
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe enantioselective addition of 1-fluoro-1-nitro(phenylsulfonyl)methane to isatin-derived ketimines†
M.
Urban
 a,
M.
Franc
a,
M.
Hofmanová
a,
I.
Císařová
b and
J.
Veselý
*a
a,
M.
Franc
a,
M.
Hofmanová
a,
I.
Císařová
b and
J.
Veselý
*a
aDepartment of Organic Chemistry, Charles University, Hlavova 8, 12843 Prague, Czech Republic. E-mail: jxvesely@natur.cuni.cz; Web: http://orgchem.cz/vesely/
bDepartment of Inorganic Chemistry, Charles University, Hlavova 8, 12843 Prague, Czech Republic
First published on 19th October 2017
Abstract
An asymmetric organocatalytic addition of fluorinated phenylsulfonylnitromethane to isatin-derived ketimines was developed. The reaction was efficiently catalyzed by a chiral tertiary amine, cinchonine. This methodology provides a new type of optically active compound with two adjacent quaternary carbon stereocenters in good yield (up to 96%), with moderate diastereoselectivity (up to 5.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and excellent enantioselectivity (up to 98/96% ee).
:1 dr) and excellent enantioselectivity (up to 98/96% ee).
Introduction
Preparation of enantiomerically pure organic compounds bearing a quaternary stereogenic center is one of the most challenging research areas in asymmetric catalysis.1 In addition, a quaternary 3-amino-2-oxindole moiety is a ubiquitous motif in natural products and pharmaceutical agents.2 Among the methods developed for the preparation of such compounds, enantioselective addition of nucleophiles to isatin-derived ketimines is straightforward and probably the most efficient approach. Easy access to N-Boc isatin imines3 together with its modulate reactivity towards nucleophiles and the ability to easily remove Boc protecting group from the adducts have made those imines as attractive substrates for a number of catalytic addition reactions.4 Not only metal-based complexes (Pd, Rh, Ni,)5 but also small organic molecules (H-bonding donors) can effectively catalyze these processes. To date a variety of organocatalytic addition reactions have been developed, including the Strecker reaction, the Mannich reaction and its vinylogous variant, the aza-Morita–Baylis–Hillman reaction, cycloadditions and others.6 Because of their impact in medicinal research and chemical biology, the development of efficient methods for the selective introduction of fluoro-containing groups into bioactive molecules is an area of high interest.7 However, the enantioselective addition reactions of fluoro-containing nucleophiles to isatin-derived ketimines have rarely been described.8 Besides α-fluoro-bis(phenylsulfonyl)methane (FBSM),9 α-fluoro-nitro(phenylsulfonyl)methane (FNSM) has also been recognized as a versatile nucleophile in various organocatalytic reactions, as demonstrated by Prakash and Olah10 and others.11 In this context, we herein report the highly enantioselective addition of α-fluoronitro(phenylsulfonyl)-methane to isatin-derived ketimines under mild organocatalytic conditions affording the fluoro-containing compounds bearing adjacent quaternary stereogenic centers.Results and discussion
At the outset we decided to study the nucleophilic addition of α-fluoro-nitro(phenylsulfonyl)methane (FNSM, 2a) to tert-butyl(1-benzyl-2-oxoindolin-3-ylidene)carbamate (1a) in the presence of cinchona alkaloids (Fig. 1). Initially, the reaction was performed with 1a and 2a in a 1:1.1 ratio at room temperature in toluene (Table 1, entry 1). It provided the desired adduct 3a/3a′ within a few hours in high yields (varying from 93 to 98%) and with moderate diastereoselectivity (up to 3.5:1). Unfortunately, the obtained enantiomeric purity of 3a was unsatisfactory, and further screening of the reaction conditions was required. Initially, we tested the influence of temperature on the course of the reaction (Table SI 1, ESI†). Cinchonine (CN, Fig. 1) has shown the most promising results in terms of effectiveness and enantiocontrol in the model reaction performed at a lower temperature (−50 °C, entry 5). Full conversion of isatin-derived ketimine 1a was reached within 18 hours and adduct 3a was isolated in high yield (85%), with moderate diastereoselectivity (3.5:1) and excellent enantioselectivity of both diastereoisomers (96/95% ee). It is noteworthy that a further decrease in the reaction temperature led to a significant drop in the yield of 3a (Table 1, entry 6).
 | ||
| Fig. 1 Bifunctional organocatalysts screened in the reaction of ketimine 1a with FNSM 2a. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Temp. [°C] | Time [h] | Yielda [%] | drb |
eec [%] |
| a Isolated yield after column chromatography. b Determined by 19F NMR of the reaction mixture. c Determined by HPLC analysis of the purified product for two diastereoisomers. | ||||||
| 1 | QN | 25 | 18 | 98 | 1.5:1 |
−3/−3 |
| 2 | QD | 25 | 4 | 94 | 1.2:1 | 19/62 |
| 3 | CN | 25 | 4 | 97 | 2:1 |
77/82 |
| 4 | CD | 25 | 4 | 93 | 1.6:1 |
−60/−70 |
| 5 | CN | 0 | 8 | 95 | 2.8:1 |
85/85 |
| 6 | CN | −50 | 18 | 82 | 3.5:1 |
96/96 |
| 7 | CN | −78 | 168 | 68 | 4:1 |
97/96 |

Next, we assessed the influence of the solvent in the reaction of 1a and 2a (Table SI 1, ESI†). As shown in Table 1, apart from toluene, the addition reaction proceeded well in several nonpolar chlorinated and etheric solvents, but with lower diastereo- and enantioselectivity and also yield (Table 2, entries 1–6). On the other hand no reaction was observed in polar protic and aprotic solvents, such as methanol and DMSO. Screening of the solvents and temperature is summarized in the ESI† (Tables SI 1) in detail.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time [h] | Yielda [%] | drb |
eec [%] |
| a Isolated yield after column chromatography. b Determined by 19F NMR of the reaction mixture. c Determined by HPLC analysis of the purified product for two diastereoisomers. d Reaction at 25 °C. | |||||
| 1 | Toluene | 18 | 85 | 3.5:1 |
96/95 |
| 2 | DCM | 19 | 57 | 2:1 |
79/86 |
| 3 | CHCl3 | 19 | 84 | 3:1 |
88/89 |
| 4 | THF | 96 | 65 | 1.9:1 |
22/35 |
| 5 | MTBE | 96 | 75 | 3:1 |
65/63 |
| 6 | EtOAc | 96 | 18 | 2.8:1 |
61/63 |
| 7 | MeOH | 96 | — | — | — |
| 8d | DMSO | 96 | — | — | — |

Next, we focused on the role of the catalyst in the enantiomeric addition of 2a to isatin-derived ketimine 1a with respect to efficiency and stereoselectivity (Table 2). Besides cinchona alkaloids, screened initially, chiral thioureas such as Takemoto's bifunctional catalyst A and Soós's catalysts B, C Sharpless bases and squaramides D, E were examined in the reaction between 1a and 2b (Fig. 1). Surprisingly, none of the above-mentioned organocatalysts catalysed the model reaction as effectively as cinchonine (Table 3, entries 1–6, more details in the ESI†). We also screened the dependence of the reaction efficiency and selectivity on the loading of catalysts. Interestingly, no significant change in selectivity and yield was observed when 5 mol% of the catalyst was used. On the other hand a lower catalyst loading dramatically influenced the reaction time (Table 3, entry 9). A reduced yield was observed, when 2.5 mol% of the catalyst was used, nevertheless, the selectivity of the reaction was retained (Table 3, entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time [h] | Yielda [%] | drb |
eec [%] |
| a Isolated yield after column chromatography. b Determined by 19F NMR of the reaction mixture. c Determined by HPLC analysis of the purified product for two diastereoisomers. | |||||
| 1 | Takemoto's A | 18 | 96 | 3.3:1 |
−27/−25 |
| 2 | Soós's with QN B | 18 | 96 | 3.7:1 |
64/56 |
| 3 | Soós's with CD D | 20 | 96 | 5.9:1 |
70/62 |
| 4 | (DHQ)2AQN | 24 | 90 | 4:1 |
35/28 |
| 5 | Rawal's with QN E | 24 | 89 | 4.3:1 |
18/47 |
| 6 | Rawal's with CN F | 168 | 76 | 4.5:1 |
−75/−16 |
| 7 | CN (10 mol%) | 24 | 80 | 3.5:1 |
96/95 |
| 8 | CN (5 mol%) | 36 | 85 | 3.7:1 |
96/96 |
| 9 | CN (2.5 mol%) | 48 | 68 | 3.6:1 |
96/95 |
| 10 | — | 168 | — | — | — |

Once we uncovered the optimized reaction conditions for the enantioselective nucleophilic addition of FNSM to isatin-derived ketimine 1a, we proceeded with the scope of the process (Scheme 1). First we studied the reactivity of FNSM towards ketimines 1a–1h having different protecting groups on the nitrogen of the 3-amino-2-oxindole moiety. The desired products 3a/3a′–3g/3g′ were prepared in good yields with high to excellent enantioselectivity (93–99% ee for the major diastereoisomer and 93–95% ee for the minor diastereoisomer). A moderate degree of diastereoselectivity (3:1 to 5:1) was obtained in the reaction of 2a with most ketimines 1a–1h. The highest diastereocontrol was observed in the reaction with Boc protected ketamine 1d (dr 5:1). On the other hand, the reaction between 2a and ketimine 1h with a tosyl protecting group proceeded with low diastereo- (dr 1:1.9) and enantioselectivity (70/75% ee); the corresponding adduct 3h was isolated in low yield (31%) as a mixture of diastereoisomers.
 | ||
| Scheme 1 Substrate scope of ketimines 1a–p and fluorinated sulfones 2a–3 in the organocatalytic alkylation reaction. Isolated yield after column chromatography. Determined by 19F NMR of the reaction mixture. Determined by HPLC analysis of the purified product for two diastereoisomers. | ||
Subsequently, we focused on ketimine derivatives with different substitutions on the aromatic ring (1i–1o). Substitution at the 5-position on the aromatic ring of ketimines led to the formation of the corresponding products 3i–3l in high yield (81–95%) and excellent enantiomeric excess for both enantiomers (major diastereoisomer 96–98% ee, minor diastereoisomer 94–96% ee). On the other hand, diastereoselectivity reached moderate values (dr 3.7:1–5.7:1). A significant change in reactivity was observed, when ketimine 1o substituted at the 4-position with bromine was used. The formation of the corresponding adduct 3o was not observed even after a prolonged reaction time (96 h). This can be probably caused due to the higher steric hindrance of the imine group with the bulky bromine substituent at the 4-position, because all other adducts 3l, 3m and 3n having bromine atoms at positions 5-,6- and 7 were obtained in high yields with good levels of enantioselectivity. Replacement of the Boc group for the Cbz group on the nitrogen of the imine moiety led to a considerable drop in enantioselectivity (75/64% ee). The corresponding product 3p was obtained in 88% yield and with good diastereoselectivity (dr 5.1:1).
Next, we turned our attention to other types of fluorinated sulfones 2b–e. The nitro group was replaced by various functional groups such as nitrile, ester, acetyl, phenylcarbonyl groups, etc. In all cases significantly lower reactivity towards 1a under the optimized reaction conditions (QN, toluene, −50 °C) was observed. Due to this reason we approached to set up the reactions with appropriate sulfones 2b–e at 25 °C. It showed up that apart from the model reaction between 1a and 2a, reactions proceeded smoothly only with 2-fluoro-2-(phenylsulfonyl)acetonitrile (2b), affording product 3q in high yield (94%) with low diastereoselectivity (dr 2.3:1) and unsatisfactory enantioselectivity (3/32% ee). Both the subsequential turn of temperature down to −50 °C and also the change of the solvent from toluene to DCM did not lead to improved selectivity of the reaction (dr 2.9:1, 30/59% ee, Scheme 1).
Next, the potential versatility of a phenylsulfanyl group as a traceless activating group was verified. The standard procedure for the removal of phenylsulfonyl group with magnesium failed.12 Nevertheless, desulfonation of enantioenriched adducts 3a was accomplished using AIBN as a radical agent in the presence of BNAH as a hydrogen transfer agent (Table 4).13 α-Fluoronitro alkane 4 was isolated in 96% yield as a mixture of diastereoisomers (dr = 1:1) with the enantioselectivity retained (95/96% ee). Furthermore, Boc deprotection of 4a and 4b was successfully performed using TFA (Table 4).14 The corresponding amines 5a and 5b were obtained in good yields 48% and 47%, respectively, with excellent enantiomeric excess (97/96%).


To determine the absolute configuration of alkylated products 3a, we performed single-crystal X-ray diffraction analysis of 3a, obtained from the reaction of ketimine 1a with fluoro-nitro(phenylsulfonyl)methane 2a (Fig. 2). As shown in Fig. 3, the C3 and C1′ stereogenic centres have the (S) and (R) absolute configuration, respectively.
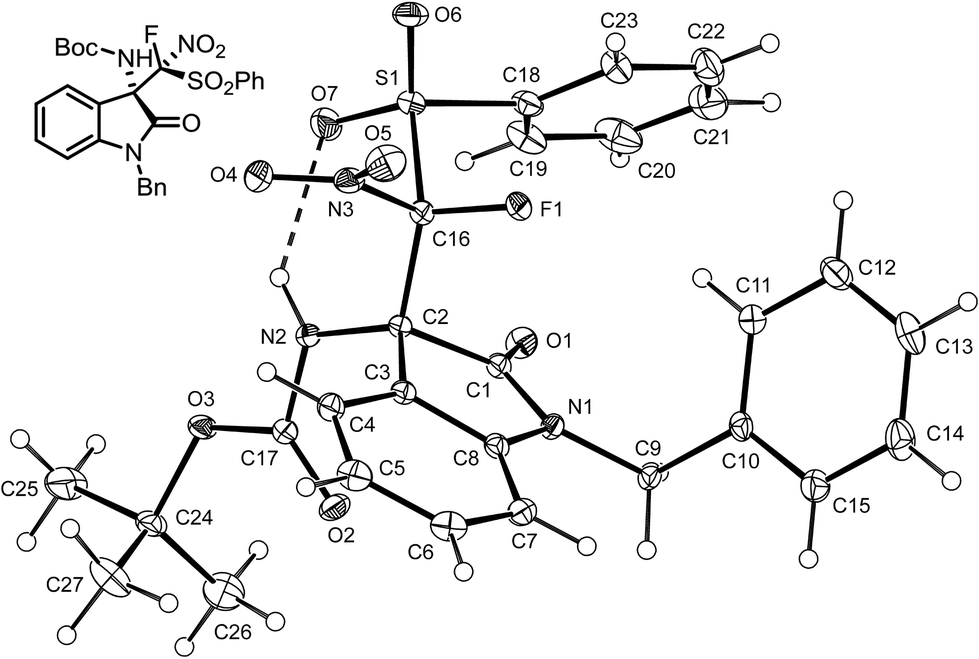 | ||
| Fig. 2 View of the molecular structure of 3a, the displacement ellipsoids at the 30% probability level. The dashed line indicates the intramolecular hydrogen bond N2–H2⋯O7, N2⋯O7 2.894(2) Å; angle on H2 138.7°.15 | ||
 | ||
| Fig. 3 Proposed transition state. | ||
On the basis of the crystallographic analysis of 3a an envisaged transition-state structure was proposed. As shown in Fig. 3, cinchonine can act as a dual catalyst and activate both reaction substrates via hydrogen bonding. The tertiary amine of the chinuclidine moiety deprotonates FNSM forming the corresponding anionic nucleophile that preferentially approaches the ketimine from the Re-face due to the H-bonding of the ketimine and hydroxyl groups of cinchonine.
Conclusion
In summary, we have explored the stereoselective fluoroalkylation of isatin-derived ketimines with α-fluoronitro(phenylsulfonyl) methane. The reaction was effectively catalyzed with cinchonine furnishing the corresponding adducts in good yields, with good diastereo- (dr up to 6:1) and high enantioselectivity (up to 98% ee). The reactivity of isatin-derived ketimines toward other fluorinated compounds was also studied. The outcome of the reported procedure was demonstrated by the preparation of rarely available β-fluoro-β-nitro amines using a desulfonylation/Boc-removal reaction sequence. Further studies and synthetic applications based on the reported methodology are currently ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Jan Veselý gratefully acknowledges the Czech Science Foundation (no. 16-23597S) for financial support. Michal Urban thanks the Charles University Grant Agency (grant number 392315) for financial support. We also thank Dr Hybelbauerová for NMR service and Dr Štícha for MS analysis.Notes and references
- For excellent reviews and perspective articles on the catalytic asymmetric construction of quaternary stereogenic centers, see: (a) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740 CrossRef CAS PubMed; (b) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed; (c) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef CAS PubMed; (d) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745 CrossRef CAS PubMed; (e) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC; (f) M. Shimizu, Angew. Chem., Int. Ed., 2011, 50, 5998 CrossRef CAS PubMed; (g) B. Wang and Y. Q. Tu, Acc. Chem. Res., 2011, 44, 1207 CrossRef CAS PubMed; (h) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC.
- (a) T. Oost, G. Backfisch, S. Bhowmik, M. M. van Gaalen, H. Geneste, W. Hornberger, W. Lubisch, A. Netz, L. Unger and W. Wernet, Bioorg. Med. Chem. Lett., 2011, 21, 3828 CrossRef CAS PubMed; (b) M. Rottmann, C. McNamara, B. S. K. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. Gonzalez-Paez, L. Lakshiminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175 CrossRef CAS PubMed; (c) V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hübel, D. Rauh and H. Waldmann, Angew. Chem. Int. Ed., 2010, 49, 5902 CrossRef CAS PubMed; (d) T. Shimazaki, M. Iijima and S. Chaki, Eur. J. Pharmacol., 2006, 543, 63 CrossRef CAS PubMed; (e) A. K. Ghosh, G. Schiltz, R. S. Perali, S. Leshchenko, S. Kay, D. E. Walters, Y. Koh, K. Maeda and H. Mitsuya, Bioorg. Med. Chem. Lett., 2006, 16, 1869 CrossRef CAS PubMed; (f) K. Bernard, S. Bogliolo and J. Ehrenfeld, Br. J. Pharmacol., 2005, 144, 1037 CrossRef CAS PubMed; (g) G. Gilles and S. L. Claudine, Stress, 2003, 6, 199 CrossRef PubMed; (h) M. Ochi, K. Kawasaki, H. Kataoka and Y. Uchio, Biochem. Biophys. Res. Commun., 2001, 283, 1118 CrossRef CAS PubMed; (i) L. Chevolot, A. M. Chevolot, M. Gajhede, C. Larsen, U. Anthoni and C. Christophersen, J. Am. Chem. Soc., 1985, 107, 4542 CrossRef CAS.
- W. J. Yan, D. Wang, J. C. Feng, P. Li, D. Zhao and R. Wang, Org. Lett., 2012, 14, 2512 CrossRef CAS PubMed.
- For recent reviews from the area of enantioselective additions to N-carbamoylimines, see: (a) J. Veselý and R. Rios, Chem. Soc. Rev., 2014, 43, 611 RSC; (b) E. Marcantoni and M. Petrini, Adv. Synth. Catal., 2016, 358, 3657 CrossRef CAS.
- (a) Z. Liu, X. Feng and H. Du, Org. Lett., 2012, 14, 3154 CrossRef CAS PubMed; (b) T. Rajasekaran, G. Karthik, B. Sridhar, S. K. Kumar and B. V. S. Reddy, Eur. J. Org. Chem., 2014, 2221 CrossRef CAS; (c) L.-Y. Mei, X.-Y. Tang and M. Shi, Org. Biomol. Chem., 2014, 12, 1149 RSC; (d) C. S. Marques and A. J. Burke, Eur. J. Org. Chem., 2016, 806 CrossRef CAS; (e) S. Nakamura, K. Hyodo, M. Nakamura, D. Nakane and H. Masuda, Chem. – Eur. J., 2013, 19, 7304 CrossRef CAS PubMed; (f) T. Arai, E. Matsumura and H. Masu, Org. Lett., 2014, 16, 2768 CrossRef CAS PubMed; (g) T. Arai, K. Tsuchiya and E. Matsumura, Org. Lett., 2015, 17, 2416 CrossRef CAS PubMed; (h) H. Zheng, X. Liu, C. Xu, Y. Xia, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 10958 CrossRef CAS PubMed.
- (a) N. Hara, S. Nakamura, M. Sano, R. Tamura, Y. Funahashi and N. Shibata, Chem. – Eur. J., 2012, 18, 9276 CrossRef CAS PubMed; (b) Y.-L. Liu and J. Zhou, Chem. Commun., 2013, 49, 4421 RSC; (c) D. Wang, J. Liang, J. Feng, K. Wang, Q. Sun, L. Zhao, D. Li, W. Yan and R. Wang, Adv. Synth. Catal., 2013, 355, 548 CAS; (d) F. L. Hu, Y. Wei, M. Shi, S. Pindi and G. Li, Org. Biomol. Chem., 2013, 11, 1921 RSC; (e) T.-Z. Li, X.-B. Wang, F. Sha and X.-Y. Wu, Tetrahedron, 2013, 69, 7314 CrossRef CAS; (f) X.-B. Wang, T.-Z. Li, F. Sha and X.-Y. Wu, Eur. J. Org. Chem., 2014, 739 CrossRef; (g) J. George, B. Sridhar and B. V. S. Reddy, Org. Biomol. Chem., 2014, 12, 1595 RSC; (h) X. Zhao, T.-Z. Li, J.-Y. Qian, F. Sha and X.-Y. Wu, Org. Biomol. Chem., 2014, 12, 8072 RSC; (i) T.-Z. Li, X.-B. Wang, F. Sha and X.-Y. Wu, J. Org. Chem., 2014, 79, 4332 CrossRef CAS PubMed; (j) Y. Guo, Y. Zhang, L. Qi, F. Tian and L. Wang, RSC Adv., 2014, 4, 27286 RSC; (k) Z. Tang, Y. Shi, H. Mao, X. Zhu, W. Li, Y. Cheng, W.-H. Zheng and Ch. Zhu, Org. Biomol. Chem., 2014, 12, 6085 RSC; (l) A. Kumar, J. Kaur, S. S. Chimni and A. K. Jassal, RSC Adv., 2014, 4, 24816 RSC; (m) Y.-H. Wang, Y. L. Liu, Z.-Y. Cao and J. Zhou, Asian J. Org. Chem., 2014, 3, 429 CrossRef CAS; (n) S. Nakamura, S. Takahashi, D. Nakane and H. Masuda, Org. Lett., 2015, 17, 106 CrossRef CAS PubMed; (o) A. Kumar, V. Sharma, J. Kaur, N. Kumar and S. S. Chimni, Org. Biomol. Chem., 2015, 13, 5629 RSC; (p) Y. Yoshida, M. Sako, K. Kishi, H. Sasai, S. Hatakeyama and S. Takizawa, Org. Biomol. Chem., 2015, 13, 9022 RSC; (q) S. Nakamura and S. Takahashi, Org. Lett., 2015, 17, 2590 CrossRef CAS PubMed; (r) O. D. Engl, S. P. Fritz and H. Wennemers, Angew. Chem., Int. Ed., 2015, 54, 8193 CrossRef CAS PubMed; (s) M. Montesinos-Magraner, C. Vila, R. Cantón, G. Blay, I. Fernández, M. C. Muñoz and J. R. Pedro, Angew. Chem., Int. Ed., 2015, 54, 6320 CrossRef CAS PubMed; (t) T. Liu, W. Liu, X. Li, F. Peng and Z. Shao, J. Org. Chem., 2015, 80, 4950 CrossRef CAS PubMed; (u) X. Bao, B. Wang, L. Cui, G. Zhu, Y. He, J. Qu and Y. Song, Org. Lett., 2015, 17, 5168 CrossRef CAS PubMed; (v) M.-X. Zhao, L. Jing, H. Zhou and M. Shi, RSC Adv., 2015, 5, 75648 RSC; (w) K. Zhao, T. Shu, J. Jia, G. Raabe and D. Enders, Chem. – Eur. J., 2015, 21, 3933 CrossRef CAS PubMed; (x) Y. Zhu, E. G. Zhang, C. Luo, X. Li and J.-P. Cheng, Tetrahedron, 2015, 71, 4090 CrossRef CAS; (y) F. I. Amr, C. Vila, G. Blay, M. C. Muñoz and J. R. Pedro, Adv. Synth. Catal., 2016, 358, 1583 CrossRef CAS; (z) C. Vila, F. I. Amr, G. Blay, M. C. Muñoz and J. R. Pedro, Chem. – Asian J., 2016, 11, 1532 CrossRef CAS PubMed; (a a) K. Zhao, Y. Zhi, X. Li, R. Puttreddy, K. Rissanen and D. Enders, Chem. Commun., 2016, 52, 2249 RSC; (a b) B. X. Feng, J.-D. Yang, J. Li and X. Li, Tetrahedron Lett., 2016, 57, 3457 CrossRef CAS; (a c) P. Cheng, W. Guo, P. Chen, Y. Liu, X. Du and C. Li, Chem. Commun., 2016, 52, 3418 RSC; (a d) M. M. Magraner, C. Vila, A. R. Patiño, G. Blay, I. Fernández, M. C. Muñoz and J. R. Pedro, ACS Catal., 2016, 6, 2689 CrossRef; (a e) W. Guo, Y. Liu and C. Li, Org. Lett., 2017, 19, 1044 CrossRef CAS PubMed; (a f) S. Hajra and B. Jana, Org. Lett., 2017, 19, 4778 CrossRef CAS PubMed; (a g) J. Dai, D. Xiong, T. Yuan, J. Liu, T. Chen and Z. Shao, Angew. Chem., Int. Ed., 2017, 56, 12697 CrossRef CAS PubMed; (a h) C. Cheng, X. Lu, L. Ge, J. Chen, W. Cao, X. Wu and G. Zhao, Org. Chem. Front., 2017, 4, 101 RSC.
- (a) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (d) Fluorine in Bioorganic and Medicinal Chemistry, ed. I. Ojima, Wiley-Blackwell, Chichester, U.K., 2009 Search PubMed; (e) D. J. O'Hagan, Fluorine Chem., 2010, 131, 1071 CrossRef; (f) G. Valero, X. Companyo and R. Rios, Chem. – Eur. J., 2011, 17, 2018 CrossRef CAS PubMed; (g) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (h) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- (a) J. Liu, L. Zhang and J. Hu, Org. Lett., 2008, 10(23), 5377 CrossRef CAS PubMed; (b) J. Liu and J. Hu, Chem. – Eur. J., 2010, 16, 11443 CrossRef CAS PubMed; (c) J.-S. Yua and J. Zhou, Org. Biomol. Chem., 2015, 13, 10968 RSC; (d) X. Liu, J. Zhang, L. Zhao, S. Ma, D. Yang, W. Yan and R. Wang, J. Org. Chem., 2015, 80, 12651 CrossRef CAS PubMed; (e) J.-S. Yu and J. Zhou, Org. Chem. Front., 2016, 3, 298 RSC; (f) X.-L. Zeng, Z.-Y. Deng, C. Liu, G. Zhao, J.-H. Lin, X. Zheng and J.-Ch. Xiao, J. Fluorine Chem., 2017, 193, 17 CrossRef CAS . For other fluoroalkylation reagents, see: Ch. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 Search PubMed.
- For selected studies, see: (a) T. Fukuzumi, N. Shibata, M. Sugiura, H. Yasui, S. Naka-mura and T. Toru, Angew. Chem., Int. Ed., 2006, 45, 4973 CrossRef CAS PubMed; (b) S. Mizuta, N. Shibata, Y. Goto, T. Furukawa, S. Nakamura and T. Toru, J. Am. Chem. Soc., 2007, 129, 6394 CrossRef CAS PubMed; (c) T. Furukawa, N. Shibata, S. Mizuta, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2008, 47, 8051 CrossRef CAS PubMed; (d) Ch. Ni, L. Zhang and J. Hu, J. Org. Chem., 2008, 73, 5699 CrossRef CAS PubMed; (e) A.-N. Alba, X. Company, A. Moyano and R. Rios, Chem. – Eur. J., 2009, 15, 7035 CrossRef CAS PubMed; (f) F. Ullah, G.-L. Zhao, L. Deiana, M. Zhu, P. Dziedzic, I. Ibrahem, P. Hammar, J. Sun and A. Córdova, Chem. – Eur. J., 2009, 15, 10013 CrossRef CAS PubMed; (g) X. Shen, L. Zhang, Y. Zhao, L. Zhu, G. Li and J. Hu, Angew. Chem., Int. Ed., 2011, 50, 2588 CrossRef CAS PubMed; (h) W. Yang, X. Wei, Y. Pan, R. Lee, B. Zhu, H. Liu, L. Yan, K.-W. Huang, Z. Jiang and Ch.-H. Tan, Chem. – Eur. J., 2011, 17, 8066 CrossRef CAS PubMed; (i) T. Furukawa, J. Kawazoe, W. Zhang, T. Nishimine, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 9684 CrossRef CAS PubMed; (j) S.-L. Zhang, H.-X. Xie, J. Zhu, H. Li, X.-S. Zhang, J. Li and W. Wang, Nat. Commun., 2011, 211(2), 1 Search PubMed; (k) W. Huang, Ch. Ni, Y. Zhao and J. Hu, New J. Chem., 2013, 37, 1684 RSC; (l) K. Matsuzaki, T. Furukawa, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Org. Lett., 2013, 15(13), 3282 CrossRef CAS PubMed.
- (a) G. K. S. Prakash, X. Zhao, S. Chacko, F. Wang, H. Vaghoo and G. A. Olah, Beilstein J. Org. Chem., 2008, 4, 17 Search PubMed; (b) G. K. S. Prakash, F. Wang, T. Stewart, T. Mathew and G. A. Olah, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4090 CrossRef CAS PubMed.
- For selected studies, see: (a) F. Ullah, G. L. Zhao, L. Deiana, M. Zhu, P. Dziedzic, I. Ibrahem, P. Hammar, J. Sun and A. Cordova, Chem. – Eur. J., 2009, 15, 10013 CrossRef CAS PubMed; (b) M. Kamlar, N. Bravo, A.-N. R. Alba, S. Hybelbauerova, I. Cisarova, J. Vesely, A. Moyano and R. Rios, Eur. J. Org. Chem., 2010, 5464 CrossRef CAS; (c) Y. Pan, Y. Zhao, T. Ma, Y. Yang, H. Liu, Z. Jiang and C.-H. Tan, Chem. – Eur. J., 2010, 16, 779 CrossRef CAS PubMed; (d) H. W. Moon and D. Y. Kim, Bull. Korean Chem. Soc., 2012, 33(9), 2845 CrossRef CAS; (e) M. Kamlar, P. Putaj and J. Veselý, Tetrahedron Lett., 2013, 54, 2097 CrossRef CAS; (f) G. K. S. Prakash, L. Gurung, P. V. Jog, S. Tanaka, T. E. Thomas, N. Ganesh, R. Haiges, T. Mathew and G. A. Olah, Chem. – Eur. J., 2013, 19, 3579 CrossRef CAS PubMed.
- Y. Kazuta, A. Matsuda and S. Shuto, J. Org. Chem., 2002, 67, 1669 CrossRef CAS PubMed.
- Y. Seo, H. Kim, D. W. Chae and Y. G. Kim, Tetrahedron: Asymmetry, 2014, 25, 625 CrossRef CAS.
- M. Montesinos-Magraner, C. Vila, R. Cantón, G. Blay, I. Fernández, M. C. Muñoz and J. R. Pedro, Angew. Chem., Int. Ed., 2015, 54, 6320 CrossRef CAS PubMed.
- X-ray crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 1575684† for 3a.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and spectral data for all prepared compounds with copies of the 1H NMR, 13C NMR and 19F NMR and HPLC chromatographs. CCDC 1575684. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob02408h |
| This journal is © The Royal Society of Chemistry 2017 |