
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Desolvation and aggregation of sterically demanding alkali metal diarylphosphides†
Keith
Izod
 *,
Peter
Evans
and
Paul G.
Waddell
*,
Peter
Evans
and
Paul G.
Waddell
Main Group Chemistry Laboratories, School of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: keith.izod@ncl.ac.uk
First published on 29th September 2017
Abstract
The reaction between (Dipp)2PH and one equivalent of n-BuLi, PhCH2Na or PhCH2K in THF gives the complexes [(Dipp)2P]Li(THF)3 (2a), {[(Dipp)2P]Na(THF)2}2 (3a) and [(Dipp)2P]K(THF)4 (4a), respectively [Dipp = 2,6-iPr2C6H3]. Exposure of these compounds to vacuum yields the alternative solvates [(Dipp)2P]Li(THF)2 (2b), [(Dipp)2P]Na(THF)1.5 (3b), and [(Dipp)2P]K (4b), respectively; the alternative adduct [(Dipp)2P]Na(PMDETA) (3c) was prepared by treatment of 3a with PMDETA. Treatment of (Dipp)(Mes)PH or (Mes)2PH with one equivalent of n-BuLi in THF gives the complexes [(Dipp)(Mes)P]Li(THF)3 (7a) and [(Mes)2P]2Li2(THF)2(OEt2) (8a) after crystallisation from diethyl ether [Mes = 2,4,6-Me3C6H2]; crystallisation of 8a from hexane gives the alternative adduct [(Mes)2P]Li(THF)3 (8b). Exposure of 7a, 8a and 8b to vacuum leads to loss of coordinated solvent, yielding the solvates [(Dipp)(Mes)P]Li(THF)2 (7b) and [(Mes)2P]Li(THF) (8c). The solid-state structures of complexes 2a, 3a, 3c, 4a, 7a, 8a, and 8b have been determined by X-ray crystallography. Variable-temperature 31P{1H} and 7Li NMR spectroscopy indicates that 2b, 3b and 7b are subject to a monomer–dimer equilibrium in solution, where the monomeric forms are favoured at low temperature. In contrast, variable-temperature 31P{1H} and 7Li NMR spectroscopy suggests that 8c is subject to a dynamic equilibrium between a dimer and a cyclic trimer in solution, where the trimer is favoured at low temperatures.
Introduction
Alkali metal phosphides are key intermediates in the synthesis of secondary and tertiary phosphine ligands and are ubiquitous metathesis reagents for the synthesis of main group, transition metal and f-element phosphide complexes. The alkali metal species are of interest in their own right and have been shown to adopt a wide variety of structural motifs in the solid state, ranging from monomers to dimers, trimers, tetramers, higher oligomers (often with ladder-like catenated P2Li2 rings), polymers, and 3-dimensional networks.1–11 The degree of oligomerisation is determined by the interplay between the size of the metal cation, the steric demands of the ligand substituents and the presence of co-ligands such as THF, TMEDA and PMDETA [TMEDA = N,N,N′,N′-tetramethylethylenediamine, PMDETA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine].1The oligomeric nature of alkali metal phosphides may be preserved in solution and, for lithium phosphides, 31P and 7Li NMR spectroscopy provide a convenient means for identifying oligomers and monitoring dynamic equilibria.3–5 In particular, coupling between these nuclei affords invaluable information on molecular connectivity; for monodentate phosphide ligands such coupling is often not observed at room temperature due to rapid exchange processes, but may be seen at low temperature, where these processes are frozen out.3 Solution-state studies of lithium phosphides have identified separated ion pairs, solvated monomers, and dimers containing a P2Li2 ring as the major species.3 In addition, NMR studies suggest that [PhHP]Li adopts a cyclic trimeric structure in Et2O at −73 °C.4 Pulsed-gradient spin-echo 7Li diffusion NMR experiments have also been used to show that [Ph2P]Li adopts a monomeric structure in THF but a dimeric structure in diethyl ether.5
As part of a wider project investigating phosphorus-substituted heavier group 14 carbene analogues,12 we have recently prepared a series of alkali metal complexes with sterically demanding diarylphosphide ligands. Herein we present the crystal structures of these compounds, comment on the frequently observed coordinated solvent loss, and describe their solution behaviour.
Results and discussion
The secondary phosphine (Dipp)2PH (1) has been reported previously by us,12a but its molecular structure has not been determined before [Dipp = 2,6-iPr2C6H3]. For the purposes of comparison, single crystals of 1 were obtained by sublimation at 100 °C/10−3 mmHg and its structure was determined by X-ray crystallography (Fig. 1). The crystal from which data were collected was twinned by pseudo-merohedry and the hydrogen atom bound to phosphorus and one isopropyl group were modelled as disordered over two positions. | ||
| Fig. 1 Molecular structure of 1 with minor disorder components and C-bound H atoms omitted for clarity. Selected bond lengths (Å) and angles (°): P(1)–C(1) 1.8536(14), P(1)–C(13) 1.8527(15), C(1)–P(1)–C(13) 104.81(7). | ||
Treatment of 1 with one equivalent of nBuLi in THF gives the corresponding lithium phosphide complex [(Dipp)2P]Li(THF)3 (2a) as orange blocks after crystallisation from diethyl ether. A similar reaction between 1 and one equivalent of either PhCH2Na or PhCH2K in THF gives the corresponding heavier alkali metal complexes {[(Dipp)2P]Na(THF)2}2 (3a) and [(Dipp)2P]K(THF)4 (4a), respectively. The coordinated THF in 2a, 3a, and 4a is rapidly lost under vacuum, yielding the alternative solvates [(Dipp)2P]Li(THF)2 (2b), [(Dipp)2P]Na(THF)1.5 (3b), and the solvent-free complex [(Dipp)2P]K (4b), respectively. Single crystals of 3a and 4a were obtained by crystallisation of the crude materials from a mixture of light petroleum and THF or diethyl ether and THF, respectively, while single crystals of the adduct [(Dipp)2P]Na(PMDETA) (3c) were obtained by crystallisation of 3a from n-hexane in the presence of one equivalent of PMDETA.
The molecular structures of 2a, 3a, 3c and 4a are shown in Fig. 2, along with selected bond lengths and angles. Compound 2a crystallises with two crystallographically independent molecules in the unit cell, the conformations of which differ only marginally. The data for 4a are of rather poor quality and so any discussion of bond lengths and angles for this compound must be rather cautious. Overall the structures of 2a, 3c and 4a are rather similar, with the alkali metal ions coordinated by the phosphide ligand and the O or N atoms of the co-ligands, resulting in distorted tetrahedral geometries at the metal centres in 2a and 3c and a distorted trigonal bipyramidal geometry at the potassium centre in 4a. In addition, there is one short Na⋯Me(N) contact in 3c [Na(1)⋯C(32) 3.121(17) Å] and two short K⋯CH2 contacts in 4a [K(1)⋯C(29) 3.505(13), K(1)⋯C(37) 3.537(13) Å]. The P–M distances [2.482(3) (2a), 2.8745(12) (3c) and 3.221(2) Å (4a)] all lie at the shorter end of the range typical for such contacts.1 For example, the Li–P distances in [(Ph2P)Li(TMEDA)]2 range from 2.574(19) to 2.629(20) Å,6 while the Na–P distances in [(CyPH)Na(PMDETA)]2 are 2.884(8) and 2.936(7) Å,7 and the K–P distances in polymeric [(Mes*PH)K]∞ range from 3.181(2) to 3.357(2) Å (Mes* = 2,4,6-tBu3C6H2).8 The phosphorus centres in 2a and 4a are close to planar [sum of angles at P = 358.13(13) and 356.73(13)° for the two independent molecules in 2a, and 359.97(20)° for 4a], whereas the phosphorus centre in 3c adopts a pyramidal geometry [sum of angles at P = 322.73(17)°].
 | ||
| Fig. 2 Molecular structures of (a) 2a, (b) 3a, (c) 3c and (d) 4a with H atoms and disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): 2a (molecule 1) Li(1)–P(1) 2.482(3), Li(1)–O(1) 1.934(3), Li(1)–O(2) 1.948(3), Li(1)–O(3) 1.960(3), P(1)–C(1) 1.8474(15), P(1)–C(13) 1.8459(15), Li(1)–P(1)–C(1) 123.05(8), Li(1)–P(1)–C(13) 132.16(8), C(1)–P(1)–C(13) 102.92(6), (molecule 2) Li(2)–P(2) 2.498(3), Li(2)–O(4) 1.978(3), Li(2)–O(5) 1.934(3), Li(2)–O(6A) 1.922(18), P(2)–C(37) 1.8506(15), P(2)–C(49) 1.8467(15), Li(2)–P(2)–C(37) 129.85(8), Li(2)–P(2)–C(49) 124.14(8), C(37)–P(2)–C(49) 102.74(6); 3a Na(1)–P(1) 2.9246(8), Na(1)–P(1A) 3.0178(8), Na(1)–O(1) 2.3231(16), Na(1)–O(1A) 2.343(4), P(1)–C(1) 1.8672(17), P(1)–C(13) 1.8622(16), Na(1)–P(1)–Na(1A) 73.83(2), P(1)–Na(1)–P(1A) 106.17(2), C(1)–P(1)–C(13)100.84(7); 3c Na(1)–P(1) 2.8745(12), Na(1)–N(1) 2.509(9), Na(1)–N(2) 2.525(10), Na(1)–N(3) 2.472(10), Na(1)⋯C(32) 3.121(17), P(1)–C(1) 1.856(3), P(1)–C(13) 1.847(3), Na(1)–P(1)–C(1) 85.70(8), Na(1)–P(1)–C(13) 132.75(9), C(1)–P(1)–C(13) 104.28(12); 4a K(1)–P(1) 3.221(2), K(1)–O(1) 2.696(7), K(1)–O(2) 2.698(7), K(1)–O(3) 2.706(5), K(1)–O(4) 2.817(6), K(1)⋯C(29) 3.505(13), K(1)⋯C(37) 3.537(13), P(1)–C(1) 1.844(3), P(1)–C(13) 1.841(3), K(1)–P(1)–C(1) 126.47(10), K(1)–P(1)–C(13) 130.00(10), C(1)–P(1)–C(13) 103.50(14). | ||
In contrast, compound 3a crystallises as centrosymmetric dimers containing a planar Na2P2 rhombus-shaped core. Each sodium ion is further coordinated by two molecules of THF in a distorted tetrahedral geometry. The Na–P distances [2.9246(8) and 3.0178(8) Å] are rather unsymmetrical, but are typical of Na–(μ-PR2) distances; for example, the Na–P distances in [EtSi{PNa(SiPri3)}3]2·PhMe range from 2.778(2) to 3.357(2) Å.9
Perhaps surprisingly, metalation of 1 has little impact on the gross structure of the (Dipp)2P framework: the P–C distances in 1 [1.8536(14) and 1.8257(15) Å] are similar to the P–C distances in 2a, 3a, 3c and 4a [range 1.841(3)–1.8672(17) Å] and the C(1)–P(1)–C(13) angle in 1 [104.81(17)°] is similar to the corresponding angles in 2a, 3a, 3c and 4a [range 100.84(3)–104.28(12)°].
Due to rapid solvent loss, it was not possible to obtain NMR spectra of 2a–4a; however, the 1H, 31P{1H} and 7Li NMR spectra of 2b and 3b suggest the operation of dynamic equilibria at room temperature (Fig. 3). At 90 °C the 31P{1H} NMR spectrum of 2b consists of a singlet at −113.7 ppm (A); as the temperature is reduced this signal broadens and begins to decoalesce, until, at room temperature, the spectrum consists of two very broad, overlapping signals at −106.0 (B) and −118.3 ppm (C). As the temperature is reduced further, these two signals sharpen and change in intensity: while at room temperature these two signals are in the approximate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5, as the temperature is reduced, the lower field signal (B) increases in intensity at the expense of the higher field signal (C), until, at −80 °C, the two signals are in the ratio 1:0.05. At this latter temperature the two signals are clearly resolved as a 1:1:1:1 quartet (JPLi = 70.7 Hz) and a 1:2:3:4:3:2:1 septet (JPLi = 67.1 Hz) at −104.0 and −122.7 ppm, respectively. Similarly, at 90 °C the 7Li NMR spectrum of 2b consists of a singlet at 2.1 ppm (D). As the temperature is reduced, this signal broadens and decoalesces, until, at 0 °C, the spectrum consists of two broad, overlapping signals at 2.5 (E) and 0.8 ppm (F) of approximately equal intensity. These signals sharpen, resolve into a triplet and a doublet, respectively, and rapidly change in intensity as the temperature is reduced further, such that, at −80 °C, the spectrum consists of a triplet (JPLi = 71.4 Hz) and a doublet (JPLi = 68.9 Hz) at 2.8 and 0.4 ppm in the approximate ratio of 0.08:1.
:1.5, as the temperature is reduced, the lower field signal (B) increases in intensity at the expense of the higher field signal (C), until, at −80 °C, the two signals are in the ratio 1:0.05. At this latter temperature the two signals are clearly resolved as a 1:1:1:1 quartet (JPLi = 70.7 Hz) and a 1:2:3:4:3:2:1 septet (JPLi = 67.1 Hz) at −104.0 and −122.7 ppm, respectively. Similarly, at 90 °C the 7Li NMR spectrum of 2b consists of a singlet at 2.1 ppm (D). As the temperature is reduced, this signal broadens and decoalesces, until, at 0 °C, the spectrum consists of two broad, overlapping signals at 2.5 (E) and 0.8 ppm (F) of approximately equal intensity. These signals sharpen, resolve into a triplet and a doublet, respectively, and rapidly change in intensity as the temperature is reduced further, such that, at −80 °C, the spectrum consists of a triplet (JPLi = 71.4 Hz) and a doublet (JPLi = 68.9 Hz) at 2.8 and 0.4 ppm in the approximate ratio of 0.08:1.
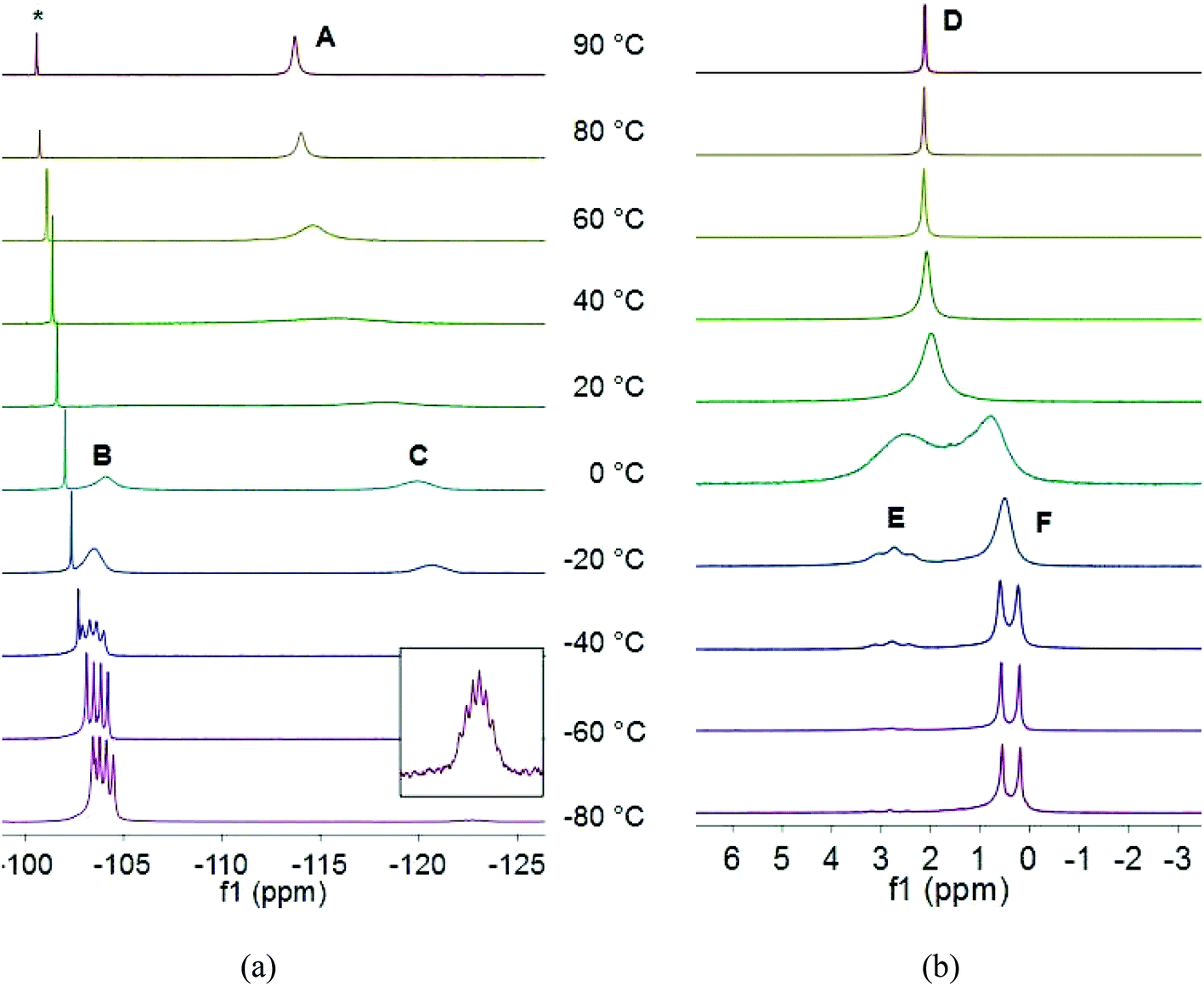 | ||
| Fig. 3 Variable-temperature (a) 31P{1H} and (b) 7Li NMR spectra of 2b in d8-toluene (* free phosphine 1). | ||
The variable-temperature 31P{1H} and 7Li NMR spectra of 2b are consistent with a dynamic equilibrium between monomeric 2b and a dimeric species of the form (THF)nLi{μ-P(Dipp)2}2Li(THF)n, which is rapid above room temperature. At lower temperatures the dimer appears to be disfavoured and the monomer predominates. The formation of a dimer is clearly associated with the loss of THF from 2a under vacuum, which would leave each lithium three-coordinate in the monomer; however, the steric demands of the (Dipp)2P ligand appear to be sufficient that such a three-coordinate monomeric species is stable. This behaviour is somewhat unusual, given that low temperatures typically favour the formation of higher aggregates.
Temperature-dependent NMR behaviour is not observed for 3c, but the variable-temperature 31P{1H} NMR spectra of 3b do exhibit behaviour consistent with the operation of one or more dynamic process(es) (Fig. 4). At 80 °C the spectrum consists of a singlet at −116.1 ppm and this peak shifts to slightly higher field as the temperature is reduced. This may be attributed to a straightforward temperature dependence of the chemical shift caused by conformational changes within the compound and changes in intermolecular interactions with the solvent (a similar temperature dependence is observed for the free phosphine impurity 1). However, below 0 °C, this peak begins to broaden and to move to lower field, such that, at −80 °C, the spectrum consists of a broad singlet at −114.1 ppm (FWHM 420 Hz). These spectra are consistent with a dynamic equilibrium between a dimer similar to that observed in the solid-state structure of 3a and a monomeric (or possibly higher oligomeric) species.
 | ||
| Fig. 4 Variable temperature 31P{1H} NMR spectra of 3b in d8-toluene (*free phosphine 1). | ||
Compound 4b has limited solubility in non-coordinating solvents and so a comparable variable-temperature NMR study is not possible; in THF solution compound 4b exhibits no evidence for dynamic behaviour.
Reducing the steric demands of the substituents at phosphorus might be expected to increase the tendency towards aggregation of the resulting lithium phosphides. In order to investigate this the lithiation of the new secondary phosphine (Dipp)(Mes)PH (5) and the known phosphine (Mes)2PH (6) was explored [Mes = 2,4,6-Me3C6H2].10 The secondary phosphine 5 was synthesised by the reaction between (Mes)PCl2 and one equivalent of DippLi(OEt2) in THF, followed by one equivalent of LiAlH4.
Treatment of 5 with one equivalent of nBuLi in THF gives the corresponding lithium phosphide [(Dipp)(Mes)P]Li(THF)3 (7a) after crystallisation from diethyl ether. It has been reported previously that the reaction between 6 and nBuLi in diethyl ether gives the dimer [(Mes)2P]2Li2(OEt2)2.10 In contrast, the reaction between 6 and nBuLi in THF gives the alterative dinuclear complex [(Mes)2P]2Li2(THF)2(OEt2) (8a) after crystallisation from diethyl ether; however, treatment of 6 with nBuLi in THF, followed by crystallisation from n-hexane gives monomeric [(Mes)2P]Li(THF)3 (8b). For 7a, 8a and 8b, exposure to vacuum leads to loss of coordinated solvent, ultimately yielding the solvates [(Dipp)(Mes)P]Li(THF)2 (7b) and [(Mes)2P]Li(THF) (8c), which appear to be stable towards further solvent loss.
The molecular structures of 7a, 8a and 8b are shown in Fig. 5, along with selected bond lengths and angles.
 | ||
| Fig. 5 Molecular structures of (a) 7a, (b) 8a and (c) 8b with H atoms and disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): 7a Li(1)–P(1) 2.572(3), Li(1)–O(1) 1.963(4), Li(1)–O(2) 1.950(4), Li(1)–O(3) 1.999(4), P(1)–C(1) 1.854(2), P(1)–C(13) 1.832(2), Li(1)–P(1)–C(1) 99.42(10), Li(1)–P(1)–C(13) 127.65(10), C(1)–P(1)–C(13) 104.34(9); 8a Li(1)–P(1) 2.609(4), Li(1)–P(2) 2.684(4), Li(2)–P(1) 2.513(4), Li(2)–P(2) 2.518(4), P(1)–C(1) 1.8365(18), P(1)–C(10) 1.836(2), P(2)–C(19) 1.8391(19), P(2)–C(28) 1.840(2), P(1)–Li(1)–P(2) 97.46(12), P(1)–Li(2)–P(2) 104.53(13), Li(1)–P(1)–Li(2) 79.69(12), Li(1)–P(2)–Li(2) 78.18(12); 8b Li(1)–P(1) 2.552(3), Li(1)–O(1) 1.944(3), Li(1)–O(2) 1.954(4), Li(1)–O(3B) 1.944(10), P(1)–C(1) 1.8164(19), P(1)–C(10) 1.829(2), Li(1)–P(1)–C(1) 122.48(10), Li(1)–P(1)–C(10) 97.81(10), C(1)–P(1)–C(10) 110.29(9). | ||
Compounds 7a and 8b crystallise as discrete monomers in which the lithium ions are coordinated by the phosphide P atom and the O atoms of three THF molecules in a distorted tetrahedral geometry. The Li–P distances in 7a and 8b [2.572(3) and 2.552(3) Å, respectively] are similar to previously reported Li–P distances;1,2 perhaps surprisingly, given the essentially isostructural nature of 2b, 7a and 8b, the Li–P distances do not correlate with the steric bulk of the phosphide ligands, decreasing in the order 7a > 8b > 2a, i.e. with the shortest Li–P distance occurring in the complex with the most sterically demanding phosphide ligand. In both 7a and 8b the phosphorus atoms adopt a pyramidal geometry [sum of angles at P = 331.41(17) (7a) and 330.58(17)° (8b)].
In contrast to 7a and 8b, 8a crystallises as a phosphide-bridged dimer with an essentially planar P2Li2 core. The two lithium ions in 8a are in distinct environments: Li(1) is coordinated by the two bridging phosphide ligands and two molecules of THF in a distorted tetrahedral geometry, while Li(2) is coordinated by the two phosphide ligands and a single molecule of diethyl ether in a trigonal planar geometry. The Li(1)–P distances [2.609(4) and 2.684(4) Å] are significantly longer than the Li(2)–P distances [2.513(4) and 2.518(4) Å] and the P(1)–Li(1)–P(2) angle [97.46(12)°] is considerably smaller than the P(1)–Li(2)–P(2) angle [104.53(13)°], consistent with the lower coordination number at Li(2).
Once again, the variable temperature 31P{1H} and 7Li NMR spectra of 7b indicate the operation of a dynamic process in solution (Fig. 6). At room temperature the 31P{1H} NMR spectrum of 7b in d8-toluene consists of a singlet at −100.7 ppm (G). As the temperature is reduced, this peak broadens and moves downfield, eventually decoalescing at −40 °C into two broad signals at −97.5 (H) and −84.4 ppm (I). Below this temperature, peak H sharpens into a well-resolved quartet (JPLi = 69.5 Hz), while peak I further decoalesces into two low intensity, broad signals at approximately −99 (J) and −94 ppm (K) (approximate ratio of H:J:K at −80 °C 12:1:1). The room temperature 7Li NMR spectrum of 7b consists of a single signal at 2.1 ppm (L), which gradually decoalesces as the temperature is reduced until, at −80 °C, the spectrum consists of a sharp doublet at 0.4 ppm (M, JPLi = 69.5 Hz) and a very broad, approximate triplet at 1.8 ppm (N, JPLi = 48 Hz).
 | ||
| Fig. 6 Variable-temperature (a) 31P{1H} and (b) 7Li NMR spectra of 7b in d8-toluene. | ||
The variable-temperature 31P{1H} and 7Li NMR spectra of 7b are consistent with a dynamic equilibrium between monomeric (H/M) and dimeric (J,K/N) species in which the monomeric species is favoured at low temperatures. We assign the two higher field signals J and K in the low temperature 31P{1H} NMR spectra to the cis and trans isomers of the dimer (Scheme 1), which appear to have equal probabilities. This behaviour is similar to that observed for 2; however, while, at room temperature, the time-averaged 31P{1H} NMR signal for 2b suggests equal proportions of monomer and dimer in equilibrium, the time-averaged room temperature 31P{1H} NMR signal for 7b lies to higher field than an arithmetical mean of the monomer and dimer signals would suggest, and so it appears that, for this compound, the dimeric form is substantially favoured at room temperature, even though this form is observed only in low concentrations at low temperatures.
 | ||
| Scheme 1 | ||
Compound 8c exhibits rather different variable-temperature NMR spectra to both 2b and 7b. At 80 °C the 31P{1H} NMR spectrum of 8c in d8-toluene consists of a singlet (O) at −90.3 ppm (Fig. 7). As the temperature is reduced, this peak broadens and moves to lower field, eventually decoalescing at −40 °C into a broad signal (P) at −86.9 ppm, a well-resolved septet (Q) at −90.0 ppm (JPLi = 60.4 Hz), and a very broad, low intensity signal (R) at approximately −95.5 ppm. As the temperature is decreased further, peak Q broadens and then sharpens again, while peak P moves to lower field and resolves into a complex multiplet, and peak R decreases in intensity, such that, at −80 °C, the spectrum consists of a complex multiplet at −84.4 and a broad septet at −91.0 ppm. The 7Li NMR spectrum of 8c at 80 °C consists of a singlet at 2.2 ppm (S) and this moves slightly downfield and resolves at −90 °C into a complex multiplet centred at approximately 2.7 ppm that appears to consist of two overlying signals (T/U).
 | ||
| Fig. 7 Variable-temperature (a) 31P{1H} and (b) 7Li NMR spectra of 8c in d8-toluene (*free phosphine 6); (c) expansion of the low-field 31P{1H} NMR signal (P) at −80 °C and (d) simulation of this peak. | ||
The low-field peak P observed in the −80 °C 31P{1H} NMR spectrum of 8c is inconsistent with a dimeric structure. While the presence of a higher oligomer cannot be ruled out completely, simulation of this peak (Fig. 7d) is consistent with a cyclic trimer with 1JPLi = 60 Hz, 2JPP = 46 Hz and 3JPLi < 5 Hz; simulations based on tetrameric oligomers did not give a successful match to the observed peak. The lineshape and calculated coupling constants for 8c are similar to those obtained for the proposed trimer [(PhPH)Li(OEt2)n]3 at −73 °C in diethyl ether, the 31P{1H} NMR spectrum of which exhibits a complex multiplet with JPLi = 39 Hz, JPP = 70 Hz.4 The variable-temperature 31P{1H} and 7Li NMR spectra of 8c are therefore consistent with a dynamic equilibrium between a dimer, a cyclic trimer (Scheme 2), and a further unidentified species which is in low concentration. For 2b and 7b the monomeric species gives rise to a quartet to significantly lower field than the peak due to the dimer, whereas peak R lies to higher field than peaks Q and P (assigned to the dimer and trimer, respectively), suggesting that peak R is due to a higher oligomer, rather than a monomeric species.
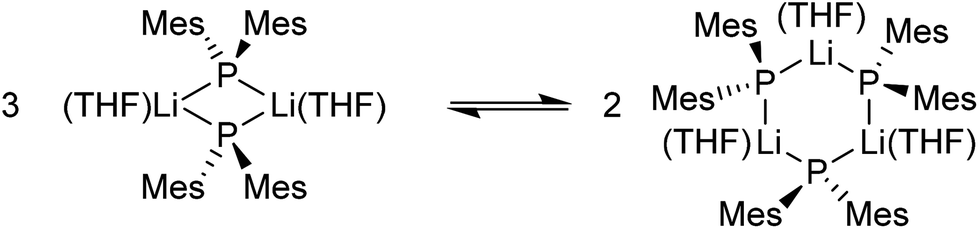 | ||
| Scheme 2 | ||
A single example of a lithium phosphide with a crystallographically characterised cyclic trimeric structure has been reported: the sterically hindered tetrasilaphosphide [[(iPr2Si)4P]Li]3 adopts a triangular structure in the solid state, but, unfortunately, the NMR data available for this compound are limited and the 31P{1H} NMR spectrum is reported as consisting of a broad singlet at room temperature, preventing comparisons with our data.12
Conclusions
The sterically-demanding alkali metal diarylphosphides 2, 3, 4, 7 and 8 are readily accessible and are potentially useful ligand transfer reagents. In the solid-state these compounds crystallise as either solvated monomers (2a, 3c, 4a, 7a, 8b) or phosphide-bridged dimers (3a, 8a); however, loss of coordinated solvent is common, leading to alternative solvates. Variable-temperature 31P{1H} and, where applicable, 7Li NMR spectroscopy indicates that these less-solvated compounds are frequently subject to dynamic processes in solution and that the species present are dependent on the substituents at phosphorus. For 2b, 3b, and 7b this dynamic behaviour may be attributed to a monomer–dimer equilibrium, where the monomer is favoured at low temperatures, whereas for 8c there appears to be a dynamic equilibrium between a dimer and a cyclic trimer, where the trimer is favoured at low temperatures. The preference exhibited by 2b, 3b, and 7b for a monomeric structure in solution at low temperatures contrasts with the behaviour of less sterically hindered diarylphosphide complexes such as Ph2PLi(THF)n3 and 8c, which favour dimeric, or higher oligomeric, structures at low temperatures.
Experimental
All manipulations were carried out using standard Schlenk or dry-box techniques under an atmosphere of dry nitrogen or argon. THF, light petroleum (b.p. 40–60 °C), diethyl ether, and n-hexane were dried prior to use by distillation under nitrogen from sodium, potassium, or sodium/potassium alloy, as appropriate. THF and was stored over activated 4 Å molecular sieves; all other solvents were stored over a potassium film. Deuterated THF and toluene were distilled from potassium and CDCl3 was distilled from CaH2 under nitrogen; all NMR solvents were deoxygenated by three freeze–pump–thaw cycles and were stored over activated 4 Å molecular sieves. Benzylpotassium,13 benzylsodium,14 DippLi·OEt2,12a (Mes)PCl215 and (Dipp)2PH12a were prepared by previously published procedures; the new adduct [(Mes)2P]2Li2(OEt2)(THF)2 (8a) was obtained by the reaction between (Mes)2PH and nBuLi in THF, followed by crystallization from cold (−35 °C) diethyl ether. n-Butyllithium was purchased from Aldrich as a 2.5 M solution in hexanes. All other compounds were used as supplied by the manufacturer.
1H NMR spectra were recorded on a Bruker Avance300 or Avance400 spectrometer operating at 300.13 and 400.17 MHz, respectively; 13C{1H}, 7Li, and 31P{1H} NMR spectra were recorded on a Bruker Avance 500 spectrometer operating at 125.78, 194.38 and 202.44 MHz, respectively. 1H and 13C chemical shifts are quoted in ppm relative to tetramethylsilane, 31P and 7Li chemical shifts are quoted in ppm relative to external 85% H3PO4 and 0.1 M LiCl, respectively. Due to the air-sensitive nature of the alkali metal phosphides, we were unable to obtain consistent elemental analyses.
Synthesis of [(Dipp)2P]Li(THF)2 (2b)
To a solution of (Dipp)2PH (0.695 g, 1.97 mmol) in THF (10 ml) was added a solution of nBuLi in hexanes (2.3 M, 0.85 ml, 2.0 mmol). The resulting red solution was stirred for 1 h. The solvent was removed in vacuo and the orange solid was dissolved in Et2O (5 ml). Storage of this solution at −25 °C for 2 days resulted in the formation of large yellow crystals of [(Dipp)2P]Li(THF)3 (2a). These crystals were washed with cold (−10 °C) light petroleum (2 × 5 ml) and residual solvent was removed in vacuo to give the alternative solvate [(Dipp)2P]Li(THF)2 (2b) as a yellow powder. Yield of 2b: 0.79 g, 80%. 1H NMR [d8-toluene, 363 K]: δ 0.99 (d, JHH = 6.9 Hz, 24H, CHMe2), 1.46 (m, 8H, THF), 3.57 (m, 8H, THF), 4.17 (m, 4H, CHMe2), 6.97 (d, JHH = 7.7 Hz, 4H, ArH), 7.04 (m, 2H, ArH). 7Li NMR [d8-toluene, 363 K]: δ 2.2 (s). 13C{1H} NMR [d8-toluene, 363 K]: δ 25.00 (CHMe2), 25.88 (THF), 33.92 (CHMe2), 68.61 (THF), 123.29 (ArH), 125.47 (ArH), 147.00 (br. d, JPC = 23.1 Hz, Ar), 152.85 (d, JPC = 6.2 Hz, Ar). 31P{1H} NMR [d8-toluene, 363 K]: δ −114.4.Synthesis of [(Dipp)2P]Na(THF)1.5 (3b)
To a solution of Dipp2PH (0.81 g, 2.28 mmol) in THF (15 ml) was added a solution of benzylsodium (0.261 g, 2.28 mmol) in THF (10 ml). The resulting orange solution was stirred for 30 min and the solvent was removed in vacuo to give a sticky yellow solid. Light petroleum (15 ml) was added to give an orange solution that spontaneously formed yellow crystals of {[(Dipp)2P]Na(THF)2}2 (3a). Further crystalline material was obtained by cooling the mixture to −25 °C for 12 h. The supernatant solution was removed by filtration and the crystals were washed with cold (−10 °C) light petroleum. The residual solvent was removed under vacuum to give the alternative solvate [(Dipp)2P]Na(THF)1.5 (3b) as a yellow powder. Yield of 3b: 0.89 g, 81%. 1H NMR [d8-toluene]: δ 1.04 (d, JHH = 6.9 Hz, 24H, CHMe2), 1.38 (m, 6H, THF), 3.50 (m, 6H, THF), 4.24 (m, 4H, CHMe2), 7.03 (d, JHH = 7.6 Hz, 4H, ArH), 7.11 (m, 2H, ArH). 13C{1H} NMR [d8-toluene]: δ 24.54 (CHMe2), 25.60 (THF), 33.57 (d, JPC = 11.7 Hz, CHMe2), 68.19 (THF), 122.95, 124.92 (ArH), 147.84 (d, JPC = 33.5 Hz, Ar), 151.98 (d, JPC = 4.8 Hz, Ar). 31P{1H} NMR [d8-toluene]: δ −118.6.Synthesis of [(Dipp)2P]Na(PMDETA) (3c)
(Dipp)2PH (0.596 g, 1.68 mmol) and benzylsodium (0.194 g, 1.70 mmol) were dissolved in THF (10 ml) and this mixture was stirred for 1 h to give a red solution. The solvent was removed in vacuo. To the resulting orange solid was added n-hexane (10 ml) and PMDETA (0.35 ml, 1.70 mmol) to give a red solution that deposited yellow crystalline material on standing. The mixture was cooled to −25 °C for 2 h, the supernatant was removed, the yellow crystalline material was washed with cold (−10 °C) light petroleum (2 × 5 ml) and residual solvent was removed in vacuo. Yield: 0.60 g, 65%. 1H NMR [d8-toluene]: δ 1.21 (d, JHH = 7.0 Hz, 24H, CHMe2), 1.67 (br. s, 8H, NCH2CH2N), 1.79 (s, 12H, NMe2), 1.81 (s, 3H, NMe), 4.68 (m, 4H, CHMe2), 7.10 (s, 6H, ArH). 13C{1H} NMR [d8-toluene]: δ 24.93 (CHMe2), 33.18 (CHMe2), 43.30 (NMe), 45.28 (NMe2), 54.03 (NCH2CH2N), 56.97 (NCH2CH2N), 122.42, 123.20 (ArH), 151.88 (d, JPC = 7.3 Hz, Ar), 152.92 (d, JPC = 49.3 Hz, Ar). 31P{1H} NMR [d8-toluene]: δ −106.7.Synthesis of [(Dipp)2P]K (4b)
To a mixture of (Dipp)2PH (1.06 g, 2.99 mmol) and benzylpotassium (0.391 g, 3.00 mmol) was added THF (15 ml) and the resulting red solution was stirred for 1 h. The solution was reduced in volume to 3 ml in vacuo and light petroleum (25 ml) was added, generating an orange precipitate. The solid was isolated by filtration, washed with light petroleum (2 × 5 ml) and dried in vacuo. Yield: 0.99 g, 84%. 1H NMR [d8-toluene/d8-THF]: δ 1.11 (d, JHH = 7.0 Hz, 24H, CHMe2), 4.59 (m, 4H, CHMe2), 6.92–6.96 (m, 6H, ArH). 13C{1H} NMR [d8-toluene/d8-THF]: δ 24.81 (CHMe2), 33.09 (d, JPC = 13.7 Hz, CHMe2), 122.27, 122.50 (ArH), 151.93 (d, JPC = 8.2 Hz, Ar), 154.68 (d, JPC = 55.8 Hz, Ar). 31P{1H} NMR [d8-toluene/d8-THF]: δ −92.3.Crystals of the solvate [(Dipp)2P]K(THF)4 (4a) suitable for characterisation by single crystal X-ray diffraction were obtained by recrystallization of 4b from an Et2O/THF mixture at −25 °C. These crystals rapidly lose THF under vacuum to give the THF-free complex [(Dipp)2P]K (4b).
Synthesis of (Dipp)(Mes)PH (5)
To a cold (−78 °C) solution of (Mes)PCl2 (2.99 g, 13.5 mmol) in Et2O (50 ml) was added a solution of DippLi·Et2O (3.28 g, 13.5 mmol) in Et2O (20 ml). The resulting yellow solution was warmed to room temperature and stirred for 1 h. The mixture was cooled to −78 °C and solid LiAlH4 (0.511 g, 13.5 mmol) was added in portions. The mixture was warmed to room temperature and stirred for 1 h. Degassed water (40 ml) was added slowly, the product was extracted into light petroleum (4 × 20 ml) and the combined extracts were dried over 4 Å molecular sieves. The solution was filtered and the solvent was removed under vacuum to yield 5 as a pale yellow solid. Yield: 3.29 g, 78%. 1H NMR [CDCl3]: δ 1.03 (d, JHH = 6.8 Hz, 6H, CHMeMe), 1.09 (d, JHH = 6.8 Hz, 6H, CHMeMe), 2.22 (s, 6H, o-Me), 2.23 (s, 3H, p-Me), 3.59 (m, 2H, CHMeMe), 5.34 (d, JPH = 233 Hz, 1H, PH), 6.80 (m, 2H, ArH), 7.11 (d, JHH = 7.7 Hz, 1H, ArH), 7.12 (d, JHH = 7.7 Hz, 1H, ArH), 7.30 (t, JHH = 7.7 Hz, 1H, ArH). 13C{1H} NMR [CDCl3]: δ 21.05 (o-Me), 23.46 (d, JPC = 9.7 Hz, p-Me), 23.86 (CHMeMe), 24.52 (CHMeMe), 32.84 (d, JPC = 13.5 Hz, CHMeMe), 123.40 (d, JPC = 3.4 Hz, ArH), 129.25 (ArH), 129.28 (m, ArH), 130.47 (d, JPC = 16.9 Hz, Ar), 131.54 (d, JPC = 16.9 Hz, Ar), 137.46 (Ar), 141.08 (d, JPC = 11.8 Hz, Ar), 153.83 (d, JPC = 11.2 Hz, Ar). 31P [CDCl3]: δ −96.9 (d, JPH = 233 Hz).Synthesis of [(Dipp)(Mes)P]Li(THF)2 (7b)
To a solution of 5 (0.55 g, 1.76 mmol) in THF (10 ml) was added a solution of nBuLi in hexanes (2.3 M, 0.8 ml, 1.84 mmol). The resulting red solution was stirred for 1 h and the solvent was removed in vacuo. The resulting sticky orange solid was crystallized from cold (−25 °C) Et2O (10 ml) to give large orange crystals of [(Dipp)(Mes)P]Li(THF)3 (7a). The crystals were isolated by filtration, washed with cold (0 °C) light petroleum (2 × 5 ml) and residual solvent was removed under vacuum to give the alternative solvate [(Dipp)(Mes)P]Li(THF)2 (7b) as a yellow solid. Yield of 7b: 0.60 g, 74%. 1H NMR [d8-toluene, 298 K]: 1.11 (d, JHH = 6.9 Hz, 12H, CHMe2), 1.37 (m, 8H, THF), 2.20 (s, 3H, p-Me), 2.34 (s, 6H, o-Me), 3.49 (m, 8H, THF), 4.19 (m, 2H, CHMe2), 6.81 (s, 2H, ArH), 7.08 (d, JHH = 7.6 Hz, 2H, ArH), 7.14 (m, 1H, ArH). 13C{1H} NMR [d8-toluene, 298 K]: 20.67 (p-Me), 24.36 (CHMe2), 24.94 (d, JPC = 10.7 Hz, o-Me), 25.23 (THF), 33.27 (d, JPC = 12.3 Hz, CHMe2), 67.84 (THF), 122.30, 124.79, 128.39 (ArH), 131.39 (Ar), 140.38 (d, JPC = 8.3 Hz, Ar), 144.39 (d, JPC = 25.5 Hz, Ar), 144.82 (d, JPC = 22.5, Ar), 151.94 (d, JPC = 6.2 Hz, Ar). 7Li NMR [d8-toluene, 298 K]: 2.1 (s). 31P{1H} NMR [d8-toluene, 298 K]: −100.7 (br. s).Crystal structure determinations of 1, 2a, 3a, 3c, 4a, 7a, 8a and 8b
Measurements were made at 150 K on an Oxford Diffraction (Agilent Technologies) Gemini A Ultra diffractometer using CuKα radiation (λ = 1.54178 Å; 1, 2a, 3c, 4a, 7a and 8a) or MoKα radiation (λ = 0.71073 Å; 3a and 8b). Cell parameters were refined from the observed positions of all strong reflections. Intensities were corrected for absorption using a multifaceted crystal model created by indexing the faces of the crystal for which data were collected for 1, 3a, 4a, 8a and 8b,16 and by empirical methods using spherical harmonics for 2a, 3c and 7a. The structures were solved by direct methods and refined on F2 values for all unique data; Table 1 gives further details. All non-hydrogen atoms were refined anisotropically, and C-bound H atoms were constrained with a riding model; U(H) was set at 1.2 (1.5 for methyl groups) times Ueq for the parent C atom. The programs used were Rigaku Oxford Diffraction CrysAlisPro for data collection and processing, and Olex2 utilising SHELXT for structure solution and SHELXL for refinement, with molecular graphics produced using SHELXTL.17| Compound | 1 | 2a | 3a | 3c | 4a | 7a | 8a | 8b |
|---|---|---|---|---|---|---|---|---|
| a R = ∑||Fo| − |Fc||/∑|Fo|; Rw = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2; S = [∑w(Fo2 − Fc2)2/(no. data − no. params)]1/2 for all data. | ||||||||
| Formula | C24H35P | C36.01H58.41LiO3P | C64H100Na2O4P2 | C33H57N3NaP | C72.01H66KO4P | C33H52LiO3P | C48H70Li2O3P2 | C30H46LiO3P |
| M w | 354.49 | 577.20 | 1041.35 | 549.77 | 680.99 | 534.65 | 770.86 | 492.58 |
| Cryst. size (mm) | 0.14 × 0.12 × 0.10 | 0.36 × 0.14 × 0.08 | 0.40 × 0.34 × 0.20 | 0.30 × 0.15 × 0.10 | 0.34 × 0.24 × 0.20 | 0.30 × 0.15 × 0.05 | 0.28 × 0.09 × 0.08 | 0.38 × 0.25 × 0.18 |
| Cryst. syst. | Monoclinic | Triclinic | Orthorhombic | Tetragonal | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca | P43212 | P21/c |
P |
P21/c | P21/c |
| a (Å) | 7.75472(8) | 11.32606(19) | 18.7669(6) | 12.67110(10) | 16.7963(5) | 9.5265(4) | 12.0982(2) | 9.0626(3) |
| b (Å) | 19.0684(2) | 17.4145(5) | 16.8927(5) | 12.67110(10) | 14.4308(3) | 9.5469(5) | 15.7006(3) | 17.2694(7) |
| c (Å) | 14.79343(17) | 19.3933(5) | 19.9516(6) | 42.8676(5) | 16.6968(5) | 19.4340(10) | 24.9514(6) | 19.2321(16) |
| α (°) | 90 | 68.346(3) | 90 | 90 | 90 | 82.213(4) | 90 | 90 |
| β (°) | 90.1101(10) | 80.5527(18) | 90 | 90 | 91.061(3) | 89.691(4) | 102.271(2) | 102.935(4) |
| γ (°) | 90 | 89.5652(16) | 90 | 90 | 90 | 65.484(5) | 90 | 90 |
| V (Å3) | 2187.51(4) | 3501.06(16) | 6325.2(3) | 6882.68(14) | 4046.36(15) | 1590.76(15) | 4631.21(17) | 2933.56(19) |
| Z | 4 | 2 | 4 | 8 | 4 | 2 | 4 | 4 |
| μ (mm−1) | 1.108 | 0.922 | 0.125 | 0.993 | 1.793 | 0.980 | 1.127 | 0.120 |
| Reflns. measd. | 31464 |
49842 |
28208 |
52251 |
56946 |
22353 |
33402 |
23834 |
| Unique reflns. | 3874 | 12397 |
7244 | 6116 | 7175 | 5634 | 8211 | 6573 |
| R int | 0.046 | 0.037 | 0.043 | 0.059 | 0.038 | 0.060 | 0.064 | 0.037 |
| Refined parameters | 249 | 856 | 378 | 387 | 486 | 350 | 514 | 367 |
| R (on F, F2 > 2σ)a | 0.034 | 0.039 | 0.050 | 0.039 | 0.085 | 0.048 | 0.044 | 0.053 |
| R w (on F2, all data)a | 0.087 | 0.105 | 0.127 | 0.104 | 0.206 | 0.131 | 0.114 | 0.149 |
| Goodness of fita | 1.036 | 1.034 | 1.026 | 1.016 | 1.052 | 1.021 | 1.010 | 1.032 |
| Max, min electron density (e Å−3) | 0.12, −0.25 | 0.27, −0.25 | 0.26, −0.20 | 0.37, −0.19 | 0.78, −0.84 | 0.43, −0.24 | 0.22, −0.22 | 0.25, −0.21 |
Acknowledgements
The authors are grateful to Newcastle University and the UK Engineering and Physical Sciences Research Council (EPSRC, Grant No. EP/L5048281) for support. We thank Prof William McFarlane (Newcastle) for simulation of the 31P{1H} NMR spectrum of 8c.References
- For a review see: K. Izod, Coord. Chem. Rev., 2000, 50, 33–108 CAS.
- For selected examples see: (a) R. A. Jones, A. L. Stuart and T. C. Wright, J. Am. Chem. Soc., 1983, 105, 7459–7460 CrossRef CAS; (b) G. W. Rabe, J. Riede and A. Schier, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1350–1352 CrossRef; (c) R. A. Bartlett, M. Olmstead and P. P. Power, Inorg. Chem., 1986, 25, 1243–1245 CrossRef CAS; (d) P. B. Hitchcock, M. F. Lappert, P. P. Power and S. J. Smith, J. Chem. Soc., Chem. Commun., 1984, 1669–1670 RSC; (e) E. Hey-Hawkins and E. Sattler, J. Chem. Soc., Chem. Commun., 1992, 775–776 RSC; (f) M. Driess and H. Pritzkow, Z. Anorg. Allg. Chem., 1996, 622, 1524–1530 CrossRef CAS; (g) G. W. Rabe, S. Kheradmandan and G. P. A. Yap, Inorg. Chem., 1998, 25, 6541–6543 CrossRef; (h) G. W. Rabe, S. Kheradmandan, L. M. Liable-Sands, I. A. Guzei and A. L. Rheingold, Angew. Chem., Int. Ed., 1998, 37, 1404–1407 CrossRef CAS; (i) F. Dornhaus, M. Bolte, H.-W. Lerner and M. Wagner, Eur. J. Inorg. Chem., 2006, 1777–1785 CrossRef CAS; (j) K. Izod, J. Stewart, E. R. Clark, W. Clegg and R. W. Harrington, Inorg. Chem., 2010, 49, 4698–4708 CrossRef CAS PubMed; (k) R. Edge, R. J. Less, V. Nasari, E. J. L. McInnes and D. S. Wright, Dalton Trans., 2008, 6454–6460 RSC; (l) I. Jevtovikj, R. Herrero, S. Gomez-Ruiz, P. Lonnecke and E. Hey-Hawkins, Inorg. Chem., 2013, 52, 4488–4493 CrossRef CAS PubMed; (m) C. von Hanisch, S. Traut and S. Stahl, Z. Anorg. Allg. Chem., 2007, 633, 2199–2204 CrossRef; (n) M. Westerhausen, T. Rotter, H. Görls, C. Birg, M. Warchold and H. Nöth, Z. Naturforsch., B: Chem. Sci., 2005, 60, 766–770 CAS.
- (a) I. J. Colquhoun, H. C. E. McFarlane and W. McFarlane, J. Chem. Soc., Chem. Commun., 1982, 220–221 RSC; (b) R. A. Bartlett, M. M. Olmstead and P. P. Power, Inorg. Chem., 1986, 25, 1243–1247 CrossRef CAS; (c) A. Zschunke, M. Riemer, H. Schmidt and K. Issleib, Phosphorus, Sulfur Relat. Elem., 1983, 17, 237–244 CrossRef CAS; (d) H. J. Reich and R. R. Dykstra, Organometallics, 1994, 13, 4578–4585 CrossRef CAS.
- I. J. Colquhoun, H. C. E. McFarlane and W. McFarlane, Phosphorus Sulfur, 1983, 18, 61–64 CrossRef CAS.
- I. Fernandez, E. Martinez-Viviente and P. S. Pregosin, Inorg. Chem., 2004, 43, 4555–4557 CrossRef CAS PubMed.
- R. E. Mulvey, K. Wade, W. Clegg and D. Reed, Polyhedron, 1987, 6, 987–993 CrossRef CAS.
- E. Hey, C. L. Raston, B. W. Skelton and A. H. White, J. Organomet. Chem., 1989, 362, 1–10 CrossRef CAS.
- G. W. Rabe, G. P. A. Yap and A. L. Rheingold, Inorg. Chem., 1997, 36, 1990–1991 CrossRef CAS PubMed.
- M. Driess, G. Huttner, N. Knopf, H. Pritzow and L. Asolnai, Angew. Chem., Int. Ed. Engl., 1995, 34, 316–318 CrossRef CAS.
- R. A. Bartlett, M. M. Olmstead, P. P. Power and G. A. Sigel, Inorg. Chem., 1987, 26, 1941–1946 CrossRef CAS.
- S. Traut, C. von Hänisch and H.-J. Kathagen, Eur. J. Inorg. Chem., 2009, 777–783 CrossRef CAS.
- (a) K. Izod, D. G. Rayner, S. M. El-Hamruni, R. W. Harrington and U. Baisch, Angew. Chem., Int. Ed., 2014, 53, 3636–3640 CrossRef CAS PubMed; (b) K. Izod, P. Evans, P. G. Waddell and M. R. Probert, Inorg. Chem., 2016, 55, 10510–10522 CrossRef CAS PubMed; (c) K. Izod, P. Evans and P. G. Waddell, Angew. Chem., Int. Ed., 2017, 56, 5593–5597 CrossRef CAS PubMed.
- L. Lochmann and J. Trekoval, J. Organomet. Chem., 1987, 326, 1–7 CrossRef CAS.
- S. Corbelin, N. P. Lorenzen, J. Kopf and E. Weiss, J. Organomet. Chem., 1991, 415, 293–313 CrossRef CAS.
- D. Bockfield, A. Doddi, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2016, 3704–3713 CrossRef.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887 CrossRef.
- (a) CrysAlisPro, Agilent Technologies, Version 1.171.36 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (d) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For 1, 2a, 3a, 3c, 4a, 7a, 8a and 8b details of structure determination, atomic coordinates, bond lengths and angles, and displacement parameters in CIF format. 1H, 13C{1H}, 7Li and 31P{1H} NMR spectra of 1, 2b, 3b, 3c, 4b, 7b, and 8b. CCDC 1557229–1557236. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02238g. Data supporting this publication is openly available under an ‘Open Data Commons Open Database License’. Additional metadata are available at DOI: 10.17634/154300-52. Please contact Newcastle Research Data Service at E-mail: rdm@ncl.ac.uk for access instructions. |
| This journal is © The Royal Society of Chemistry 2017 |