
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sky-blue emitting bridged diiridium complexes: beneficial effects of intramolecular π–π stacking†
Daniel G.
Congrave
 ,
Yu-Ting
Hsu
,
Andrei S.
Batsanov
,
Andrew
Beeby
and
Martin R.
Bryce
*
,
Yu-Ting
Hsu
,
Andrei S.
Batsanov
,
Andrew
Beeby
and
Martin R.
Bryce
*
Department of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 22nd January 2018
Abstract
The potential of intramolecular π–π interactions to influence the photophysical properties of diiridium complexes is an unexplored topic, and provides the motivation for the present study. A series of diarylhydrazide-bridged diiridium complexes functionalised with phenylpyridine (ppy)-based cyclometalating ligands is reported. It is shown by NMR studies in solution and single crystal X-ray analysis that intramolecular π–π interactions between the bridging and cyclometalating ligands rigidify the complexes leading to high luminescence quantum efficiencies in solution and in doped films. Fluorine substituents on the phenyl rings of the bridge promote the intramolecular π–π interactions. Notably, these non-covalent interactions are harnessed in the rational design and synthesis of the first examples of highly emissive sky-blue diiridium complexes featuring conjugated bridging ligands, for which they play a vital role in the structural and photophysical properties. Experimental results are supported by computational studies.
Introduction
Iridium(III) complexes possess rich metal–ligand based photochemistry, typically with high luminescence quantum efficiency (Φ) and short excited state lifetimes (τp). They are widely employed in applications1 such as photocatalysis,2 biological labelling,3 sensing4 and as emissive dopants in phosphorescent organic light-emissive devices (PhOLEDs).5,6 Their emission colour can be tuned across the entire visible spectrum by systematic variation of the ligands.7Unlike their monometallic analogues, diiridium complexes are rarely studied for luminescence applications due to their generally low photoluminescence quantum yields (PLQYs) and limited colour range.8–17 However, there are examples where the favourable luminescent properties of monoiridium complexes are retained in diiridium complexes by the careful choice of conjugated bridging ligands.18–27 Moreover, bridging ligands offer scope for increased structural variation compared to monoiridium analogues, and allow tuning of the electronic communication between the iridium centres which may lead to interesting photophysical properties, such as improved spin–orbit coupling effects,24,26 or dual emission. Diiridium complexes are known with efficient emission from red to green;18–26 however, we are not aware of any blue/sky-blue diiridium complexes featuring conjugated bridging ligands.28
Recently, we described diarylhydrazide-bridged diiridium complexes functionalised with phenylpyridine (ppy)-based cyclometalating ligands.22 These complexes are highly emissive in the green region when doped into rigid poly(methylmethacrylate) (PMMA) films, but are practically nonemissive in solution, presumably due to the flexibility of their non-ancillary bridging units which leads to non-radiative decay via intramolecular motion. An interesting structural feature was observed: the pendant aryl rings on the bridge engage in intramolecular face-to-face π–π stacking with the cyclometalating phenyl ligands in the solid state (complex 1, Fig. 1).
 | ||
| Fig. 1 Representative iridium complexes which display intramolecular π–π stacking interactions, highlighted by the coloured rings. D = centroid–centroid distance determined by X-ray diffraction for the same-coloured rings. D* = distance between the centroid of the bridge aryl ring and the plane of the cyclometalating ligand. | ||
Intramolecular π–π stacking between aryl and heteroaryl rings has been reported in a few specific monoiridium complexes (e.g.2–6, Fig. 1), particularly in charged derivatives.29–33 For example, in complex 2 intramolecular π–π stacking between a cyclometalating ligand and a pendant pentafluorophenyl group leads to an order of magnitude increase in solution PLQY, due to a reduction in the non-radiative rate constant (knr).31 Intramolecular π–π stacking in complex 3 leads to increased operational stability of light-emitting electrochemical cells (LEECs).29 Nonetheless, the potential of intramolecular π–π interactions to influence the photophysical properties of diiridium complexes remains unexplored, and provides the motivation for the present study.
We now show that intramolecular π–π stacking can be exploited to rigidify diiridium complexes and to obtain high luminescence quantum efficiencies in solution and in doped films. We also present the first examples of highly emissive sky-blue diiridium complexes featuring conjugated bridging ligands, for which the π–π interactions play an important role in the structural and photophysical properties.
Results and discussion
Design, synthesis and characterisation
The structural versatility of 1 and analogues22 provides an ideal opportunity to explore how intramolecular π–π interactions between the bridging and cyclometalating ligands can influence the photophysical properties of diiridium systems. Benzene is well known to stack with hexafluorobenzene in a slipped face-to-face configuration in the solid state.34–36 Complexes 7–9 (Fig. 2) with an increasing number of fluorine substituents on the phenyl rings of the bridge, were, therefore, designed with the aim of promoting intramolecular π–π interactions. Methoxy derivative 10 was also included based on calculations (discussed below) which predict the bridge of 10 to be non-ancillary despite the highly fluorinated aryl rings (in contrast to 8 and 9). The analogues 12 and 14, featuring CF3 substituents instead of perfluoroaryl rings, were studied as model compounds for which π–π interactions involving the bridge are not possible. For derivatives 11–15, the substituents on the pyridyl rings serve to enhance solubility. For 13–15 the difluorophenyl rings of the ppy ligands were chosen to blue shift the emission, based on monoiridium precedents.37,38 | ||
| Fig. 2 (Top) Structures for the diiridium complexes studied in this work. (Bottom) Structures for the bridging and cyclometalating ligands. Complexes were studied as diastereomeric mixtures unless otherwise stated. * Complexes 14 and 15 were isolated as single diastereomers; their absolute configurations are unknown. | ||
The diarylhydrazide bridges 17a–d (Fig. 2) were synthesised (Scheme S1†) by condensation of hydrazine monohydrate with the corresponding benzoyl chlorides, which were either commercially available or prepared from the corresponding benzoic acid (16a–d). The bridge units were heated in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio with [Ir(ppy)2μ-Cl]2 in either 2-ethoxyethanol (17a) or dry diglyme (17b–d) in the presence of K2CO3, to obtain the complexes 7–10 as diastereomeric mixtures (meso ΛΔ and rac ΛΛ/ΔΔ) (Fig. 2). In previous investigations, the diastereomers of analogous phenylpyridine-functionalised diiridium systems were separated and minimal differences were observed in the photophysical properties of the two diastereomers.21,22 Therefore, complexes 7–10 were characterised as diastereomeric mixtures. The complexes were unambiguously identified by 1H, 19F and 13C (where solubility allowed) NMR spectroscopy, MALDI-TOF mass spectrometry and elemental analysis. NMR peak assignments were aided by 1H–1H COSY, 1H–1H NOESY, 1H–1H ROESY, 1H–13C HSQC, 1H–13C HMBC and 19F–19F COSY 2D NMR experiments.
:1 molar ratio with [Ir(ppy)2μ-Cl]2 in either 2-ethoxyethanol (17a) or dry diglyme (17b–d) in the presence of K2CO3, to obtain the complexes 7–10 as diastereomeric mixtures (meso ΛΔ and rac ΛΛ/ΔΔ) (Fig. 2). In previous investigations, the diastereomers of analogous phenylpyridine-functionalised diiridium systems were separated and minimal differences were observed in the photophysical properties of the two diastereomers.21,22 Therefore, complexes 7–10 were characterised as diastereomeric mixtures. The complexes were unambiguously identified by 1H, 19F and 13C (where solubility allowed) NMR spectroscopy, MALDI-TOF mass spectrometry and elemental analysis. NMR peak assignments were aided by 1H–1H COSY, 1H–1H NOESY, 1H–1H ROESY, 1H–13C HSQC, 1H–13C HMBC and 19F–19F COSY 2D NMR experiments.
For complexes 7–10 the 19F NMR data are of particular interest. For the bis(difluorophenyl)hydrazide-bridged complex 7, a single peak is observed in the 19F spectrum of the diastereomeric mixture (Fig. S2†), analogous to the spectrum of the free bridge (17a) (Fig. S74†). This indicates that the 19F environments are very similar for each diastereomer of 7 and that the bridging phenyl rings are freely rotating in solution on the NMR timescale.
This contrasts with the data for the bis(pentafluorophenyl)hydrazide-bridged complex 9. The ligand 17c features 3 distinct environments in its 19F NMR spectrum as expected (Fig. S80†), whereas the 19F NMR spectrum of meso9 features 5 well-resolved distinct environments (Fig. 3 and Fig. S15†) due to an apparent breakdown in symmetry, suggesting that rotation of the bridging pentafluorophenyl rings is restricted at room temperature in solution. This was confirmed when meso9 was further studied by 19F–19F COSY NMR (Fig. 3). This is because, although only ortho (3J ≈ 23 Hz) and para (5J ≈ 6 Hz) couplings are observed (in agreement with the multiplicities of the signals in the 1D spectrum), the data indicate that all 5 fluorine environments are on the same ring. meta (4J) 19F–19F coupling constants that are considerably smaller than those for ortho and para coupling (or even absent) have been commonly reported for heavily fluorinated aryl systems.39–43 It has been suggested that this is because π-conjugation contributes significantly to 19F–19F coupling in aromatics.39,43
 | ||
| Fig. 3 (Top) 19F NMR spectrum of the diastereomeric mixture of 9 (ca. 5:4 molar ratio of meso (ΛΔ) and rac (ΛΛ/ΔΔ)). (Middle) 19F NMR spectrum of meso9. (Bottom) 19F–19F COSY NMR spectrum of meso9. Chemical shifts are in ppm. | ||
We propose that this restriction of rotation is due to intramolecular π–π interactions. Steric restriction alone is unlikely to explain such well-resolved 19F NMR signals, considering that fluorine atoms exert similar steric effects as protons,44 and that the analogous difluoro complex 7 does not exhibit this effect. The 19F NMR spectra of complexes 8, 10, 11, 13 and 15 also show this feature (Fig. S5, S18, S24, S42, S51 and S68†). These observations indicate that a bridge tetrafluorophenyl group is sufficient to promote strong intramolecular π–π interactions in solution, and that fluorine atoms on the cyclometalating phenyl rings of ppy ligands (13 and 15) do not suppress them.
The bis(trifluoromethyl) bridge 1845 (Fig. 2) was also investigated, as although it is strongly electron withdrawing like the perfluoroaryl bridge 17c,46 it cannot engage in intramolecular π–π stacking. Attempts to isolate a complex analogous to 9 by reacting the bridge 18 with [Ir(ppy)2μ-Cl]2 were unsuccessful, due to its extremely poor solubility (mass spectra suggested the complex had formed). As an alternative, complex 12 was synthesised (Fig. 2), which features 4-mesityl-2-phenylpyridine (20) cyclometalating ligands. Mesityl groups are known to improve the solubility of cyclometalated iridium complexes while exerting minimal influence on their photophysical properties.47–49 Complex 12 was isolated as a diastereomerically pure meso sample (confirmed by X-ray diffraction, Fig. S102†) in 61% yield. No rac diastereomer was detected in the crude reaction mixture. This stereoselectivity is surprising as DFT calculations predict the rac diastereomer to be the more thermodynamically stable, as is usually the case for diiridium systems.21,22,50 Attempts to isomerise 12 thermally or photochemically were unsuccessful, as previously reported for other diiridium diastereomers.22
To allow a direct comparison with complex 12, complex 11 (the mesityl-functionalised analogue of complex 9) (Fig. 2), was also synthesised. Interestingly, the presence of mesityl groups leads to a larger difference in the solubilities of the diastereomers of 11 compared to 9, making them trivial to separate by column chromatography. However, the extremely poor solubility of meso11 prevented its purification and so only rac11 is studied here (stereochemistry confirmed by X-ray diffraction, Fig. S101†). It is noteworthy that meso11 is less soluble than complex 9 despite the presence of mesityl groups, in contrast to the expectation based on previous reports.47,48,50 A tentative explanation is based on the symmetry of the complex.51
We have previously shown that colour tuning of the emission of diarylhydrazide-bridged diiridium complexes within the range λmax 520–490 nm can be achieved through functionalisation of either the bridge or cyclometallating phenyl rings with electron withdrawing groups.21,22 We reasoned, therefore, that simultaneous functionalisation of both moieties with electron withdrawing groups might afford blue/sky-blue diiridium complexes, which to date remain elusive.
Initial attempts to obtain diiridium complexes through a combination of 2-(2,4-difluorophenyl)pyridine (dfppy) or 2-(2,4-difluorophenyl)-4-mesitylpyridine48 with the bis(pentafluorophenyl)/(trifluoromethyl) bridges 17c and 18 (Fig. 2) were unsuccessful due to the extremely poor solubility of the products. To enhance solubility the new dfppy derivative 21 (Fig. 2) was synthesised (Scheme S1†), wherein the mesityl group is replaced by a methylenecyclohexylether-functionalised xylyl group. The methylenecyclohexyl group provides the beneficial solubilising properties of a branched alkyl group while being achiral. Additionally, the xylyl spacer in 21 is a rigid non-conjugated linker to limit the electronic influence of the electron-donating ether group. The ligand 22 (Fig. 2) was also synthesised (Scheme S1†) to investigate the effect of directly functionalising the pyridyl moiety with the methylenecyclohexylether group, which is expected to destabilise the lowest unoccupied molecular orbital (LUMO) and further blue shift emission.
As observed for 12, the bis(trifluoromethyl) bridge 18 resulted in only a single diastereomer for complex 14 (Fig. 2). These two examples (12 and 14) suggest that bis(alkyl)hydrazide bridges afford diiridium complexes from racemic μ-dichloro dimers without the formation of diastereomeric mixtures. This is complementary to using enantiomerically pure dichloro-bridged dimers, as reported for other systems.49,52
Analogous to the mesityl-functionalised complex 11, the diastereomers rac13 (stereochemistry confirmed by X-ray diffraction, Fig. 4) and meso13 were easily separated. The improved solubility imparted by the methylenecyclohexylether groups allowed both diastereomers to be fully characterised. Complex 15 was isolated as a single diastereomer: the absolute configuration is unknown, although it is probably the meso structure from inspection of the 1H NMR spectrum (Fig. S66†). A second diastereomer was observed by NMR but could not be isolated.
 | ||
| Fig. 4 X-ray molecular structures of meso7, meso9 and the core part of rac13 (ΔΔ) with the xylyl substituents (R) omitted. Thermal ellipsoids are drawn at 50% probability level. H atoms are omitted for clarity. Vector D identifies intramolecular π–π interactions, meso7 = 3.32 Å, meso9 = 3.24 Å, rac13 = 3.27, 3.19 Å. | ||
Thermal gravimetric analysis (TGA) shows that all the complexes 7–15 possess good thermal stability (Fig. S144–S153†).
X-Ray molecular structures
Complexes 7 and 9–13 (Fig. 4 and S97–S103†) were characterised by single-crystal X-ray crystallography. Relevant parameters are listed in Table S1.† All structures except 9 and 10 contained disordered CH2Cl2 or CD2Cl2 of crystallisation.In meso complexes 7, 9 and 12, the molecule possesses a crystallographic inversion centre (located at the midpoint of the N–N bond) relating the Λ and Δ metal centres. The rac complexes 10, 11 and 13 all crystallise in centrosymmetric space groups, thus each molecule is chiral (ΛΛ or ΔΔ) but the crystal is racemic. Two solvent-free polymorphs of 10 formed concomitantly; in α-10 the molecule lies on a crystallographic twofold axis while in β-10 (as in 11 and 13) it has no crystallographic symmetry. Each Ir atom has distorted octahedral coordination, involving one N and one O atom of the bridging hydrazide (OCNNCO) ligand, and two C^N cyclometalating ligands. As usual, the N atoms of the latter occupy axial positions, trans to one another.6,21 As reported earlier,22 in meso complexes the hydrazide moiety is planar, while in rac isomers it is variously (by 7 to 24°) folded along the central N–N bond into two planar OCNN chelating fragments. The chelated Ir atoms can be coplanar with, or displaced from, their planes, but this does not affect the bonding pattern significantly. Each aryl substituent (A) at the bridging ligand is oriented approximately perpendicular to the hydrazide plane (thus precluding π-conjugation) and is stacked face-to-face (π–π) with a cyclometalating ligand, essentially with its phenyl ring (B) (Fig. 4, S98–S101 and S103†). This will shorten the effective conjugation length of the bridge and is beneficial for shifting emission towards the blue (see below).
Generally, the stacking is closer and more parallel than in previously studied analogues with t-Bu and CF3-substituents.21,22 To the best of our knowledge the systems studied here demonstrate the closest intramolecular π–π stacking reported for cyclometallated iridium complexes.22,29–33 Comparison of the two polymorphs of 10 shows that different crystal packing has limited effect on the molecular conformation: in α-10 both rings A in a molecule are eclipsed with corresponding rings B, in β-10 one pair is nearly eclipsed and the other shows a quasi-graphitic overlap, ring A shifting towards the pyridyl ring of the C^N ligand. Interestingly, molecule 12, which lacks intramolecular stacking, is much less rigid – note the different conformations of two crystallographically non-equivalent molecules in the crystal (Fig. S102†).
Computational study
The optimised ground state S0 geometries for the complexes were calculated at the B3LYP/LANL2DZ:3-21G* level with the LANL2DZ pseudopotential for the iridium atoms and the 3-21G* basis set for other atoms. This model chemistry was selected on the basis of previous computational studies,50,53 and ensures that these calculations are directly comparable with those reported for other diiridium complexes (such as complex 1).21,22 For the complexes 13–15 the methylene cyclohexylether groups were substituted for methoxy groups to shorten calculation times. The geometries of the central hydrazide fragments are in good agreement with the XRD results discussed above.Molecular orbital calculations provided insight into the localisation of the frontier molecular orbitals (FMOs). Reasonable agreement is observed between diastereomers for all complexes. The LUMOs are localised on the cyclometalating ligands, particularly the pyridyl moieties.21,22 However, the localisation of the highest occupied molecular orbitals (HOMOs) varies more significantly between complexes: in some cases the HOMO contribution from the bridge centre is high (≥30%) (complexes 7, 10, 13 and 15) whereas in other cases the bridging ligands display ancillary character (complexes 8, 9, 11, 12 and 14). In this study, if the average HOMO contribution from the bridge centre for both diastereomers is <15%, the bridge is considered ancillary. This is summarised in Table S2.† FMO plots for complexes 7, 9, 12 and 13 are given in Fig. 5 as representative examples. FMO plots for the other complexes are shown in Fig. S126–S143.†
 | ||
| Fig. 5 Molecular orbital compositions for complexes rac7, rac9, meso12 and rac13. The stated ratios represent the atom/group contributions in percentages. Bridge = central OCNNCO fragment. | ||
For complex 7 the HOMO has significant contributions from the Ir centres, the central component of the hydrazide bridge and the cyclometalating phenyl moieties, as in complex 1.21,22 Further fluorination of the bridging aryl rings decreases the bridge HOMO contributions for complexes 8 (octafluoro) and 9 (decafluoro), so their HOMOs are primarily localised on the Ir centres and the cyclometalating phenyl groups, with their bridges expected to behave as ancillary ligands. As complex 10 also features methoxy groups on the bridging unit, the effect of the electron withdrawing fluorine atoms is somewhat negated and the bridge still features notable HOMO localisation (32% average). Calculations predict very similar HOMO contributions for complexes 9 and 11, indicating that the mesityl groups have a negligible electronic effect, as expected.47,48 Lowering the π orbital energy of the cyclometalating ligands of complexes 13 and 15 through fluorination strongly shifts their HOMOs onto the bridging ligands so that the cyclometalating phenyl moieties have very low HOMO contributions (average of both diastereomers <15% for both complexes). There is negligible frontier orbital (HOMO or LUMO) contribution from the bridge aryl rings for all complexes featuring diarylhydrazide bridges, even upon perfluorination.
For complexes 12 and 14 the bridging ligands are ancillary with negligible HOMO contributions (average of both diastereomers = 4% for both complexes), regardless of cyclometalating ligand fluorination. This is indicative of the shorter conjugation length of the bis(trifluoromethyl) bridge 18 compared to the diarylhydrazide bridges studied here.
Electrochemistry
Complexes 7–15 (Fig. 2) were studied by cyclic voltammetry (CV) to obtain their oxidation and reduction potentials. The data are listed in Table 1 and voltammograms are shown in Fig. S104–S125.† All complexes display two electrochemically reversible oxidation waves. These represent sequential oxidation of the iridium centres (Ir3+/Ir4+ redox couples), which are electronically coupled via the conjugated bridging units and so are electrochemically inequivalent. For complexes 11 and 15 as representative examples, both oxidation processes were shown to be chemically reversible over 10 cycles (Fig. S114 and S115†).| Complex | Isomer | E ox(1)/V | E ox(2)/V | ΔE1/2a/V |
E
redonsetb/V |
HOMOc/eV | LUMOd/eV |
|---|---|---|---|---|---|---|---|
| E pa/Epc [E1/2] | E pa/Epc [E1/2] | ||||||
| a Peak splitting between Eox(1) and Eox(2). b All reductions are electrochemically irreversible. c HOMO levels calculated from CV potentials by HOMO = −4.8 + (−Eox(1)1/2), using ferrocene as the standard. d LUMO levels calculated from CV potentials by LUMO = −4.8 + (−Eredonset), using ferrocene as the standard. e Complexes 14 and 15 were isolated as single diastereomers; their absolute configurations are unknown. | |||||||
| 7 | Mixture | 0.53/0.31 [0.42] | 0.77/0.58 [0.67] | 0.25 | −2.38 | −5.22 | −2.42 |
| 8 | Mixture | 0.56/0.49 [0.52] | 0.81/0.74 [0.77] | 0.25 | −2.18 | −5.32 | −2.62 |
| 9 | Mixture | 0.61/0.52 [0.56] | 0.85/0.76 [0.81] | 0.25 | −2.37 | −5.36 | −2.43 |
| 10 | Mixture | 0.54/0.46 [0.50] | 0.80/0.72 [0.76] | 0.26 | −2.29 | −5.30 | −2.51 |
| 11 | rac | 0.66/0.49 [0.58] | 0.96/0.84 [0.90] | 0.32 | −2.37 | −5.38 | −2.43 |
| 12 | meso | 0.67/0.57 [0.62] | 0.85/0.72 [0.78] | 0.16 | −2.44 | −5.42 | −2.36 |
| 13 | meso | 0.96/0.90 [0.93] | 1.36/1.21 [1.28] | 0.35 | −2.16 | −5.73 | −2.66 |
| rac | 1.00/0.93 [0.97] | 1.43/1.23 [1.33] | 0.36 | −2.14 | −5.77 | −2.64 | |
| 14 | 0.99/0.91 [0.95] | 1.18/1.07 [1.12] | 0.17 | −2.15 | −5.75 | −2.65 | |
| 15 | 0.87/0.75 [0.81] | 1.24/1.12 [1.18] | 0.37 | −2.19 | −5.61 | −2.61 | |
Complex 7, which features 4 fluorine atoms on the bridging unit, displays the lowest first oxidation potential (Eox(1)). As expected, increasing to 8 (complex 8) and 10 fluorine atoms (complex 9) leads to successively higher oxidation potentials. Due to the addition of electron-rich methoxy groups to the octafluoro bridging unit, the oxidation potential of complex 10 is slightly decreased by 0.02 V compared to complex 9. A relatively small variation in oxidation potentials (0.04 V) across the series 7–10 supports DFT predictions that the bridges in 8 and 9 behave as ancillary ligands. Complexes 7–10, which vary only in the extent of bridge fluorination, all feature very similar peak splittings (ΔE1/2ca. 0.25 V), indicating similar electronic coupling between the Ir centres for this series.
Functionalising the ppy ligands of complex 11 with mesityl groups does not significantly influence Eox(1) (an increase of only 0.02 V is observed compared to complex 9), indicating that they have minimal electronic effect.47,48 However, it is interesting that the second oxidation potential (Eox(2)) of 11 is shifted to a significantly higher potential compared to complex 9 (0.90 V vs. 0.81 V) leading to a larger ΔE1/2 value of 0.32 V for 11 compared to 0.25 V for 9. A tentative explanation is that the mesityl groups, could sterically interact over the bridging unit (Fig. S101†). This would lower the molecular flexibility and could hinder structural rearrangement to the dication, thereby increasing Eox(2) of 11 compared to the more flexible complex 9.
The oxidation potential of 12 is higher than that of 11 by 0.04 V, suggesting that the bis(trifluoromethyl)-functionalised bridge (18) is more strongly electron withdrawing than the bis(pentafluorophenyl) bridge (17c).46 The ΔE1/2 value obtained for 12 (0.16 V) is also half of that observed for 11, implying weak communication between the two iridium centres. This is in line with the ancillary nature of the bridge and in agreement with DFT (Table S2†). The addition of fluorinated cyclometalating ligands to complexes meso13 and rac13 further shifts their oxidation potentials to more positive values, as expected from DFT, which predicts high HOMO contributions from the cyclometalating phenyl rings of complex 11 (Table S2†). The ΔE1/2 values for meso13 and rac13 are also greater than for complex 11 (by 0.03/0.04 V) which may be due to the reduced ancillary character of the bis(pentafluorophenyl) bridge in these complexes, also in line with DFT predictions.
Complex 14 has an oxidation potential almost identical to meso13 and rac13, indicating very similar HOMO energies. Analogous to the relationship between complexes 11 and 12, complex 14 displays a much lower ΔE1/2 value than either diastereomer of complex 13, which suggests a higher ancillary character of the bis(trifluoromethyl) bridge (and so weaker Ir⋯Ir communication), as inferred by DFT.
The first oxidation potential of 15 is cathodically shifted compared to complexes 13 (by ca. 0.1 V). This is due to the absence of the xylyl spacer which electronically decouples the electron donating methylenecyclohexylether group from the ppy ligands. Complex 15 also has the largest ΔE1/2 value (0.37 V), in agreement with DFT which predicts the bridging unit to be the least ancillary of the series (Table S2†).
The reduction potentials for 7–15 were also estimated by CV. The data for the reduction scans are included in Table 1 and the voltammograms are shown in Fig. S116–125.† All complexes display irreversible reductions. This adds significant error to their accurate determination, complicating the detailed analysis of any trends. A similar situation has been previously encountered in the study of monoiridium complexes by Baranoff and Nazeeruddin et al.54 Nevertheless, the reduction onsets for the complexes 7–15 are in the range of −2.1 to −2.4 V vs. FcH/FcH+, which is a reasonable fit with their emission energies (discussed below) and are similar to those reported for ppy-based monoiridium complexes.55 Generally, functionalisation of the cyclometallating ligands of 13–15 with electron-withdrawing fluorine atoms decreases their reduction potentials compared to those of complexes 7–12 as expected.55 The reduction potential for 15 is marginally greater than for 13 and 14 (−2.19 V vs. −2.14/−2.16 V and −2.15 V), which is expected from the DFT data upon direct functionalisation of the LUMO-bearing pyridyl moieties with electron-donating methylenecyclohexyl ether groups.
Photophysical data
The emission spectra for the complexes are shown in Fig. 6–9 and Fig. S155–S157† and the key photophysical data are given in Table 2. Absorption data are presented in Fig. S154 and Table S3.† Complex 7 is nonemissive in DCM solution at room temperature, while being highly emissive (PLQY = 61 ± 10%) when doped into a rigid poly(methyl methacrylate) (PMMA) matrix. This is consistent with the data for complex 1,22 for which the flexible central bridging unit (that DFT predicts to have significant HOMO character) can provide a pathway for non-radiative quenching of the excited state in solution, which can be inhibited by doping the complex into a rigid host matrix.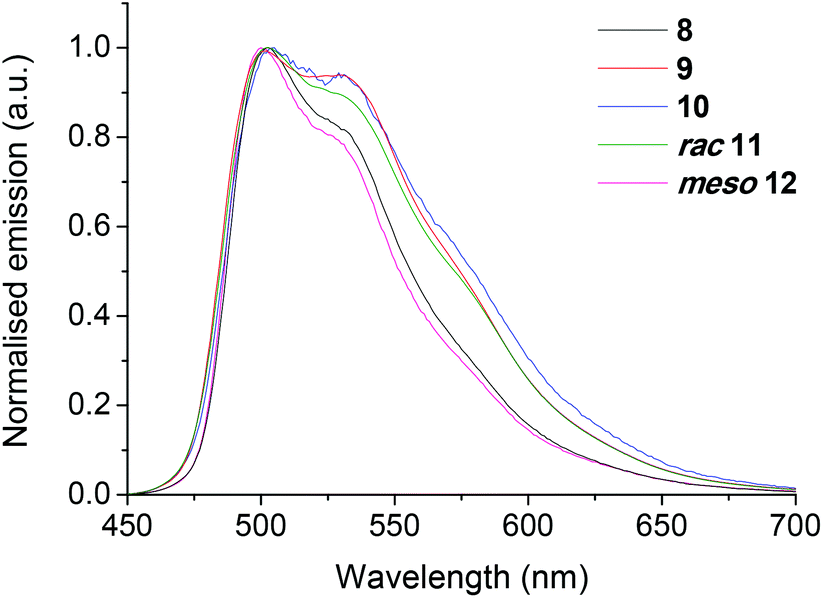 | ||
| Fig. 6 Normalised emission spectra of complexes 8–12 in degassed DCM solutions at room temperature (λexc 355 nm). | ||
 | ||
| Fig. 7 Normalised emission spectra of complexes 7–12 doped into PMMA at 1 wt% at room temperature (λexc 355 nm). Inset: photograph of emission from a doped PMMA film (left) and degassed DCM solution (right) of rac11 under irradiation from a 365 nm UV lamp. | ||
 | ||
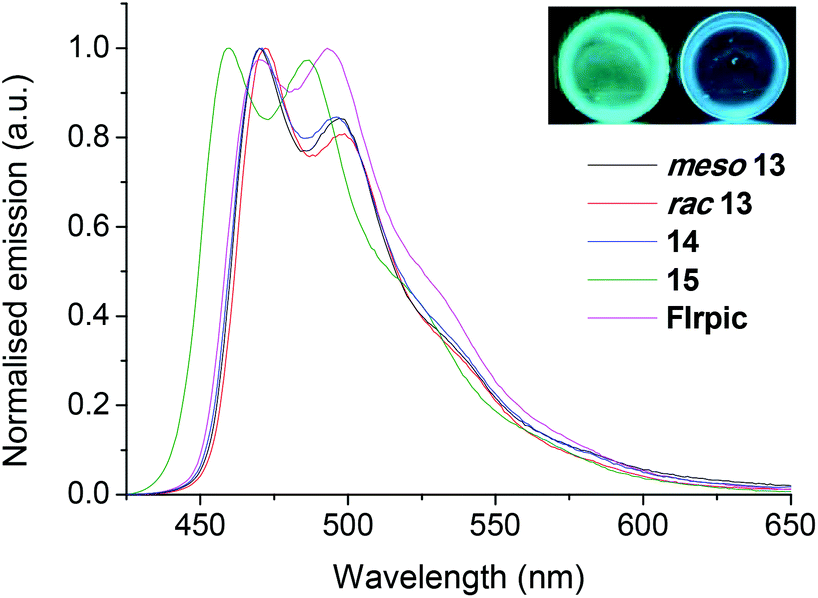
| Fig. 8 Normalised emission spectra of complexes 13–15 and FIrpic in degassed DCM solutions at room temperature (λexc 355 nm). The emission spectrum of 15 is poorly resolved due to a low solution PLQY. Inset: (left) chemical structure of FIrpic. (Right) photograph of emission from a doped PMMA film and degassed DCM solution of rac13 under irradiation from a 365 nm UV lamp. | ||
 | ||
| Fig. 9 Normalised emission spectra of complexes 13–15 and FIrpic doped into PMMA at 1 wt% at room temperature (λexc 355 nm). Inset: photograph of the emission from doped PMMA films of rac13 (left) and 15 (right) under irradiation from a 365 nm UV lamp. | ||
| DCM solutionb | 2-MeTHF glassc | Doped into PMMA 1 wt%d | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Complex | Isomer | λ max em/nm [CIExy] | PLQY/% (±5%) | τ p/μs | k r/× 105 s−1 | k nr/× 105 s−1 | λ max em/nm (λ10% em/nm)e[ET/eV]f | τ p/μs | λ max em/nm [CIExy] | PLQY/% (±10%) | τ p/μs | k r/× 105 s−1 | k nr/× 105 s−1 |
| a Single diastereomer of unknown absolute configuration. sh = shoulder. b Solution photoluminescence measurements were recorded in degassed DCM solutions at ca. 20 °C with an excitation wavelength of 355 nm with quinine sulfate in 0.5 M H2SO4 as standard (Φ = 0.546).61 c Measured at 77 K using an excitation wavelength of 355 nm. d Measured in an integrating sphere under air using an excitation wavelength of 355 nm. e Wavelength at 10% intensity on the blue edge of the spectrum obtained at 77 K. f Estimated using ET = hc/λ10% em. g Non-emissive is defined as PLQY <0.05%. h Error = ±4%. i Error = ±2%. j All FIrpic data were obtained in-house for direct comparison unless otherwise stated. k Values taken from ref. 62. τp = 1/knr + kr. | |||||||||||||
| 7 | Mixture | Non-emissiveg | 500 (490) [2.53] | 3.62 | 516 [0.28, 0.64] | 61 | 1.81 | 3.37 | 2.15 | ||||
| 8 | Mixture | 503 [0.27, 0.61] | 66 | 1.84 | 3.61 | 1.83 | 492 (484) [2.56] | 3.41 | 503 [0.25, 0.62] | 59 | 2.00 | 2.95 | 2.05 |
| 9 | Mixture | 499 [0.30, 0.58] | 76 | 2.24 | 3.40 | 1.07 | 492 (482) [2.57] | 3.55 | 503 [0.25, 0.62] | 71 | 2.08 | 3.41 | 1.39 |
| 10 | Mixture | 505 [0.31, 0.58] | 78 | 2.09 | 3.73 | 1.05 | 493 (485) [2.56] | 3.33 | 507 [0.25, 0.63] | 66 | 2.02 | 3.27 | 1.68 |
| 11 | rac | 502 [0.30, 0.58] | 88 | 1.66 | 5.30 | 0.72 | 494 (485) [2.56] | 2.67 | 507 [0.25, 0.63] | 72 | 1.39 | 5.18 | 2.01 |
| 12 | meso | 500 [0.26, 0.60] | 22 | 0.34 | 6.41 | 22.7 | 491 (483) [2.57] | 2.30 | 504 [0.25, 0.63] | 66 | 1.14 | 5.79 | 2.98 |
| 13 | meso | 470 [0.18, 0.36] | 48 | 0.69 | 6.93 | 7.48 | 461 (455) [2.72] | 2.24 | 470 [0.16, 0.33] | 65 | 1.19 | 5.46 | 2.94 |
| rac | 470 [0.18, 0.36] | 47 | 0.73 | 6.49 | 7.23 | 463 (456) [2.72] | 1.78 | 472 [0.15, 0.33] | 60 | 1.18 | 5.51 | 3.39 | |
| 14 | 470 [0.16, 0.33] | 4h | 0.07 | 5.77 | 135 | 462 (454) [2.73] | 1.92 | 471 [0.15, 0.33] | 46 | 1.12 | 4.11 | 4.82 | |
| 15 | 459 [0.20, 0.28] | 2i | 0.11 | 1.64 | 89.3 | 451 (441) [2.81] | 2.24 | 460 [0.15, 0.24] | 69 | 1.62 | 4.26 | 1.91 | |
| FIrpicj | — | 468 [0.19, 0.37] | 73 | 1.85 | 3.95 | 1.46 | 463 [2.62]k | 2.24k | 470sh, 493 [0.15, 0.33] | 74 | 1.69 | 4.38 | 1.54 |
Complexes 8–10 have significantly different photophysical properties than 7, in that they are highly emissive in solution and in PMMA, with very similar PLQY values in both media. This is consistent with rigidification of 8–10 by intramolecular π–π stacking, which restricts rotation of the bridge aryl rings. This is observed in the solution 19F NMR spectra of 8–10 (Fig. 3, S5, S9, S15 and S18†) and removes the requirement to impede bridge flexibility by using a rigid matrix such as PMMA.
Another possible explanation is that for complexes with an ancillary bridging unit (Table S2†) such as 8 and 9, motion of the bridge does not provide as efficient a non-radiative pathway to the ground state in solution. However, as complex 10 features a non-ancillary bridge with notable HOMO character (Table S2†) while still exhibiting a high solution PLQY (78 ± 5%), it is evident that intramolecular π–π stacking is the main reason for high solution PLQYs in highly fluorinated diarylhydrazide-bridged diiridium complexes.
The emission spectra of 8–10 are blue shifted compared to 7 (by ca. 10 nm in PMMA) (Fig. 7). This is a result of HOMO stabilisation through further fluorination of the bridging units (in agreement with electrochemical data – Table 1). Complexes 8–10 exhibit near identical Commission Internationale de L′Éclairage (CIExy) colour coordinates in PMMA of (0.25, 0.62/0.63) in the green region of the spectrum. The triplet energies (ET) for 8–10 (obtained from emission spectra recorded in 2-MeTHF at 77 K, Fig. S156†) are also nearly identical (2.56–2.57 eV). These data provide additional experimental support for the DFT prediction that the bridges in 8 and 9 behave as ancillary ligands.
The mesityl groups in rac11 result in a significant increase in the radiative rate constant (kr) compared to complex 9 in DCM solution (5.30 vs. 3.40 × 105 s−1) and in PMMA (5.18 vs. 4.41 × 105 s−1). This leads to a small increase in solution PLQY (88 ± 5% for rac11vs. 76 ± 5% for complex 9), whereas the PLQYs in PMMA for 9 and rac11 are very similar (71 ± 10% and 72 ± 10%, respectively). The incorporation of mesityl groups is known to increase PLQYs and kr values in monoiridium systems.47,48 As mesityl groups have a negligible electronic effect, the CIExy coordinates (in both DCM an PMMA) and ET values for 9 and rac11 are nearly identical.47,48
Complex meso12 is moderately emissive in DCM solution (PLQY = 22 ± 5%) and is highly emissive in PMMA (PLQY = 66 ± 10%). This is due to an order of magnitude decrease in knr upon doping the complex into PMMA (Table 2), which can be attributed to higher molecular flexibility inferred from the XRD data (discussed above, Fig. S102†). Although meso12 is not rigidified by intramolecular π–π interactions, it is still emissive in solution, albeit to a lesser extent than rac11. This may be related to the ancillary nature of the bridging ligand (predicted by DFT), which may reduce the efficiency of non-radiative quenching through bridge motion, as mentioned above.
Other than their solution PLQY values and the presence/absence of intramolecular π–π interactions, complexes rac11 and meso12 display similar theoretical (Table S2†), electrochemical (Table 1) and photophysical (Table 2) properties. A direct comparison therefore serves as good evidence that intramolecular π–π interactions contribute significantly to the high solution PLQYs of the diarylhydrazide-bridged complexes.
Incorporation of the fluorinated cyclometalating ligand 21 into the diastereomers meso13 and rac13 shifts their emission energies into the sky-blue region (Fig. 8 and 9). In DCM both meso13 and rac13 have PLQYs of 47/48 ± 5% with CIExy coordinates (0.18, 0.36) marginally lower than the archetypal sky-blue emitter FIrpic (Fig. 8)38,56 (0.19, 0.37), even though their λmax values are red shifted compared to FIrpic by 2 nm. This is related to their narrower full width at half maximum (FWHM) values because of diminished v0,1 vibronic shoulders: FWHM FIrpic = 82 nm, meso13 = 63 nm, rac13 = 69 nm. This is again consistent with higher molecular rigidity, due to the intramolecular π–π interactions (observed in the 19F NMR spectra of meso13 and rac13 – Fig. S42 and S51†).
Molecular rigidity also influences the Huang-Rhys factor (SM), which is proportional to the degree of structural distortion which occurs in the excited state of a molecule relative to the ground state.57SM values were estimated for FIrpic, meso13 and rac13 from the relative heights of the v0,0 and v0,1 peaks in their 77 K emission spectra (Fig. S157,† FIrpic spectrum obtained from ref. 56).57,58 The following values were obtained: FIrpic = 0.7, meso13 = 0.4, rac13 = 0.5 (1 s.f.). These values indicate a lower intensity vibronic progression for the rigid diiridium complexes compared to FIrpic, which is vital for obtaining high colour purity.
Similarly, favourable photophysical properties are also observed for meso13 and rac13 when doped into PMMA: high PLQYs of 60/65 ± 10% (FIrpic 74 ± 10%) and comparatively narrow FWHM values of 55/56 nm (FIrpic 67 nm) (Fig. 9).
These comparatively narrow emission spectra are significant as the complexes are predicted to feature non-ancillary bridging ligands (see the DFT discussed above), which will likely lead to excited states with noteworthy interligand charge transfer (ILCT) character. ILCT character leads to broader, less structured emission due to more diffusely localised excited states.58–60 It is expected that the rigidifying effect of the intramolecular π–π interactions counteracts this, promoting sharper emission bands. These data indicate that diiridium complexes show promise as a platform for developing blue phosphors with good colour purity.
meso 13 and rac13 feature higher kr values than FIrpic (by ∼20–40%) under directly comparable conditions in both DCM solution and PMMA. This may be related to the strong Ir⋯Ir coupling observed in the electrochemistry (Table 1), and results in notably shorter τp values in PMMA of 1.18/1.19 μs (vs. 1.69 μs for FIrpic).
Enhanced radiative rate constants compared to monoiridium analogues have been reported for green to red diiridium complexes, which may be due to augmented spin–orbit coupling.23,24,26,50,63 Blue phosphors tend to possess excited states with more LC character than green emitting complexes,64–66 which is an indication of poorer LC/MLCT state mixing (lower MLCT character) and can lead to inherently lower kr values and so longer τp. The observations presented here indicate that diiridium complexes are promising systems for developing blue phosphors with higher kr values and therefore shorter τp which is a highly sought-after property.67
In a similar manner to the relationship between rac11 and meso12, complex 14 is an analogue of 13 which cannot exhibit intramolecular π–π interactions between the cyclometalating and bridging ligands. As a result, 14 displays a low solution PLQY of 4 ± 4%. In PMMA the PLQY of 14 increases to 46 ± 10%, which is ascribed to a restriction of intramolecular motion, evident from the substantial decrease in knr (Table 2). The PLQY of 14 in PMMA is, however, significantly lower than those for either diastereomer of 13 (60/65 ± 10%). This is due to: (1) a substantially higher knr value, which crucially indicates that intramolecular π–π interactions are also beneficial for obtaining high solid state PLQY values in diiridium complexes, and (2) a lower kr value (Table 2), which may be related to the smaller Ir⋯Ir coupling in 14 observed in the electrochemistry (Table 1).
Despite the lack of rigidifying intramolecular π–π interactions, 14 exhibits sharp emission similar to 13 (FWHM in PMMA = 57 nm) (Fig. 9). This is consistent with the ancillary nature of the bis(trifluoromethyl) bridge 18, which is expected to limit the ILCT character of the excited state. The estimated SM value for 14 is 0.6 (1 s.f.): larger than for either diastereomer of 13, but still smaller than for FIrpic. These data indicate that designing diiridium complexes with highly ancillary bridges could be a way to obtain sharp emission from such systems.
The emission from complex 15 is shifted deeper into the blue than for 13 or 14. This is attributed to the LUMO-destabilising methylenecyclohexylether groups. As well as being tentatively observed in the reduction potentials above (Table 1), this can also be concluded from the more reliable oxidation potential data which indicate that the HOMO of 15 is shallower than for 13 or 14. When doped into PMMA, 15 displays a high PLQY of 69 ± 10%. This is comparable to the value obtained for FIrpic under the same experimental conditions, while the colour is notably superior: 15 emits at a λmax of 460 nm, pushing the CIExy coordinates to a total value below 0.4 (0.15, 0.24). Complex 15 also displays a τp of 1.62 μs in PMMA, which is short in a doped film for an Ir complex with total CIExy < 0.4/λmax ≤ 460 nm and a high PLQY.47,68–71 This can be attributed to the high kr, which is likely related to the dinuclear nature of the complex as mentioned above.
Despite the presence of rigidifying intramolecular π–π interactions (observed in the 19F NMR spectrum – Fig. S68†), the PLQY for 15 in DCM solution is low (2 ± 2%). This fits a trend of decreasing solution PLQY with increasing emission energy in the complexes rac11 (λmax = 502 nm, PLQY = 88 ± 5%), 13 (λmax = 470 nm, PLQY = 47/48 ± 5%) and 15 (λmax = 459 nm, PLQY = 2 ± 2%) due to incremental order of magnitude increases in their knr values (0.72, 7.23/7.48 and 89.3 × 105 s−1). In contrast, all three complexes exhibit high PLQYs (>60%) and similar knr values (1.91–3.39 × 105 s−1) when doped into PMMA. Therefore, it appears that as the excited state energy increases, the rigidifying effect of the intramolecular π–π interactions is overcome and their capability to promote emission in solution is reduced.
Emission in the sky-blue region from diiridium complexes with conjugated bridging ligands is unprecedented. It has been accomplished by the synergistic choice of bridging and cyclometalating ligands. The key role of the bridge is clear as there are reports of diiridium complexes bearing dfppy-type peripheral ligands for which sky-blue emission was not achieved.8,16,72–74 Although diiridium systems have shown promise as high performing phosphors in the lower energy range (from red through to green),21–24,26,27,50,75 to the best of our knowledge no complex displaying λmax (PL) below ca. 490 nm at room temperature has been reported thus far.22 Mazzanti and co-workers reported a fluorinated diiridium complex with a vibronic sideband at 477 nm, but the λmax is ca. 510 nm and the emission extends to 800 nm.16 The results presented here considerably extend the diiridium complex literature, and indicate that if the complexes are correctly designed, their colour versatility is potentially comparable to monoiridium systems.
Conclusions
We have developed new concepts in the chemistry of diiridium complexes with the synthesis, structural and optoelectronic characterisation of a series of highly fluorinated hydrazide-bridged complexes.Complexes 7–12 represent an ideal platform for investigating intramolecular π–π interactions between aryl and perfluoroaryl rings in organometallic systems, both in the solid state (by XRD) and in solution (by 19F NMR spectroscopy). These interactions are shown to be an innovative way to rigidify diiridium complexes, leading to significant and advantageous effects on their photophysical properties. Electrochemical and computational studies have further extended the understanding of these systems. This knowledge has been applied to the rational design and synthesis of the first reported sky-blue emitting diiridium complexes 13–15. Their favourable photophysical properties are a consequence of both the dinuclear nature of the complexes and the beneficial intramolecular π–π interactions. They possess high PLQYs, λmax as low as 460 nm (CIEx+y < 0.4), high kr, relatively short τp, and in some cases, notably sharp emission. The results presented here greatly extend the versatility of luminescent diiridium complexes by shifting phosphorescence into the sky-blue region of the visible spectrum with the aid of tailored non-covalent interactions. It is now a challenge to design and implement further structural modifications that could shift the emission of diiridium complexes deeper in the blue region.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Alan Kenwright and Dr Juan Aguilar-Malavia are acknowledged for their help in devising, running and interpreting 2D NMR experiments. Prof. Graham Sandford is acknowledged for helpful 19F NMR discussion. We thank EPSRC grant EP/L02621X/1 for funding.Notes and references
- Z. Q. Chen, Z. Q. Bian and C. H. Huang, Adv. Mater., 2010, 22, 1534–1539 CrossRef CAS PubMed.
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- K. K.-W. Lo, K. H.-K. Tsang, K.-S. Sze, C.-K. Chung, T. K.-M. Lee, K. Y. Zhang, W.-K. Hui, C.-K. Li, J. S.-Y. Lau, D. C.-M. Ng and N. Zhu, Coord. Chem. Rev., 2007, 251, 2292–2310 CrossRef CAS.
- R. Gao, D. G. Ho, B. Hernandez, M. Selke, D. Murphy, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 14828–14829 CrossRef CAS PubMed.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- X. Yuan, S. Zhang and Y. Ding, Inorg. Chem. Commun., 2012, 17, 26–29 CrossRef CAS.
- L. Donato, C. E. McCusker, F. N. Castellano and E. Zysman-Colman, Inorg. Chem., 2013, 52, 8495–8504 CrossRef CAS PubMed.
- A. Tsuboyama, T. Takiguchi, S. Okada, M. Osawa, M. Hoshino and K. Ueno, Dalton Trans., 2004, 1115–1116 RSC.
- E. A. Plummer, W. Hofstraat and L. De Cola, Dalton Trans., 2003, 2080–2084 RSC.
- A. Auffrant, A. Barbieri, F. Barigelletti, J. Lacour, P. Mobian, J.-P. Collin, J.-P. Sauvage and B. Ventura, Inorg. Chem., 2007, 46, 6911–6919 CrossRef CAS PubMed.
- V. L. Whittle and J. A. G. Williams, Inorg. Chem., 2008, 47, 6596–6607 CrossRef CAS PubMed.
- W.-J. Xu, S.-J. Liu, X. Zhao, N. Zhao, Z.-Q. Liu, H. Xu, H. Liang, Q. Zhao, X.-Q. Yu and W. Huang, Chem. – Eur. J., 2013, 19, 621–629 CrossRef CAS PubMed.
- V. Chandrasekhar, B. Mahanti, P. Bandipalli and K. Bhanuprakash, Inorg. Chem., 2012, 51, 10536–10547 CrossRef CAS PubMed.
- E. S. Andreiadis, D. Imbert, J. Pécaut, A. Calborean, I. Ciofini, C. Adamo, R. Demadrille and M. Mazzanti, Inorg. Chem., 2011, 50, 8197–8206 CrossRef CAS PubMed.
- A. G. Tennyson, E. L. Rosen, M. S. Collins, V. M. Lynch and C. W. Bielawski, Inorg. Chem., 2009, 48, 6924–6933 CrossRef CAS PubMed.
- T. Hajra, A. J. K. Bera and V. Chandrasekhar, Aust. J. Chem., 2011, 64, 561–566 CAS.
- A. M. Prokhorov, A. Santoro, J. A. G. Williams and D. W. Bruce, Angew. Chem., Int. Ed., 2012, 51, 95–98 CrossRef CAS PubMed.
- M. Graf, R. Czerwieniec and K. Sünkel, Z. Anorg. Allg. Chem., 2013, 639, 1090–1094 CrossRef CAS.
- Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, K. Abdullah, V. Jankus, M. R. Bryce and A. P. Monkman, Angew. Chem., Int. Ed., 2014, 53, 11616–11619 CrossRef CAS PubMed.
- D. G. Congrave, Y.-t. Hsu, A. S. Batsanov, A. Beeby and M. R. Bryce, Organometallics, 2017, 36, 981–993 CrossRef CAS.
- G. Li, Y. Wu, G. Shan, W. Che, D. Zhu, B. Song, L. Yan, Z. Su and M. R. Bryce, Chem. Commun., 2014, 50, 6977–6980 RSC.
- P.-H. Lanoë, C. M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6831–6834 RSC.
- V. Chandrasekhar, T. Hajra, J. K. Bera, S. M. W. Rahaman, N. Satumtira, O. Elbjeirami and M. A. Omary, Inorg. Chem., 2012, 51, 1319–1329 CrossRef CAS PubMed.
- R. E. Daniels, S. Culham, M. Hunter, M. C. Durrant, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Dalton Trans., 2016, 45, 6949–6962 RSC.
- X. Yang, Z. Feng, J. Zhao, J.-S. Dang, B. Liu, K. Zhang and G. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 33874–33887 CAS.
- Z. Hao, H. Jiang, Y. Liu, Y. Zhang, J. Yu, Y. Wang, H. Tan, S. Su and W. Zhu, Tetrahedron, 2016, 72, 8542–8549 CrossRef CAS . Although emission of a similar colour to FIrpic is reported, the two Ir centres are non-conjugated.
- A. M. Bünzli, E. C. Constable, C. E. Housecroft, A. Prescimone, J. A. Zampese, G. Longo, L. Gil-Escrig, A. Pertegás, E. Ortí and H. J. Bolink, Chem. Sci., 2015, 6, 2843–2852 RSC.
- E. C. Constable, C. E. Housecroft, P. Kopecky, C. J. Martin, I. A. Wright, J. A. Zampese, H. J. Bolink and A. Pertegas, Dalton Trans., 2013, 42, 8086–8103 RSC.
- L. He, D. Ma, L. Duan, Y. Wei, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, Inorg. Chem., 2012, 51, 4502–4510 CrossRef CAS PubMed.
- S. Kumar, Y. Hisamatsu, Y. Tamaki, O. Ishitani and S. Aoki, Inorg. Chem., 2016, 55, 3829–3843 CrossRef CAS PubMed.
- P. Li, G. G. Shan, H. T. Cao, D. X. Zhu, Z. M. Su, R. Jitchati and M. R. Bryce, Eur. J. Inorg. Chem., 2014, 2376–2382 CrossRef CAS.
- C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CrossRef CAS.
- V. J. H. Williams, J. K. Cockcroft and N. Fitch, Angew. Chem., 1992, 104, 1666–1669 CrossRef.
- J. H. Williams, Acc. Chem. Res., 1993, 26, 593–598 CrossRef CAS.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed.
- A. F. Rausch, M. E. Thompson and H. Yersin, J. Phys. Chem. A, 2009, 113, 5927–5932 CrossRef CAS PubMed.
- S. Berger, S. Braun and H.-O. Kalinowski, NMR Spectroscopy of the Non-Metallic Elements, Wiley, 1st edn, 1997 Search PubMed.
- R. E. Banks and R. N. Haszeldine, J. Chem. Soc., 1967, 1822–1826 CAS.
- E. Klauke, L. Oehlmann and B. Baasner, J. Fluorine Chem., 1982, 21, 495–513 CrossRef CAS.
- J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS PubMed.
- R. J. Abraham, D. B. Macdonald and E. S. Pepper, J. Am. Chem. Soc., 1968, 90, 147–153 CrossRef CAS.
- J. Wang, J. Luis, C. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- R. Jana, B. Sarkar, D. Bubrin, J. Fiedler and W. Kaim, Inorg. Chem. Commun., 2010, 13, 1160–1162 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS . The σmeta, σpara and inductive/field factor (F) values for C6F5 and CF3 are 0.26, 0.27, 0.27 and 0.43, 0.54, 0.38, respectively. Therefore, CF3 is expected to be the stronger electron-withdrawing group.
- A. F. Henwood, A. K. Bansal, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2016, 4, 3726–3737 RSC.
- V. N. Kozhevnikov, Y. Zheng, M. Clough, H. A. Al-Attar, G. C. Griffiths, K. Abdullah, S. Raisys, V. Jankus, M. R. Bryce and A. P. Monkman, Chem. Mater., 2013, 25, 2352–2358 CrossRef CAS.
- D. R. Martir, C. Momblona, A. Pertegás, D. B. Cordes, A. M. Z. Slawin, H. J. Bolink and E. Zysman-Colman, ACS Appl. Mater. Interfaces, 2016, 8, 33907–33915 CAS.
- A. M′hamedi, M. A. Fox, A. S. Batsanov, H. A. Al-Attar, A. P. Monkman and M. R. Bryce, J. Mater. Chem. C, 2017, 5, 6777–6789 RSC.
- Calculations predict the structure of meso11 (Fig. S124†) to be much less folded than rac11 (Fig. S125†) and ‘rod-like’. This increased aspect ratio may facilitate solid state packing more strongly than for complex 9. DFT gas phase optimised structures in both this study and for previous analogues have been in good agreement with XRD data.21,22.
- S.-Y. Yao, Y.-L. Ou and B.-H. Ye, Inorg. Chem., 2016, 55, 6018–6026 CrossRef CAS PubMed.
- A. M′hamedi, A. S. Batsanov, M. A. Fox, M. R. Bryce, K. Abdullah, H. A. Al-Attar and A. P. Monkman, J. Mater. Chem., 2012, 22, 13529–13540 RSC.
- E. Baranoff, S. Fantacci, F. De Angelis, X. Zhang, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2011, 50, 451–462 CrossRef CAS PubMed.
- J. Frey, B. F. E. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2014, 43, 5667–5679 RSC.
- E. Baranoff and B. F. E. Curchod, Dalton Trans., 2015, 44, 8318–8329 RSC.
- J. Li, P. I. Djurovich, B. D. Alleyne, M. Yousufuddin, N. N. Ho, J. C. Thomas, J. C. Peters, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 1713–1727 CrossRef CAS PubMed.
- G. Li, T. Fleetham, E. Turner, X. C. Hang and J. Li, Adv. Opt. Mater., 2015, 3, 390–397 CrossRef CAS.
- K. Dedeian, J. Shi, N. Shepherd, E. Forsythe and D. C. Morton, Inorg. Chem., 2005, 44, 4445–4447 CrossRef CAS PubMed.
- T. Fleetham, G. Li, Z. Q. Zhu and J. Li, SID Int. Symp. Dig. Tech. Pap., 2015, 46, 411–414 CrossRef CAS.
- H. Benjamin, Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, A. P. Monkman and M. R. Bryce, Inorg. Chem., 2016, 55, 8612–8627 CrossRef CAS PubMed.
- E. Baranoff, B. F. E. Curchod, F. Monti, F. Steimer, G. Accorsi, I. Tavernelli, U. Rothlisberger, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2012, 51, 799–811 CrossRef CAS PubMed.
- The kr values reported here, although comparatively high for sky blue phosphors, are not as high as those of lower energy diiridium emitters.24,26 This is not surprising due to the lower MLCT/higher LC character excited states commonly exhibited by blue complexes, evident from their more highly structured emission spectra.65,66.
- M. A. Baldo, S. R. Forrest and M. E. Thompson, in Organic Electroluminescence, ed. Z. H. Kafafi, CRC and SPIE Press, 2005 Search PubMed.
- L. Yang, F. Okuda, K. Kobayashi, K. Nozaki, Y. Tanabe, Y. Ishii and M.-A. Haga, Inorg. Chem., 2008, 47, 7154–7165 CrossRef CAS PubMed.
- G. Li, A. Wolfe, J. Brooks, Z. Zhu and J. Li, Inorg. Chem., 2017, 56, 8244–8256 CrossRef CAS PubMed.
- S. Haneder, E. Da Como, J. Feldmann, J. M. Lupton, C. Lennartz, P. Erk, E. Fuchs, O. Molt, I. Münster, C. Schildknecht and G. Wagenblast, Adv. Mater., 2008, 20, 3325–3330 CrossRef CAS.
- C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Frohlich and L. De Cola, Chem. Mater., 2012, 24, 3684–3695 CrossRef CAS.
- T. Duan, T.-K. Chang, Y. Chi, J.-Y. Wang, Z.-N. Chen, W.-Y. Hung, C.-H. Chen and G.-H. Lee, Dalton Trans., 2015, 44, 14613–14624 RSC.
- H. J. Park, J. N. Kim, H. Yoo, K. Wee, S. O. Kang and D. W. Cho, J. Org. Chem., 2013, 78, 8054–8064 CrossRef CAS PubMed.
- T. B. Fleetham, L. Huang, K. Klimes, J. Brooks and J. Li, Chem. Mater., 2016, 28, 3276–3282 CrossRef CAS.
- G. Nasr, A. Guerlin, F. Dumur, L. Beouch, E. Dumas, G. Clavier, F. Miomandre, F. Goubard, D. Gigmes, D. Bertin, G. Wantz and C. R. Mayer, Chem. Commun., 2011, 47, 10698–10700 RSC.
- F. Lafolet, S. Welter, Z. Popović and L. De Cola, J. Mater. Chem., 2005, 15, 2820–2828 RSC.
- R. D. Costa, G. Fernandez, L. Sanchez, N. Martín, E. Ortí and H. J. Bolink, Chem. – Eur. J., 2010, 16, 9855–9863 CrossRef CAS PubMed.
- X. Yang, X. Xu, J. Dang, G. Zhou, C.-L. Ho and W.-Y. Wong, Inorg. Chem., 2016, 55, 1720–1727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, NMR spectra, X-ray data, electrochemistry, thermal gravimetric analysis; computations and photophysics. CCDC 1576081–1576084 and 1576093–1576095. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04201a |
| This journal is © The Royal Society of Chemistry 2018 |