtrate of Irinotecan hydrochloride with DMSO about 1000 times. The UV-vis absorption spectrum showed that the conjugate have the similar water solubilities with Irinotecan hydrochloride. As a prodrug, Irinotecan have better water solubility than the parent drug, SN-38, it is a real fact. So our conjugate also has better water solubility than SN-38.
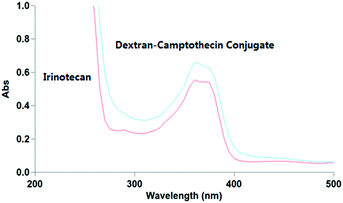 |
| | Fig. 2 The UV-vis absorption spectrum of Irinotecan hydrochloride and dextran–camptothecin conjugate solution. | |
Sizes distribution
In the presence of dextran, the dextran–camptothecin conjugate had a better water solubility than the parent drug SN-38. According to the use of dextran in drug delivery system, the similar structure could self-assembly to micelles, so did the dextran–camptothecin conjugate. The size distribution of the micelles was measured on a dynamic light scattering (DLS) (ZetasizerNano ZS, Malvern Instruments, UK). As shown in Fig. 3, the main hydrodynamic diameter of particles ranged from 91.3 to 295 nm with PDI = 0.356, and the Z-average was about 326.6 nm. The Dex-Mal solution was also measured on a dynamic light scattering, when the concentration was 1 mg mL−1, the main hydrodynamic diameter was ranged from 10.1 to 32.7 nm (PDI = 0.295). The size data suggested the conjugate might have an effective passive targeting potential to tumor tissue owing to EPR effect.
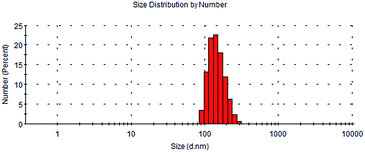 |
| | Fig. 3 The size distribution of dextran–camptothecin conjugate. | |
CTB-activated drug release
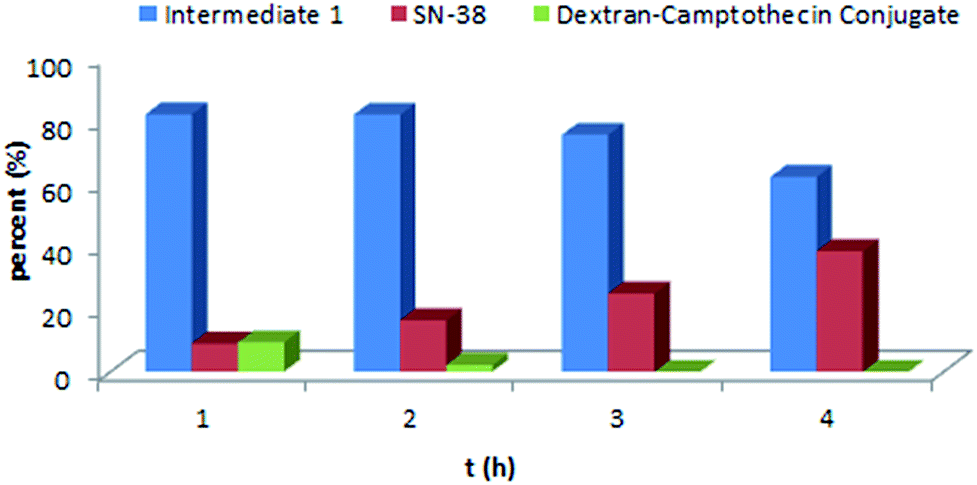
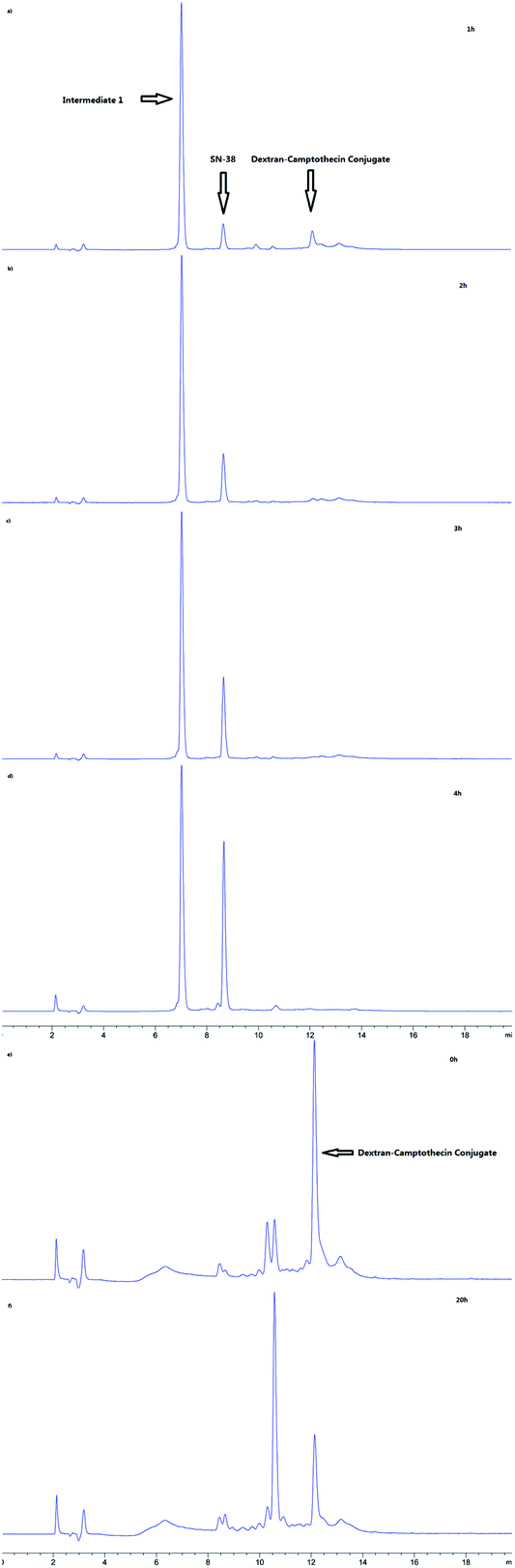
As the Val-Cit and PABOH linkers were used, we predicted the release mechanism of the conjugate (Fig. 4). The enzyme-sensitivity of the dextran–camptothecin conjugate was evaluated at the presence of CTB in acetic acid buffer (pH = 5.0), the process was monitored by HPLC (Fig. 5). Obviously, all the conjugate completely transferred into SN-38 and intermediate 1 which indicated that the releasing mechanism was just as expected (Fig. 6a–d). By contrast, there was no SN-38 and intermediate 1 released without CTB (Fig. 6e and f), however the conjugate gradually disappeared possibly due to the unstability of the maleimide.
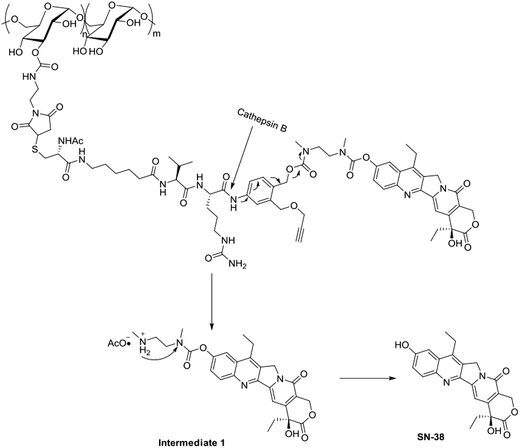 |
| | Fig. 4 The release mechanism of SN-38 from the dextran–camptothecin conjugate. | |
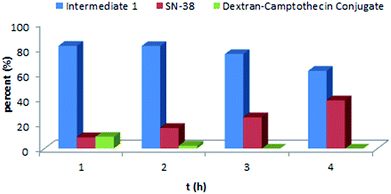 |
| | Fig. 5 Enzyme-sensitivity of the dextran–camptothecin conjugate. | |
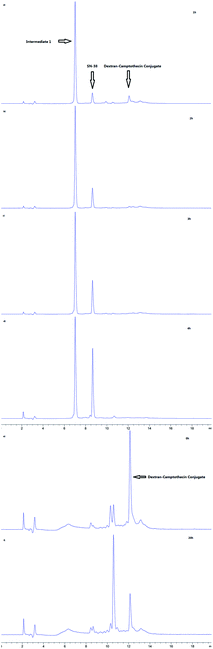 |
| | Fig. 6 HPLC results of the stability of dextran–camptothecin conjugate. (a–d) HPLC results of the stability of dextran–camptothecin conjugate with capthecin B (a, 1 h; b, 2 h; c, 3 h; d, 4 h). (e and f) HPLC results of the stability of dextran–camptothecin conjugate without capthecin B (e, 0 h; f, 20 h). | |
In vitro cytotoxicity evaluation
With the exciting results above, the antiproliferative activity of dextran–camptothecin conjugate was investigated together with Irinotecan on Hela, HepG2 and HCT-116 cell lines. As shown in Table 1, the conjugate had similar cytotoxicity with Irinotecan hydrochloride on all cell lines, and the cytotoxicity against HepG2 cell line was even higher than that of Irinotecan hydrochloride.
Table 1 In vitro cytotoxicity assay (mean ± SD, n = 3)
| Cell line |
IC50a (μM) |
| Hela |
HepG2 |
HCT-116 |
| The concentration was equivalent to SN-38. |
| Irinotecan hydrochloride |
3.66 ± 0.28 |
5.37 ± 0.67 |
0.37 ± 0.05 |
| Dextran–camptothecin conjugate |
3.78 ± 0.11 |
2.11 ± 0.25 |
1.11 ± 0.33 |
Conclusions
In summary, a new dextran derivative Dex-Mal was synthesized and was used in designing the dextran–camptothecin conjugate which contained a CTB-sensitive linker. This conjugate could efficiently release therapeutic drug SN-38 in the presence of cathepsin B. Meanwhile, the conjugate showed similar antiproliferative activity to the approved drug Irinotecan hydrochloride and a similar water solubility. Furthermore, the conjugate could form into particles in water and exhibited a narrow sizes distribution from 91.3 to 295 nm. Compared to the PEG, dextran has many hydroxyl groups which are potential for other modification, and the great biocompatibility, low immunogenicity made dextran a better material than PEG. The maleimidation of dextran showed a helpful way for dextran drug delivery system. With the help of bioorthogonal reaction, the conjugation was more efficiently and mild, and it can be used in a lot of substrates. Therefore, newly developed dextran–camptothecin conjugate, the application of maleimidation of dextran, might be a promising strategy to form an appropriate drug delivery system to overcome the obstacles that traditional anticancer drugs have meet.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support for Shanghai Science and Technology Council (16DZ2280100).
Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 7–30 CrossRef PubMed.
- M. E. Wall, Med. Res. Rev., 1998, 18, 299–314 CrossRef CAS PubMed.
- H. I. Chang and M. K. Yeh, Int. J. Nanomed., 2012, 7, 49–60 CAS.
- J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed.
- Paramjot, N. M. Khan, H. Kapahi, S. Kumar, T. R. Bhardwaj, S. Arora and N. Mishra, J. Drug Targeting, 2015, 23, 387–416 CrossRef CAS PubMed.
- J. Kopeček, P. Kopečková, T. Minko and Z. Lu, Eur. J. Pharm. Biopharm., 2000, 50, 61–81 CrossRef.
- C. Li, Adv. Drug Delivery Rev., 2002, 54(5), 695–713 CrossRef CAS PubMed.
- R. B. Greenwald, J. Controlled Release, 2001, 74, 159–171 CrossRef CAS PubMed.
- K. Greish, T. Sawa, J. Fang, T. Akaike and H. Maeda, J. Controlled Release, 2004, 97, 219–230 CrossRef CAS PubMed.
- J. Varshosaz, Expert Opin. Drug Delivery, 2012, 9, 509–523 CrossRef CAS PubMed.
- R. D. Hamstra, M. H. Block and A. L. Schocket, JAMA, 1980, 243, 1726–1731 CrossRef CAS PubMed.
- Y. Z. Du, Q. Weng, H. Yuan and F. Q. Hu, ACS Nano, 2010, 4, 6894–6902 CrossRef CAS PubMed.
- K. E. Broaders, S. Grandhe and J. M. J. Frechet, J. Am. Chem. Soc., 2011, 133, 756–758 CrossRef CAS PubMed.
- M. Böcher, T. Böldicke, M. Kiess and U. Bilitewski, J. Immunol. Methods, 1997, 208, 191–202 CrossRef.
- R. Axén, J. Porath and S. Ernback, Nature, 1967, 214, 1302 CrossRef.
- E. Dolci, V. Froidevaux, C. Joly-Duhamel, R. Auvergne, B. Boutevin and S. Caillol, Polym. Rev., 2016, 56, 512–556 CrossRef CAS.
- A. M. Sochaj, K. W. Świderska and J. Otlewski, Biotechnol. Adv., 2015, 33, 775–784 CrossRef CAS PubMed.
- S. G. Lévesque and M. S. Shoichet, Bioconjugate Chem., 2007, 18, 874–885 CrossRef PubMed.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2015, 116, 1309–1352 CrossRef PubMed.
- M. K. Houseweart, L. A. Pennacchio, A. Vilaythong, C. Peters, J. L. Noebels and R. M. Myers, Dev. Neurobiol., 2003, 56, 315 CrossRef CAS PubMed.
- A. T. Chan, Y. Baba, K. Shima, K. Nosho, D. C. Chung, K. E. Hung, U. Mahmood, K. Madden, K. Poss and A. Ranieri, Cancer Epidemiol., Biomarkers Prev., 2010, 19, 2777–2785 CrossRef CAS PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855–869 CrossRef CAS PubMed.
- B. Bao, Y. Liu, L. Wang and W. Lu, RSC Adv., 2016, 6, 69540–69545 RSC.
- X. Zhang, K. Tang, H. Wang, Y. Liu, B. Bao, Y. Fang, X. Zhang and W. Lu, Bioconjugate Chem., 2016, 27, 1267–1275 CrossRef CAS PubMed.
- R. J. Sanderson, M. A. Hering, S. F. James, M. M. Sun, S. O. Doronina, A. W. Siadak, P. D. Senter and A. F. Wahl, Clin. Cancer Res., 2005, 11, 843 CAS.
- D. C. Alberto, R. Gébleux, P. Murer, A. Soltermann and D. Neri, J. Controlled Release, 2017, 264, 211–218 CrossRef PubMed.
- L. Christina, C. B. Pascale, N. R. Marie, P. Pascal, M. M. Monique and M. M. Jean, Org. Biomol. Chem., 2011, 9, 2209 Search PubMed.
- V. Pichler, J. Mavr and P. Heffeter, et al., Chem. Commun., 2013, 49, 2249–2251 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectrum of the corresponding compounds. See DOI: 10.1039/c7ra12954h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*