
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into iron induced fouling of ion-exchange membranes revealed by a quartz crystal microbalance with dissipation monitoring†
Mei Chena,
Jinxing Ma *b,
Zhiwei Wanga,
Xingran Zhanga and
Zhichao Wua
*b,
Zhiwei Wanga,
Xingran Zhanga and
Zhichao Wua
aState Key Laboratory of Pollution Control and Resources Reuse, College of Environmental Science and Engineering, Tongji University, Shanghai, 200092, PR China
bSchool of Civil and Environmental Engineering, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: jinxing.ma@unsw.edu.au; Tel: +61 2 93859501
First published on 24th July 2017
Abstract
Understanding the mechanisms of multivalent iron interacting with ion-exchange membranes (IEMs) is crucial for the prediction of membrane fouling as well as the development of control strategies. In this study, the adsorption and desorption behaviors of Fe(III) species on a typical IEM, Nafion, were investigated using a quartz crystal microbalance with dissipation monitoring (QCM-D). The Nafion thin film formed on the crystal sensor surface via a sedimentation method showed a non-rigid structure with a Voigt-based mass concentration of ∼500 ng cm−2 at a Nafion solution injection time of 10 min. Adsorption of Fe(III) species was first assessed using a 10 mM Fe(III) solution, followed by rinsing under different conditions to induce the structural transformation and/or release of Fe(III) from the Nafion film. The QCM-D results suggested that there was a rapid deposition of Fe(III) at the initial stage. It has been found that the ongoing adsorption process exhibiting pseudo-first-order kinetics was associated with the interaction between Fe(III) and the surface functional sites (–SO3H) of Nafion, which consequently retarded the proton transfer. Compared to the rinse with an acidic solution (HCl in ultrapure water, pH of 2.37), the QCM-D results of neutral elution (ultrapure water, pH of 6.50) indicated the hydrolysis of Fe(III) and/or structural transformation of the μ-oxo bridged, Fe–O–Fe.
1. Introduction
Ion-exchange membranes (IEMs) are insoluble matrices generally made from polymeric materials attached to charged ion groups with a preference to conduct dissolved cations or anions (e.g., Na+ and Cl−) whilst being much less permeable to gases and electrolytes.1 The conventional applications of IEMs lie in the use of an electrical potential or concentration gradient to selectively transport cations or anions. For example, when applied in chlor-alkali production, IEMs are typically arranged between the electrodes with a galvanic potential provided as a voltage generated at the electrodes. An emerging interest in IEMs is the incorporation into water/wastewater treatment technologies with more recent innovations including microbial fuel cells (MFCs), microbial desalination cells (MDCs), capacitive deionization (CDI) and capacitive mixing (CapMix).2–6Despite the prominent progress in environmental applications of IEMs, crucial concerns remain with regard to (i) the non-target ion transfer and (ii) fouling that leads to the deterioration of the membrane performance.7–9 For instance, it has been well recognized that the metal cations are of great abundance in the substrates of MFCs and MDCs, which can transfer across the IEMs as non-target ions though some IEM types possess a preferential conductivity for protons such as 3.4 for H+/Na+ and 6.2 for H+/K+ in Nafion 117.10 The influences of non-proton transfer are profound as this would result in (i) pH excursion inhibiting the biocatalysts activity and (ii) the loss of thermodynamic cell potential.7,8,11 Moreover, it has been reported that the movement of multivalent cations and/or their polynuclear species across the IEMs can further cause strong interaction with the charged groups (e.g., sulfonic acid groups (–SO3H)),7,9 which retards the “hop” of subsequent ions from one site to the adjacent sites and consequently increases the internal resistance.12
Iron (Fe) is an important element involved in metabolism and redox chemistry. In surface and ground waters, Fe presents at varying concentration levels, ranging from 0.5 to 50 mg L−1.13 In the anode chambers of MFCs and MDCs, the origin of aqueous Fe(II) and Fe(III) can be associated with the reductive dissolution of Fe-(oxyhydr)oxides in addition to the authigenic dissolved species. Moreover, Fe(II)/Fe(III) can be directly employed as an electron mediator.14 When using IEMs in water/wastewater treatment, considerations should be given to the interfacial behaviors of Fe species on IEMs since recent evidence has indicated that Fe(III) species are related with the mineral precipitation on IEMs with their further interaction with macromolecules resulting in biofouling that deteriorates the process performance.8,9 Although previous studies using K-edge extended X-ray absorption fine structure (EXAFS) spectroscopy have implied that an oxo-bridged dimer, Fe–O–Fe, dominates the Fe speciation of the Fe(III) neutralized membranes (Nafion 125 IEM) followed by a structural transformation to edge-sharing FeO6 octahedra,15,16 there are still many characteristics of this process that remain unclear. For example, (i) what kind of behavior is prevailing during the adsorption and/or precipitation of Fe(III) on IEMs? (ii) How is this process influenced by the environmental factors (e.g., pH)? (iii) Will this lead to significant impacts on the performance of IEMs?
In this study, we have investigated the nature of iron induced fouling of a commercial IEM, Nafion 117, by using a quartz crystal microbalance with dissipation (QCM-D). QCM-D has been suggested as a useful method to obtain information on adherence and fouling propensity of foulants on membrane surfaces.17–19 Coating of Nafion film on gold-plated crystal sensors was conducted via a sedimentation method. The overarching goals of this work are, therefore, to (i) form Nafion thin film on crystal sensor surface, (ii) elucidate the behaviors of Fe(III) on the resultant film and (ii) evaluate the impacts of Fe precipitation on the membrane properties. An adequate understanding of the Fe induced fouling mechanisms of IEMs is expected to be conducive to advancing control strategies for IEM-related processes.
2. Materials and methods
2.1. Reagents
All chemicals were analytical reagent grade and used as received unless otherwise stated. All solutions were prepared using 18.2 MΩ cm Milli-Q water (Millipore). Glassware was soaked in a 5% v/v HCl for at least three days and rinsed prior to use. Nafion® 117 membrane (Product no. 274674) and perfluorinated resin solution (Nafion solution, Product no. 274704) were purchased from Sigma-Aldrich. 1 M NaOH and 1 M HCl were used for the adjustment of solution pH when necessary. Stock solution of 10 mM ferric chloride hexahydrate (FeCl3·6H2O) was prepared for the following experiments, and according to the calculation using an equilibrium speciation program Visual Minteq, dominant Fe(III) species are Fe3+ (39.2%), FeOH2+ (32.9%), Fe2(OH)24+ (14.7%) and FeCl2+ (10.5%) at the equilibrium pH of 2.37.2.2. Quartz crystal microbalance with dissipation monitoring
In this study, a quartz crystal microbalance with dissipation monitoring (QCM-D, E4, Q-Sense, Västra Frölunda, Sweden) system was used to monitor the sedimentation of Nafion on the crystal sensor surface and to investigate Fe(III) induced fouling of Nafion. The E4 system has four modules that allow a parallel configuration, and each module holds a 5 MHz AT-cut quartz crystal sensor. Gold-plated crystal sensors (Q-Sense) were utilized for the following studies. Prior to use, the crystal sensors were subject to the cleaning protocols documented by the QCM-D manufacturer, with the flow modules cleaned with a 2.0% sodium dodecyl sulfate (SDS) solution followed by rinse with copious amount of ultrapure water. Prepared solutions were degassed through ultrasonication for 10 min and all experiments were performed at a constant flow rate of 150 μL min−1 using a peristaltic pump (IsmaTec, IDEX). Solution temperature inside the chamber was maintained at about 25 °C.2.3. Sedimentation of Nafion on QCM-D crystal sensors
At the initial stage of the experiment, the frequency and dissipation responses of the crystal sensors at n overtone (n = 3, 5, 7, 9 and 11) were monitored whilst the system was rinsed with ultrapure water (pH = 6.50) for stabilization (step I in Fig. 1). Stable baseline was considered to be achieved when the drift of normalized frequency signals at the 5th overtone (i.e., Δf5), which was chosen based on the best signal-to-noise ratio, was decreased below 0.5 Hz in a period of 10 min. Subsequently the Nafion solution was introduced into the modules for the sedimentation of Nafion on crystal sensors (step II in Fig. 1), resulting in the decrease in the frequency (i.e., an increase of |Δf|) due to the change of solution properties and/or adsorption of Nafion on the surface of crystal sensors. The sensors were then rinsed with ultrapure water (step III in Fig. 1) until stable frequency and dissipation signals of the 5th overtone were achieved.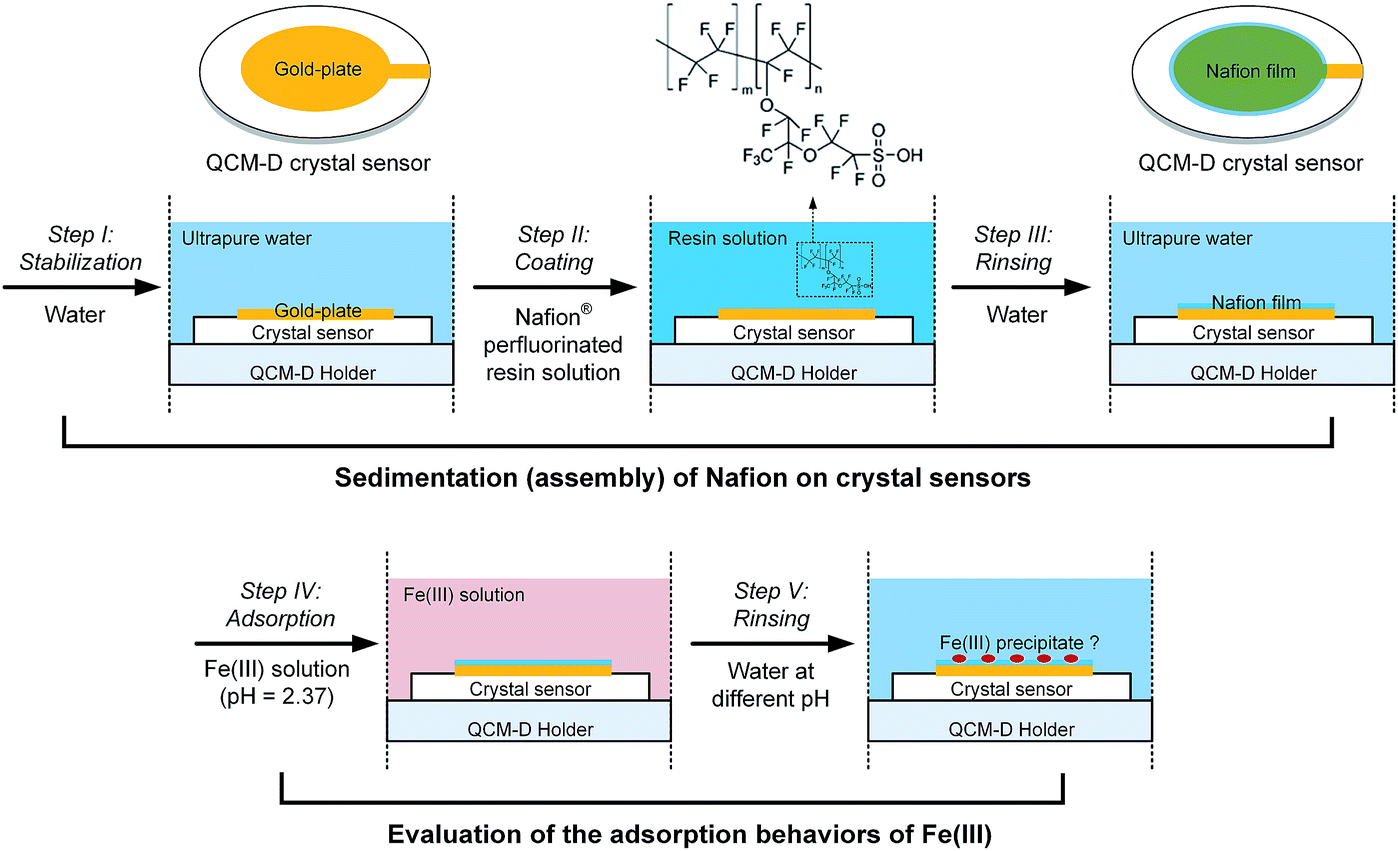 | ||
| Fig. 1 Experimental workflow for the self-assembly of Nafion film on QCM-D crystal sensors and evaluation of the interfacial behavior of Fe(III) species. | ||
The Voigt-based model20,21 was introduced herein to quantify the mass concentrations of Nafion on the QCM-D crystal surfaces. Briefly, the normalized frequency shifts (Δf) and dissipation shifts (ΔD) at the 3rd, 5th, 7th, 9th and 11th overtone collected from the experiments were used for model fitting using the Q-Tools 3.0 (Q-Sense), obtaining the surface concentration (mass and thickness), viscosity and shear modulus of the Nafion film as a function of time. The fluid density and viscosity of the Nafion solution were fixed at 0.87 × 103 kg m−3 and 6.0 mPa s according to the manufacturer,22 and those parameters for other solutions including ultrapure water and electrolyte were fixed at 1.00 × 103 kg m−3 and 1.0 mPa s, respectively. According to a previous work on water-sorption properties of Nafion 117H,23 the density of Nafion layers was about 1.10 × 103 kg m−3, which was employed for the Voigt-based model fitting in this study.
2.4. Evaluation of adsorption of Fe(III)
Following the assembly and stabilization of the Nafion film, a 10 mM Fe(III) solution was injected into the sensor modules for 20 min (step IV in Fig. 1), with control tests carried out under the same conditions but using pristine gold-plated crystal sensors. These sensors were then subject to rinse with ultrapure water (pH of 6.50) or acid solution (HCl in ultrapure water, pH of 2.37), respectively (step V in Fig. 1). The normalized frequency signals of the Nafion-coated crystals were obtained by deducting the initial Δf of Nafion film. All experiments were conducted in triplicate with average value reported.The relationship between the QCM-D results and Nafion membrane performance was further evaluated by measuring the increase in membrane ohmic resistance as a result of Fe(III) fouling. An IEM of the same material, Nafion® 117 membrane, was used to evaluate the consequent membrane performance after fouling. Briefly, 2 cm × 2 cm Nafion coupons were soaked in the vessels containing 100 mL of 10 mM Fe(III) solution on an orbital shaker table at 150 rpm. Membranes were withdrawn at pre-determined time intervals. To evaluate the impacts of Fe(III) hydrolysis and/or structural transformation on Nafion performance, parallel experiments were conducted according to the procedure described above, except for the rinse of the withdrawn coupons with copious amount of ultrapure water (pH of 6.50). Ohmic resistances of the membranes (Rm) were measured according to a protocol outlined previously.9 10 mM Fe(III) was added into the working electrolyte to avoid the possible desorption of Fe(III) from the IEMs during the measurement in 0.1 M KCl solution.
2.5. Other analytical methods
The equivalent weight (EW) of the Nafion-H type materials was determined to be 1100 ± 21 by back titration of the hydrogen ion that was exchanged from Nafion with 2 M NaOH solution over 24 hours at ambient temperature (∼25 °C). Ion exchange capacity (IEC) was calculated as the reciprocal of EW, i.e., IEC = 1/EW.7 To evaluate the relationship between the loss of IEC and aqueous pH, pre-weighed Nafion 1100EW membranes were first soaked in ultrapure water that was added with 1 M HCl or 1 M NaOH solution to obtain a series of initial pH from 2.83 to 11.41. After either 2 or 24 h of reaction, membranes were gently rinsed with copious amount of ultrapure water and then IEC were measured according to the protocol described above.Functional groups of the pristine and fouled Nafion membranes were analysed on an attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy. The ATR-FTIR spectra were collected using a Nicolet 5700 spectrometer (Thermo Electron Co., U.S.) at a 4 cm−1 resolution in the range of 400 to 4000 cm−1. Prior to the measurement, the surface of the membranes were dried with the use of lens wiping paper following withdrawal from the solution. The membrane topography was also characterized by the atomic force microscope (AFM) technology (Multimode IV, Bruker Nano Surface, USA) with the Nanoscope® control software used for image acquisition.
3. Results and discussion
3.1. Characteristics of the Nafion film
Fig. 2a shows the time course results of Δf5 and ΔD5 during the sedimentation (assembly) of Nafion films on the gold-plated crystal sensors. Injection of the Nafion solution led to a decrease in Δf5 (i.e., an increase in |Δf5|) (step II in Fig. 1) as a result of (i) the change of the properties of the fluid and/or (ii) the deposition of Nafion. It can be observed that the steady state Δf5 following the injection of the Nafion solution was not increased with prolonging the injection time, suggesting that a dynamic adsorption balance could be established in a relatively short injection time.20 The subsequent rinse with ultrapure water (step III in Fig. 1) significantly decreased the resonance frequencies (i.e., a dramatic decrease in |Δf5|). As can be seen from Fig. 2a, it is likely that a longer injection time for step II (Fig. 1) is in favor of the interaction between the Nafion and crystal surface: i.e., following the rinse, the residual Δf5 (−16.7 Hz) at an injection time of 10 min is similar with that of 20 min (−17.0 Hz) whilst exhibiting a much higher absolute value than that of 5 min (−7.9 Hz) (the inset figure of Fig. 2a). The time-resolved behavior of ΔD5 was comparable with that of Δf5; after the rinse with ultrapure water for 1000 s, the crystal sensors showed residual ΔD5 of 2.6, 5.0 and 4.9 × 10−6 at an initial injection of Nafion solution for 5, 10 and 20 min, respectively.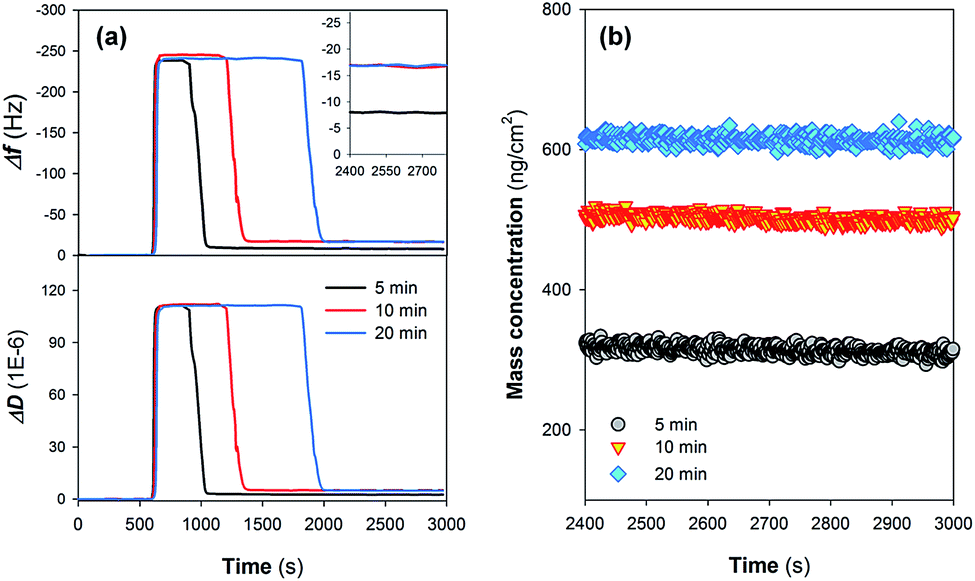 | ||
| Fig. 2 Time course results of (a) representative frequency shift (Δf5) and dissipation shift (ΔD5) describing the assembly of Nafion films on crystal sensors. Residual Δf5 of the films following rinse with ultrapure water (step III in Fig. 1) are specifically shown in the inset. (b) Mass concentrations of the Nafion films as a function of Nafion injection time. | ||
After stabilization of the residual response signals (following step III in Fig. 1), Δf and ΔD at the 3rd, 5th, 7th, 9th and 11th overtone were collected from the experiments and utilized for model fitting. Despite the injection of Nafion solution for 10 and 20 min resulting in similar residual Δf5 (Fig. 2a), the Voigt-based mass concentrations of Nafion films were different, suggesting that the deposited Nafion did not form a homogeneous rigid layer on the crystal surface (Fig. 2b). Moreover, the effects of the rinsing process on the properties of Nafion films were evaluated by quantifying the loss of IEC of the Nafion material as a function of the solution pH. Fig. S1 in the ESI† shows that the loss of IEC is not significant in the pH range of 2.0–8.0, implying that the rinsing process using ultrapure water (pH = 6.50) should not change the acid sites of the Nafion film.
An interpretation of the ΔD/Δf from QCM-D results can provide insights into the viscoelastic properties of the adsorbed layers. As can be seen from Fig. 3a, |ΔD5/Δf5| values for all the three kinds of Nafion films are >0.2 × 10−6. Upon rinsing with ultrapure water, |ΔD5/Δf5| values were all decreased. The large decrease in energy dissipation (Fig. 2a) is typical for a structural transformation from a dissipative non-rigid structure to a stiffer and most likely more compact structure, as observed in several earlier model systems.24–26 It is generally expected that the assembly of heterogeneous Nafion film on the crystal sensors might begin with the interaction between the hydrophobic body parts of Nafion and crystal surface that leads to the formation of an adsorptive monolayer,27 followed by the further decoration of the undeformed Nafion resin as a result of the affinity and steric hindrance (Fig. 3b). After rinse, the loosely-adsorbed Nafion were likely removed from the film with partial leaching of the water-soluble LAA.28 As such, the layer became more rapid (lower |ΔD5/Δf5| values).29
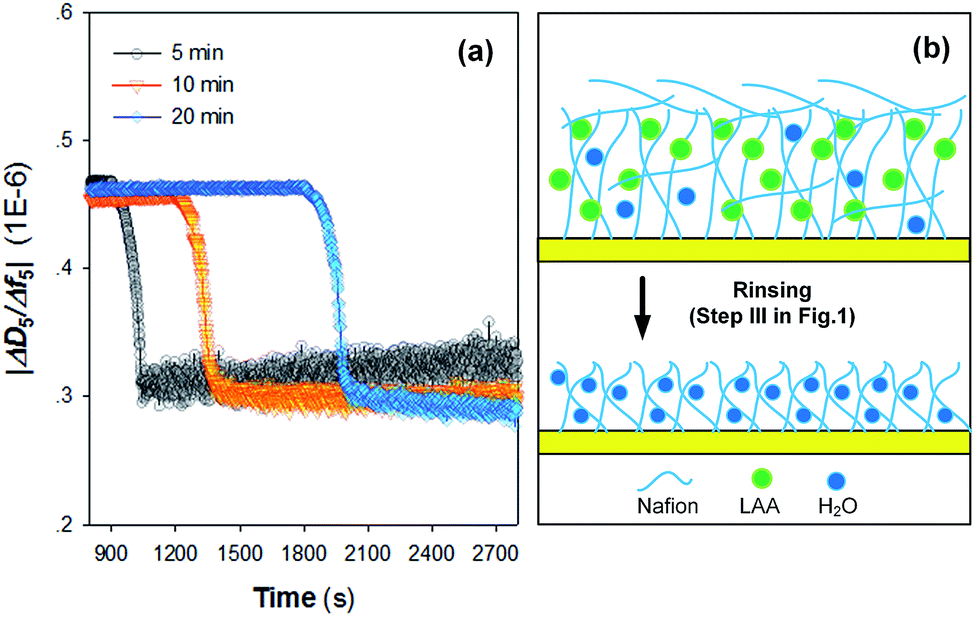 | ||
| Fig. 3 (a) Time course results of |ΔD5/Δf5| changes during assembly of Nafion films on crystal sensors as a function of Nafion injection time; (b) schematic presentation of the structural transformation of the Nafion film upon rinsing with ultrapure water. LAA represents lower aliphatic alcohols. | ||
Fig. 3a suggests that the injection time also affects the variation of viscoelastic properties of the Nafion film, i.e., an injection time of Nafion solution over 10 min resulted in a more rigid film after rinsing. In view of the obtained results of this study, a Nafion injection time of 10 min was finally chosen for the assembly of Nafion film on the QCM-D crystal sensors.
3.2. Adsorption behaviors of Fe(III) species
Fig. 4 shows the adsorption and desorption behaviors of Fe(III) species on different surfaces. Two pH values (i.e., pH = 2.37 and 6.50) were evaluated in this study as they are typical for electrochemical systems operated in acidic and circumneutral conditions. It can be observed that the magnitude of frequency changes (Δf5) is highly associated with the nature of the sensor surfaces; i.e., after the injection of 10 mM Fe(III) for 20 min (step IV in Fig. 1), Δf5 were changed by −15.1 and −20.1 Hz on the gold-plated crystal and Nafion film surface respectively, indicating the stronger interaction between Nafion and Fe(III) species. Specifically, a sharp increase in |Δf5| was noticed initially on the Nafion film (Fig. 4) followed by a process exhibiting pseudo-first-order growth with an apparent rate constant (kapp) of 0.0017 s−1 (details could be found in Fig. S2 in the ESI†), attributed to the limited surface site number of Nafion and the constant Fe(III) concentration. Release behaviors of Fe(III) species from the pristine crystal surface and Nafion film were then evaluated at different pH. When rinsed with neutral ultrapure water (Fig. 4a and b), |Δf5| of the Nafion film underwent a transient decrease followed by a notable jump and subsequent decline over time. In comparison, the jump of |Δf5| was not noticed during the rinsing process on the gold-plated crystal surface. Furthermore, it can be noted from Fig. 4c that continuous decline of |Δf5| occurred when the Fe(III) bound Nafion film subject to acidic elution (pH of 2.37), with the observation of a rapid mass loss followed by a pseudo-first-order decay giving kapp of 0.0007 s−1.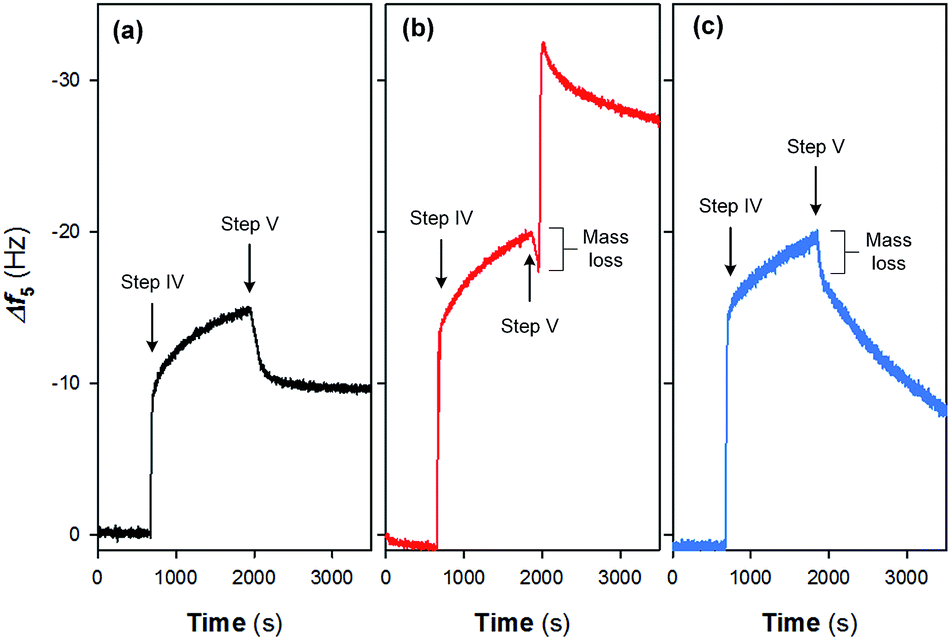 | ||
| Fig. 4 Interfacial behaviors of Fe(III) species on (a) pristine crystal surface followed by the rinse with ultrapure water (pH = 6.50) and Nafion film surface followed by the rinse with (b) ultrapure water and (c) acid solution (pH = 2.37). Experimental conditions: injection time for step IV = 20 min; [Fe(III)] = 10 mM. | ||
In environmental applications of the IEMs, although it is acknowledged that the interaction of Fe(III) species from the electrolyte with the functional groups (e.g., –SO3H) leads to serious consequences such as the inhibition of ion transfer and increase in system resistance,7,8 synthesis of the adsorption behaviors of Fe(III) on IEM surfaces is still scarce. Based on the obtained results of this study and past literature,15,16 the following reaction pathways are therefore proposed (as shown schematically in Fig. 5):
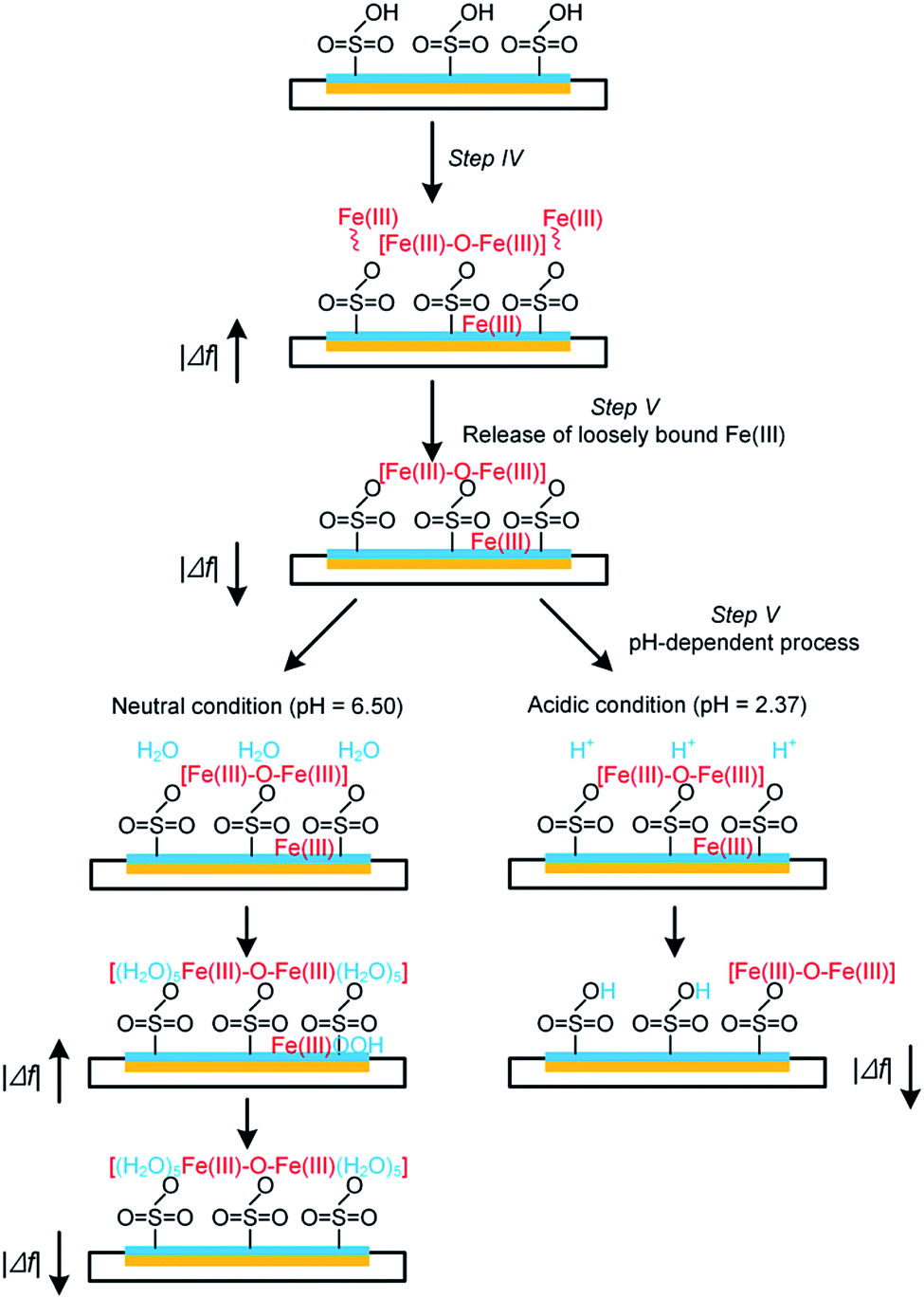 | ||
| Fig. 5 A schematic presentation of Fe(III) interfacial behaviors on Nafion film surface at different pH. | ||
Initially, a heterogeneous fouling layer is likely developed on the surface with exposure to water/wastewater containing Fe(III). The formation of Fe(III) fouling can be associated with (i) the loosely binding of Fe(III) species on Nafion film surface, (ii) iron exchange of Fe(III) with –SO3H groups and/or (iii) trapping of Fe(III) species (e.g., Fe-(oxyhydr)oxides) in the cluster-channel.29 As can be seen from Fig. 4b and c, upon elution under either neutral or acidic condition, a rapid mass loss occurs at the initial stage of step V (Fig. 1), which suggests the ease of removing loosely bound Fe(III) species on the boundary of the fouling layer as a result of the back-diffusion. After this, release of Fe(III) exhibits a pH-dependent manner (Fig. 5): i.e., under the neutral condition, hydrolysis of the bound and/or trapped Fe(III) is generally expected due to the low solubility product of FeOOH.30 As such, a jump of |Δf5| is observed in Fig. 4b. In contrast, Fe(III) species could readily be removed from the Nafion surface under the acidic condition. The lower kapp (0.0007 s−1) for the release process compared to the Fe(III) adsorption process (0.0017 s−1) implied the strong interaction between Fe(III) and sulfonic groups. With prolonging of the rinsing time, decline of |Δf5| was also noted on the Nafion film under the neutral condition. Plausible explanations for this phenomenon might include (i) the leaching of the precipitates in the channel and/or (ii) the fretting of the Nafion film.
3.3. Correlation of the QCM-D results with Nafion membrane performance
The relationship of the QCM-D results with Nafion membrane performance was further evaluated by measuring the changes of functional groups and ohmic resistance of Nafion membranes as a result of Fe(III) deposition. Fig. 6a provides the ATR-FTIR spectra of pristine Nafion 117 film and the fouled one upon exposure to 10 mM Fe(III) solution for 60 min. Although the two kinds of membranes have similar ATR-FTIR bands such as shoulder peaks at 970 and 982 cm−1 assigned to C–O–C linkages, symmetric SO3− stretching vibration at 1056 cm−1 and shoulder peaks at 1146 and 1203 cm−1 representing symmetric and asymmetric CF2 vibrations,31,32 a major difference is the appearance of Fe–O–Fe antisymmetric stretching mode of the μ-oxo bridged dimer at 870 cm−1 on the Fe(III) fouled membrane.15 Fig. S3 in the ESI† shows the changes in the membrane morphology following the binding of Fe(III) species. Small bumps were observed on the surface of the fouled Nafion 117 membrane with the average roughness increased.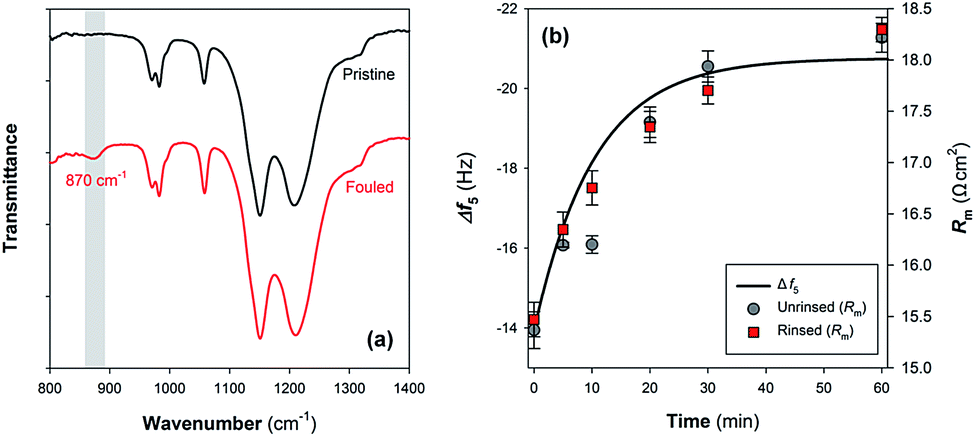 | ||
| Fig. 6 (a) ATR-FTIR spectra of the pristine Nafion 117 film and the fouled one upon exposure to 10 mM Fe(III) solution for 60 min. (b) Kinetics modeling of Fe(III) adsorption on the Nafion film and its prediction on membrane ohmic resistance (Rm). In (b), the grey circles represent the Rm of Nafion membrane measured following the exposure to 10 mM Fe(III) solution without rinse, and the red squares the Rm measured after the rinse with copious amount of ultrapure water (pH of 6.50). | ||
As can be seen from Fig. 6b, the membrane ohmic resistance (Rm) is increased from 15.4 ± 0.2 Ω cm2 to 18.2 ± 0.1 Ω cm2 upon fouling in 10 mM Fe(III) solution for 60 min. Considering that the initially rapid jump of |Δf5| that might be associated with (i) the change of the properties of the fluid and/or (ii) loosely binding of Fe(III) species on Nafion film surface did not cause a transient variation of ohmic resistance (Fig. 4b and 6b), part of |Δf5| exhibiting a pseudo-first-order growth (Fig. S2 in the ESI†) was then used to predict the time course results of Rm. Fig. 6b shows that the interaction between aqueous Fe(III) and Nafion surface functional groups should be responsible for the deterioration of membrane performance. A similar conclusion could be drawn in view of the change of specific resistance (ρm) of the membranes (Fig. S4 in the ESI†). The QCM-D results can provide a reasonable interpretation for the temporal change of Rm.
Furthermore, it has been reported that Fe(III) precipitation due to pH variation is an important factor contributing to inorganic and complex fouling in membrane filtration processes.33,34 While hydrolysis of Fe(III) and/or structural transformation of μ-oxo bridged dimer to edge-sharing FeO6 octahedra are generally expected when providing neutral elution (Fig. 4b and 5), this seems not to cause a further increase of Rm: i.e., there is no significant difference between the measured results using unrinsed and rinsed samples (Fig. 6b). As such, we can preliminarily conclude that on condition that Nafion surface sites are occupied by multivalent metal ions, the membrane conductivity might not be lowered due to the structural transformation of these species, despite the reservation remaining that crystallization of Fe(III) can cause changes of membrane surface physicochemical properties (e.g., roughness) resulting in possible interaction with other foulants and deterioration of membrane performance.9,35
In the present work, we present a novel approach to analyze the chemical scaling on IEMs. Membrane thin films can be formed on the crystal surfaces via sedimentation and/or spin-coating methods,36 with the potentially expanded applications in electrochemical membrane process including (i) the measurement of molecular adsorption and/or interactions taking place on the surface (ii) development and evaluation of novel, antifouling conductive materials. Compared to the ex situ characterizing technologies (e.g., SEM and ATR-FTIR), QCM-D could provide a real-time analysis of the interfacial behavior of ions and biopolymers on the IEM surface with the interpretation of ΔD and Δf revealing the viscoelastic properties of the fouling layers.25,37 Consideration could also be given to the effects of different environmental parameters (e.g., temperature and ionic strength) on the fouling behavior and performance of IEMs under real conditions. Moreover, of particular interest is a recent advance of Q-Sense module, QEM 401 that enables simultaneous QCM-D and electrochemical impedance spectroscopy measurements.37 There is an illumination that QCM-D experiments can provide real-time information on structure and interfacial charge transfer of thin films in the presence of electric field, from which important rate coefficients can be retrieved and used for (i) the modelling and/or (ii) prediction of the transport of concerned foulants on conductive membranes in future research.
4. Conclusions
In this study, Nafion thin film was successfully formed on the crystal surface using a sedimentation method. The Nafion film exhibited a non-rigid structure after the rinse, with a Voigt-based mass concentration of ∼500 ng cm−2 at a resin injection time of Nafion solution for 10 min. The QCM-D results evidenced the adsorption behaviors of Fe(III) on Nafion film surface: i.e., a transient deposition of Fe(III) likely occurred at the initial stage, followed by a pseudo-first-order ongoing process probably as a result of the interaction between Fe(III) and Nafion surface sites that retarded the proton transfer consequently. Although the variation pattern of Δf5 upon neutral elution indicated the hydrolysis of Fe(III) and/or structural transformation of μ-oxo bridged dimer, the electrochemical measurements indicated that this did not cause a further deterioration of Nafion performance.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is financially supported by National Natural Science Foundation of China (51378371 & 51422811) of China. Dr Jinxing Ma is a recipient of the Vice-Chancellor's Postdoctoral Research Fellow of UNSW Australia (RG152482).References
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4611 CrossRef CAS PubMed.
- X. Cao, X. Huang, P. Liang, K. Xiao, Y. Zhou, X. Zhang and B. E. Logan, Environ. Sci. Technol., 2009, 43, 7148–7152 CrossRef CAS PubMed.
- S. Porada, R. Zhao, A. Van Der Wal, V. Presser and P. Biesheuvel, Prog. Mater Sci., 2013, 58, 1388–1442 CrossRef CAS.
- S. Porada, D. Weingarth, H. V. M. Hamelers, M. Bryjak, V. Presser and P. M. Biesheuvel, J. Mater. Chem. A, 2014, 2, 9313–9321 CAS.
- Z. Wang, J. Ma, Y. Xu, H. Yu and Z. Wu, J. Power Sources, 2013, 235, 280–288 CrossRef CAS.
- J. Ma, D. He, W. Tang, P. Kovalsky, C. He, C. Zhang and T. D. Waite, Environ. Sci. Technol., 2016, 50, 13495–13501 CrossRef CAS PubMed.
- J. Xu, G.-P. Sheng, H.-W. Luo, W.-W. Li, L.-F. Wang and H.-Q. Yu, Water Res., 2012, 46, 1817–1824 CrossRef CAS PubMed.
- M.-J. Choi, K.-J. Chae, F. F. Ajayi, K.-Y. Kim, H.-W. Yu, C.-w. Kim and I. S. Kim, Bioresour. Technol., 2011, 102, 298–303 CrossRef CAS PubMed.
- J. Ma, Z. Wang, D. Suor, S. Liu, J. Li and Z. Wu, J. Power Sources, 2014, 272, 24–33 CrossRef CAS.
- I. A. Stenina, P. Sistat, A. I. Rebrov, G. Pourcelly and A. B. Yaroslavtsev, Desalin. Water Treat., 2004, 170, 49–57 CAS.
- F. Harnisch, U. Schröder and F. Scholz, Environ. Sci. Technol., 2008, 42, 1740–1746 CrossRef CAS PubMed.
- T. Okada, Y. Ayato, M. Yuasa and I. Sekine, J. Phys. Chem. B, 1999, 103, 3315–3322 CrossRef CAS.
- M. Ben Sik Ali, D. Jellouli Ennigrou and B. Hamrouni, Environ. Technol., 2013, 34, 2521–2529 CrossRef PubMed.
- A. ter Heijne, H. V. M. Hamelers, V. de Wilde, R. A. Rozendal and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5200–5205 CrossRef CAS PubMed.
- M. Yamaguchi and A. Ohira, Comput. Theor. Chem., 2015, 1071, 61–67 CrossRef CAS.
- H. K. Pan, A. Meagher, M. Pineri, G. S. Knapp and S. L. Cooper, J. Chem. Phys., 1985, 82, 1529–1538 CrossRef CAS.
- A. Sweity, W. Ying, S. Belfer, G. Oron and M. Herzberg, J. Membr. Sci., 2011, 378, 186–193 CrossRef CAS.
- M. Hashino, K. Hirami, T. Ishigami, Y. Ohmukai, T. Maruyama, N. Kubota and H. Matsuyama, J. Membr. Sci., 2011, 384, 157–165 CrossRef CAS.
- Z. Wang, F. Huang, X. Mei, Q. Wang, H. Song, C. Zhu and Z. Wu, J. Membr. Sci., 2014, 471, 258–264 CrossRef CAS.
- K. D. Kwon, H. Green, P. Bjöörn and J. D. Kubicki, Environ. Sci. Technol., 2006, 40, 7739–7744 CrossRef CAS PubMed.
- P. Yi and K. L. Chen, Environ. Sci. Technol., 2013, 47, 12211–12218 CrossRef CAS PubMed.
- H. Chen, J. D. Snyder and Y. A. Elabd, Macromolecules, 2008, 41, 128–135 CrossRef CAS.
- D. R. Morris and X. Sun, J. Appl. Polym. Sci., 1993, 50, 1445–1452 CrossRef CAS.
- F. Hook, B. Kasemo, T. Nylander, C. Fant, K. Sott and H. Elwing, Anal. Chem., 2001, 73, 5796–5804 CrossRef CAS PubMed.
- A. E. Contreras, Z. Steiner, J. Miao, R. Kasher and Q. L. Li, Environ. Sci. Technol., 2011, 45, 6309–6315 CrossRef CAS PubMed.
- O. Furman, S. Usenko and B. L. T. Lau, Environ. Sci. Technol., 2013, 47, 1349–1356 CAS.
- G. C. Abuin, M. Cecilia Fuertes and H. R. Corti, J. Membr. Sci., 2013, 428, 507–515 CrossRef CAS.
- G. Raj, C. Swalus, M. Delcroix, M. Devillers, C. Dupont-Gillain and E. M. Gaigneaux, J. Colloid Interface Sci., 2015, 445, 24–30 CrossRef CAS PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4585 CrossRef CAS PubMed.
- A. N. Pham, A. L. Rose, A. J. Feitz and T. D. Waite, Geochim. Cosmochim. Acta, 2006, 70, 640–650 CrossRef CAS.
- M. Danilczuk, L. Lin, S. Schlick, S. J. Hamrock and M. S. Schaberg, J. Power Sources, 2011, 196, 8216–8224 CrossRef CAS.
- Z. Liang, W. Chen, J. Liu, S. Wang, Z. Zhou, W. Li, G. Sun and Q. Xin, J. Membr. Sci., 2004, 233, 39–44 CrossRef CAS.
- Z. Wang, J. Ma, C. Y. Tang, K. Kimura, Q. Wang and X. Han, J. Membr. Sci., 2014, 468, 276–307 CrossRef CAS.
- P. Le-Clech, V. Chen and T. A. Fane, J. Membr. Sci., 2006, 284, 17–53 CrossRef CAS.
- F. Meng, B. Liao, S. Liang, F. Yang, H. Zhang and L. Song, J. Membr. Sci., 2010, 361, 1–14 CrossRef CAS.
- S. Iwamori, K. Yoshino, H. Matsumoto, K. Noda and I. Nishiyama, Sens. Actuators, B, 2012, 171–172, 769–776 CrossRef CAS.
- E. Briand, M. Zäch, S. Svedhem, B. Kasemo and S. Petronis, Analyst, 2010, 135, 343–350 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05510b |
| This journal is © The Royal Society of Chemistry 2017 |