
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolylysine-grafted Au144 nanoclusters: birth and growth of a healthy surface-plasmon-resonance-like band†
Ivan
Guryanov
*a,
Federico
Polo
 b,
Evgeniy V.
Ubyvovk
c,
Evgenia
Korzhikova-Vlakh
a,
Tatiana
Tennikova
a,
Armin T.
Rad
d,
Mu-Ping
Nieh
ef and
Flavio
Maran
*bg
b,
Evgeniy V.
Ubyvovk
c,
Evgenia
Korzhikova-Vlakh
a,
Tatiana
Tennikova
a,
Armin T.
Rad
d,
Mu-Ping
Nieh
ef and
Flavio
Maran
*bg
aInstitute of Chemistry, St. Petersburg State University, 26 Universitetskij Pr., 198504 Petrodvorets, St. Petersburg, Russia. E-mail: ivan.guryanov1@gmail.com
bDepartment of Chemistry, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: flavio.maran@unipd.it
cDepartment of Physics, St. Petersburg State University, 3 Ulyanovskaya, 198504 Petrodvorets, St. Petersburg, Russia
dDepartment of Biomedical Engineering, University of Connecticut, 260 Glenbrook Road, Storrs, Connecticut 06269, USA
ePolymer Program, Institute of Materials Science, University of Connecticut, 97 N. Eagleville Rd, Storrs, Connecticut 06269, USA
fDepartment of Chemical & Biomolecular Engineering, University of Connecticut, 191 Auditorium Rd, Storrs, Connecticut 06269, USA
gDepartment of Chemistry, University of Connecticut, 55 North Eagleville Road, Storrs, 06269 Connecticut, USA
First published on 2nd February 2017
Abstract
Poly(amino acid)-coated gold nanoparticles hold promise in biomedical applications, particularly because they combine the unique physicochemical properties of the gold core, excellent biocompatibility, and easy functionalization of the poly(amino acid)-capping shell. Here we report a novel method for the preparation of robust hybrid core–shell nanosystems consisting of a Au144 cluster and a densely grafted polylysine layer. Linear polylysine chains were grown by direct N-carboxyanhydride (NCA) polymerization onto ligands capping the gold nanocluster. The density of the polylysine chains and the thickness of the polymer layer strongly depend on the amount and concentration of the NCA monomer and the initiator. The optical spectra of the so-obtained core–shell nanosystems show a strong surface plasmon resonance (SPR)-like band at 531 nm. In fact, despite maintenance of the gold cluster size and the absence of interparticle aggregation, the polylysine-capped clusters behave as if they have a diameter nearly 4 times larger. To the best of our knowledge, this is the first observation of the growth of a fully developed, very stable SPR-like band for a gold nanocluster of such dimensions. The robust polylysine protective shell makes the nanoparticles very stable under conditions of chemical etching, in the presence of glutathione, and at different pH values, without gold core deshielding or alteration of the SPR-like band. This polymerization method can conceivably be extended to prepare core–shell nanosystems based on other mono- or co-poly(amino acids).
Introduction
Nanoparticles (NPs) have been widely used to prepare systems capable of displaying specific properties of interest in biomedicine applications, such as tissue engineering, drug delivery, medical diagnostics, and bioimaging.1–3 Generally, however, the use of NPs can be severely limited by various factors, such as their aggregation and precipitation, short half-life in the bloodstream caused by opsonization by plasma proteins, non-specific targeting, and the possible risk of cell and tissue damage.4 Devising proper surface modification is indeed essential for eliminating or at least minimizing these negative factors and making the NPs more biocompatible and stable in biological environments. Many NPs are small enough to pass through biological barriers and are considered to be very promising materials for biomedical applications. Due to their biocompatibility and unique physicochemical properties, Au NPs constitute one of the most useful nanostructured platforms in nanomedicine.5–8 The choice and functionalization of NP systems is of crucial importance for imparting a desired biological response. Moreover, the size, shape, charge and degree of hydrophobicity of NPs influence their ability to cross different biological barriers and affect their stability and retention time in biological fluids. Surface-modification strategies have been developed to prepare Au NPs displaying a required set of such characteristics.6–8Monolayer protected gold clusters (MPCs) are Au NPs in which a small, well-defined gold core is stabilized by a monolayer formed of Au atoms and capping ligands, typically organothiolates.9 Clusters containing 144 Au atoms or less can be prepared with atomic precision, as a consequence of refined synthetic methods and accurate mass-spectrometry analysis.9,10 Functionalization of the monolayer also requires strategies suitable for preparing the sought-after MPCs in a controlled fashion.11 This can be achieved by preparing the MPCs directly from a mixture of appropriate thiols, by reactions of the reactive groups of the capping monolayer itself, or via ligand place-exchange reactions. In the latter, thiolates of preformed MPCs are exchanged with exogenous thiols.12 Post-synthesis exchange reactions are particularly useful, e.g., to modify the monolayer of an MPC of known size, when solubility issues would prevent controlling the MPCs’ size under direct synthesis conditions, or when the exogenous thiol is not available in a sufficiently large amount. Exchange reactions have also been carried out using thiolated amino acids and peptides.13,14
The addition of polymeric coatings can drastically improve the stability of NPs, including Au NPs. Polymers can trigger the formation of core–shell nanostructures or be adsorbed/synthesized on the surface of preformed NPs via grafting strategies.15–18 Poly(amino acids) are of particular interest because of their biocompatibility, easy functionalization, and intrinsic biological activity. These properties can be employed to develop systems for drug delivery, gene therapy, disease treatment, and to construct ultrasensitive biosensors.19–24 To prepare Au NPs capped by water-soluble poly(amino acids), a wide range of encapsulation and ligation approaches have been developed. The in situ fabrication of stable Au NPs can be achieved by the direct reduction of tetrachloroauric ions using amino acid homo- and heteropolymers25–28 or via the electrostatic interaction of preformed Au NPs with poly(amino acids).29,30 Electrostatic or hydrophobic interactions between Au NPs capped by poly(amino acids) can lead to reversible assembly/disassembly and flocculation, as found for example for polylysine coated Au NPs.31,32 These phenomena occur in response to an external stimulus, such as a change of pH or temperature, with a consequent change of the conformational state of polylysine. At neutral pH, the polylysine chains of the outer shell are mostly in random coil and β-turn conformations, whereas an increase of pH causes the deprotonation of polylysine and the chains adopt a more ordered structure with a large extent of β-sheets, which display a propensity to trigger aggregation. The latter makes the Au NPs behave collectively, as evidenced by a red shift of the surface plasmon resonance (SPR) band.
Core–shell NPs obtained by complexation display a relatively high stability in biological environments, but the polymer coating can be disrupted after internalization in cells.33 Grafting-to or grafting-from methods, i.e., when the polymer is either grafted to the substrate surface or grafting proceeds by polymerization from the surface, rely on covalent fixation and thus lead to a higher stability of the core–shell nanosystem compared to methods based on non-covalent attachment.34 They also allow for controlling the characteristics of the forming polymer, as well as the density and length of the surface-tethered polymer chains.35 The grafting-to method was used, for example, for the covalent bonding of a preformed polylysine polymer to Au NPs followed by complexation with DNA to assemble Au NP nanowires,36 and to coat Au NPs with polysarcosine or polyacryloylvaline.38 Concerning the grafting-from method, a few examples have been reported.39–44 However, we are not aware of direct polymerization methods for the preparation of core–shell Au NP/poly(amino acid) systems. This approach would be of particular interest because the covalent bonding of poly(amino acid) chains to the gold core would grant a high stability to these systems in physiological liquids, and also in view of achieving a high grafting density on the NP’s surface.
Here we describe a novel grafting-from approach for the preparation of hybrid biocompatible star-like core–shell nanosystems consisting of a Au144 cluster capped by polylysine. This method works very smoothly and efficiently and could provide a versatile approach for grafting other poly(amino acids) from Au MPCs. Equally important, in the process we discovered that despite the ultrasmall size of this specific cluster, which does not change during the functionalization steps, the so-obtained core–shell nanosystems display a sizable SPR-like band. It is well known that when capped by common thiolates, Au144 only shows a trace of what for larger Au clusters and NPs becomes a true SPR band.45–47 To the best of our knowledge, this is the first observation of a ∼2 nm cluster that upon functionalization apparently behaves as if its diameter is nearly 4 times larger.
Results and discussion
Au144(SR)60 is a particularly stable MPC that for several years has attracted attention from both fundamental and applied viewpoints.12,13,48–56 Previous work showed that its stability is also unaffected under conditions of ligand exchange with exogenous thiols, including thiolated peptides.12,13,48,52 Monodisperse Au144(SCH2CH2Ph)60 was prepared as described previously.13,52 Briefly, AuCl4− was transferred from an aqueous solution to toluene using tetraoctylammonium bromide. Phenylethanethiol was added to the resulting red solution, which became milky white as a consequence of Au(III) to Au(I) reduction. The quick addition of excess NaBH4 caused the formation of a black solution, indicative of the presence of Au MPCs. After 24 h, the reaction mixture was purified to yield Au144(SCH2CH2Ph)60. The cluster had the expected UV-vis optical spectrum (Fig. 1), thermogravimetry pattern (Fig. S1†), matrix-assisted laser desorption ionization time-of-flight mass spectrum (Fig. S2†), and electrochemical behavior.13,51 The MPC size was determined by scanning transmission electron microscopy (STEM) to be 2.0 ± 0.5 nm (Fig. S3†).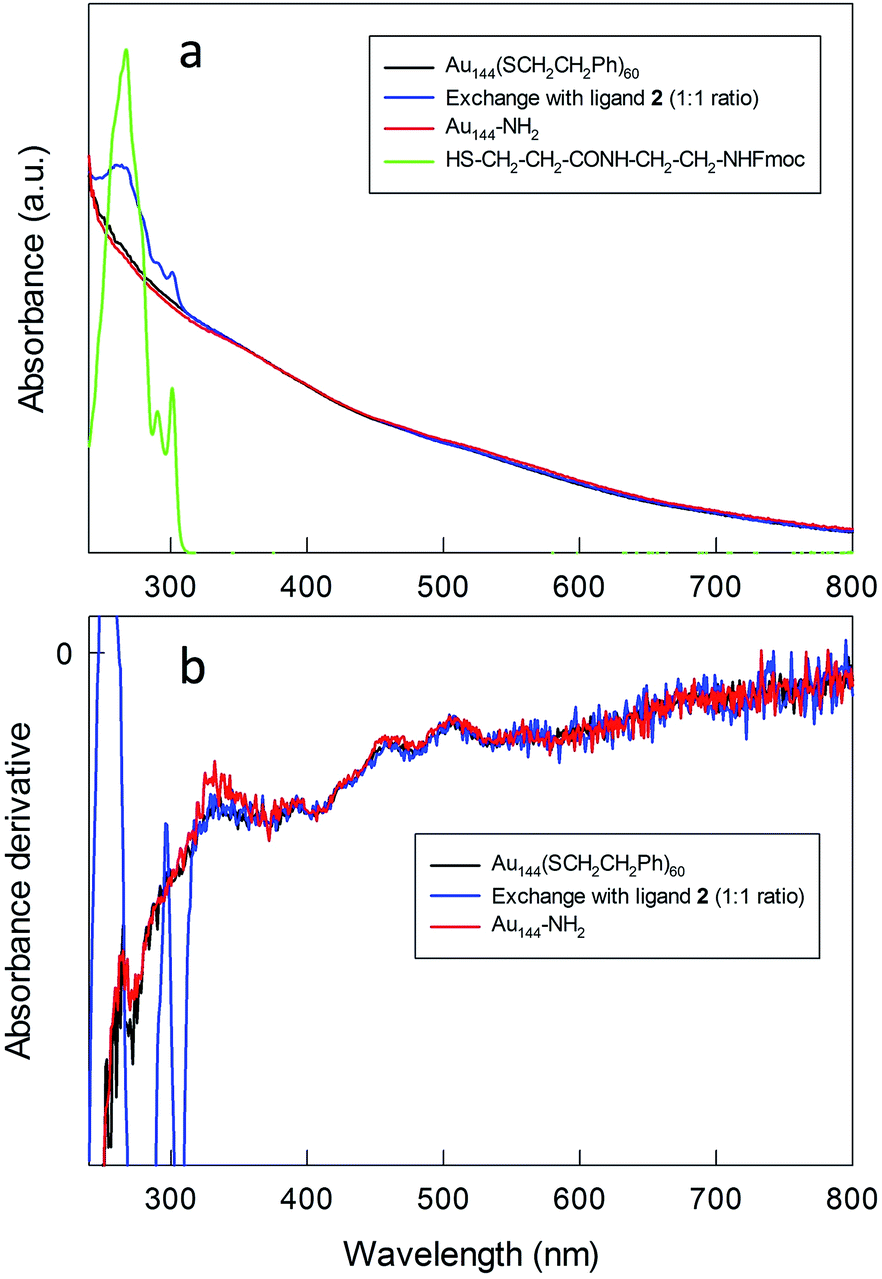 | ||
| Fig. 1 (a) UV-vis absorption and (b) corresponding derivative spectra for the relevant species before, during, and after MPC modification (CHCl3). | ||
To introduce into the Au144 monolayer structure groups suitable for initiating polymerization, we used the ligand place-exchange approach (Scheme 1). A thiolated group was linked to ethylenediamine to form thiols containing either a protected or a free amino group (Scheme 2). Both thiols were designed to be sufficiently long to make the amino group protrude from the phenylethanethiolate monolayer.
 | ||
| Scheme 1 Scheme for the preparation of the initiator for the N-carboxyanhydride polymerization. | ||
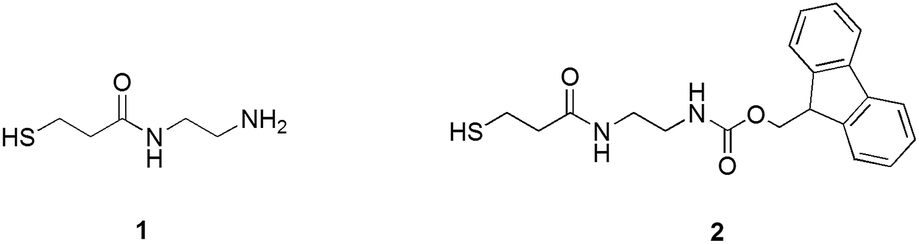 | ||
| Scheme 2 Structures of ligands 1 and 2. | ||
Attempts to use thiol 1 for the ligand exchange, however, led to aggregation of the gold nanoclusters and the formation of an insoluble precipitate. When the molar ligand/MPC ratio was increased, the amount of precipitate also increased, probably because of the interaction of both thiol moieties and free amino groups with the gold core. We thus focused on using thiol 2 protected by the fluorenylmethyloxycarbonyl (Fmoc) group, which can be removed under mild basic conditions without affecting the integrity of the Au144 cluster. The exchange reaction was carried out in dichloromethane for 24 h by using molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1.5:1 of 2 and the phenylethanethiolate ligands. This ensured that intermediate and heavier substitutions could be achieved (for details, see the Experimental section and ESI†). The incorporation of 2 into the MPC monolayer leads to the desired product in high yield. The UV-vis spectra of the purified clusters confirm the presence of the Fmoc-protected ligand in the MPC structure (Fig. 1), as indicated by the additional presence of the typical bands (267, 290, and 301 nm) of the Fmoc group.
:1 and 1.5:1 of 2 and the phenylethanethiolate ligands. This ensured that intermediate and heavier substitutions could be achieved (for details, see the Experimental section and ESI†). The incorporation of 2 into the MPC monolayer leads to the desired product in high yield. The UV-vis spectra of the purified clusters confirm the presence of the Fmoc-protected ligand in the MPC structure (Fig. 1), as indicated by the additional presence of the typical bands (267, 290, and 301 nm) of the Fmoc group.
The spectra obtained before and after exchange show the same fine features (shoulders at 458, 510, 555, and 673 nm) typical of Au144(SR)60, which is particularly evident in the derivative spectra (Fig. 1b). The average composition of the monolayers of the exchanged clusters was determined by 1H NMR spectroscopy after oxidatively decomposing the MPC with excess iodine followed by analysis of the molar ratio of the liberated ligands via quantitative comparison of the integrals of well-resolved peaks. NMR spectrum analysis (Fig. S4–S6†) indicates that for the 1.5:1 and 1:1 initial ratios the extents of exchange are about 50% (30 new thiolates) and 35% (21–22 new thiolates), respectively. These numbers are in line with previous exchanges on the same pristine cluster under comparable conditions.52
To obtain Au144 nanoclusters bearing free amino groups in the protective shell (for simplicity, herein referred to as Au144-NH2), the Fmoc protective groups were removed using piperidine. For the heavily exchanged nanoclusters (50%), deprotection yields the formation of an insoluble product, presumably due to the aggregation caused by the intercluster interaction of the surface amino groups. On the other hand, the nanoclusters carrying a lower load of amino-group-containing ligands (35%) are stable in both solution and the solid state. After deprotection, the UV-vis spectrum and its derivative maintain the same features as those of the pristine Au144(SCH2CH2Ph)60 cluster (Fig. 1), and this indicates that the deprotection conditions do not affect the gold core. Thermogravimetric analysis (TGA) (Fig. S1†) shows that, compared to Au144(SCH2CH2Ph)60, Au144-NH2 fully decomposes at a higher temperature, which can be attributed to the additional stabilization of the monolayer caused by hydrogen bond formation between the newly added ligands. The composition of the protective shell calculated from the TGA weight loss corresponds precisely to that obtained by NMR analysis of the Fmoc-protected Au MPCs; in this context, it is worth noting that for cases in which both approaches can be followed, NMR and TGA analyses give the same outcome.13,52
The Au144-NH2 clusters were used as an initiator for the ring opening polymerization of ε-Fmoc-lysine N-carboxyanhydride, NCA (Scheme 3). The polymerization of NCA was carried out at different temperatures, monomer-to-initiator ratios, and concentrations of monomer and initiator (Table 1). We focused on NCA/amine ratios of 50, 100 and 150 (hereafter, the ensuing clusters will be referred to as Au(50), Au(100) and Au(150), respectively), while keeping constant the concentration of either N-carboxyanhydride (condition 1, entries 2–4) or Au144-NH2 initiator (condition 2, entries 5–7). At low concentrations of initiator, the possible inclusion of other nanoclusters into the network of the forming polymer can be neglected. Polymerization was allowed to proceed for 4 days and was then stopped by the addition of diethyl ether. The resulting precipitate was washed several times with diethyl ether and methanol, to remove unreacted monomer and possible low molecular weight impurities.
 | ||
| Scheme 3 NCA polymerization on the Au144-NH2 nanocluster. | ||
| Entry | Sample | T (oC) | Molar ratio NCA/NH2 | C NH2 (mM) | C NCA (mM) | Appearance of reaction mixture | TGA weight loss% |
|---|---|---|---|---|---|---|---|
| a Subscript 4 indicates that polymerization was carried out at 4 °C. b Experimental condition 1 refers to a constant concentration of NCA. c Experimental condition 2 refers to a constant concentration of amino groups. d Concentration refers to the total amount of amino groups in solution, and thus to a value 35 times larger than that of the exchanged clusters. | |||||||
| 1 | Au(50)4a | 4 | 50 | 3 | 150 | Liquid | — |
| 2 | Au(50)-1b | 30 | 50 | 3 | 150 | Gel | 85 |
| 3 | Au(100)-1b | 30 | 100 | 1.5 | 150 | Viscous liquid | 90 |
| 4 | Au(150)-1b | 30 | 150 | 1 | 150 | Viscous liquid | 91 |
| 5 | Au(50)-2c | 30 | 50 | 0.15 | 7.5 | Viscous liquid | 66 |
| 6 | Au(100)-2c | 30 | 100 | 0.15 | 15 | Viscous liquid | 71 |
| 7 | Au(150)-2c | 30 | 150 | 0.15 | 22.5 | Viscous liquid | 74 |
The formation of a polymer is observed for all molar combinations. After the completion of polymerization, just before quenching, the physical appearance of the reaction mixture depends on the concentrations of the monomer and initiator, and on the temperature. At 4 °C, only low molecular weight products, partially soluble in diethyl ether, form (Table 1, entry 1). At 30 °C polymerization leads to the formation of brown viscous solutions; the only exception is found for Au(50)-1, which is the case with particularly high concentrations of both initiator and monomer, and thus particularly high polymerization rates (Table 1, entry 2).
The TGA profiles of the Fmoc-deprotected polylysine coated MPCs confirm extensive polymer formation on the surface of the MPCs (Fig. 2). The amount of polylysine coating strongly depends on the ratio of monomer to initiator, as well as their concentrations in the reaction mixture. For high concentrations of MPC and NCA (Table 1, entries 2–4), the loss of organic material is significantly larger than for reactions carried out at a low concentration of the initiator (entries 5–7). This indicates that for the latter a lower polylysine loading and thus a thinner shell is attained, which can be explained by a slower polymerization rate. The feeding of the reaction with NCA also influences the rate of polymerization in the sense that the amount of formed polylysine increases with an increasing initiator/monomer ratio. In the case of ratios 1/100 and 1/150 (entries 3 and 4, respectively), however, the final content of polylysine in the polymeric shell is almost identical: most probably, for Au(150)-1 in which the NCA monomer is highly concentrated relative to the initiator (Table 1, entry 4), a fast polymerization feed causes fast growth of the polylysine chains with the consequence of hindering some of the available sites for polymerization (due to steric crowding), thereby limiting an even larger increase of the polymeric mass. At low concentrations of both Au144-NH2 and NCA (entry 5, Au(50)-2), the overall polymerization reaction proceeds particularly slowly and thus the amount of polylysine is the lowest among the experimental conditions tested. By assuming that all the amino groups on the cluster monolayer reacted, the average chain lengths are calculated as corresponding to a number of Lys units varying from 57 to 95 or 16 to 27 for series 1 or 2, respectively.
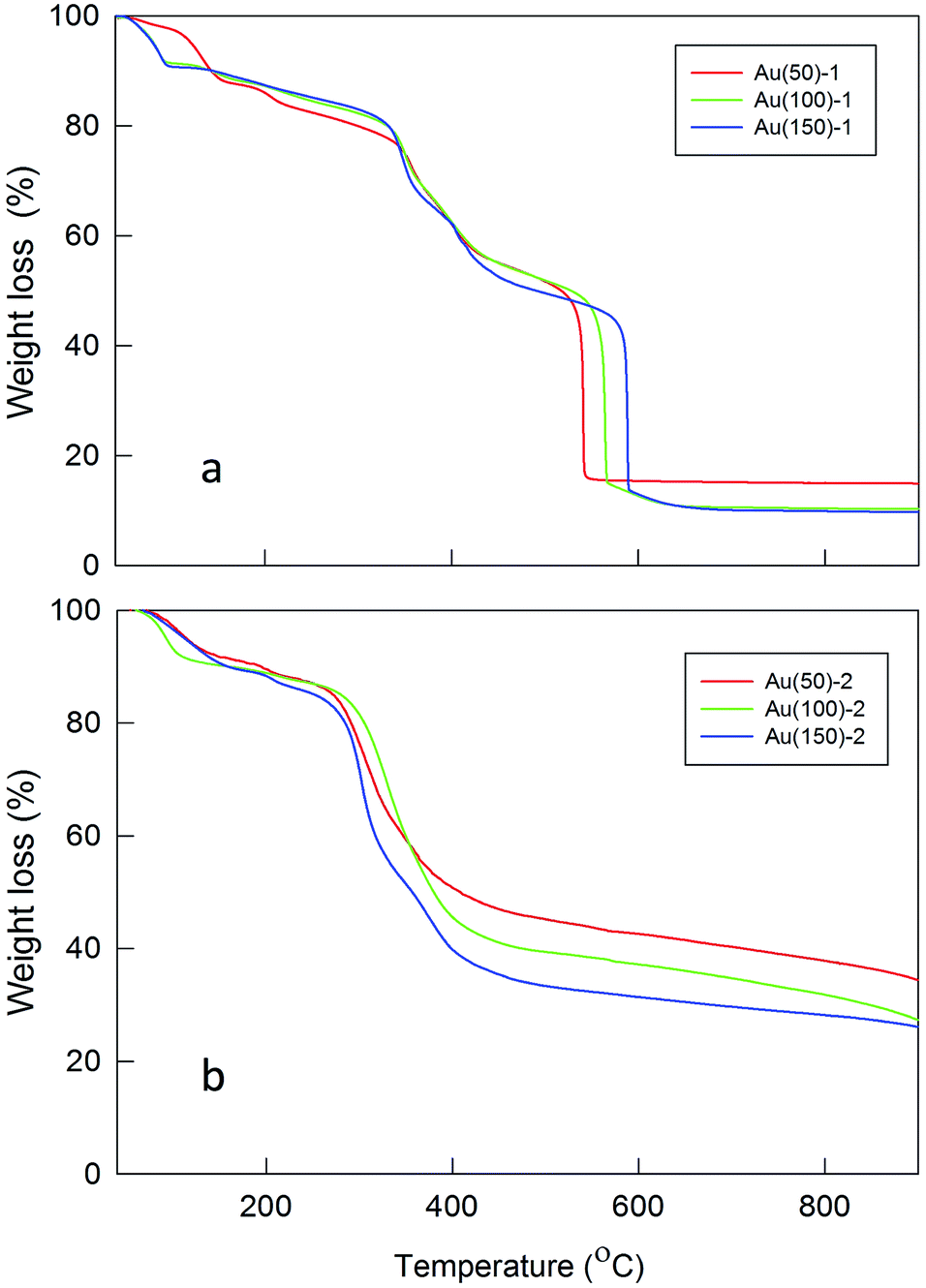 | ||
| Fig. 2 TGA plots for polylysine coated Au MPCs: (a) condition 1; (b) condition 2. | ||
TGA analysis also reveals a significant increase in the thermal stability of the core–shell NPs (Fig. 2) with respect to Au144-NH2 or Au144(SCH2CH2Ph)60 (Fig. S1†). Overall, the temperature of total decomposition increases from ca. 200 °C for the pristine Au144(SCH2CH2Ph)60 to ca. 600 °C for Au(150)-1. The decomposition temperature also depends on the amount of polymer forming the shell: in particular, a comparison of the TGA profiles of the two series of NPs shows that the lower content of polylysine in the latter facilitates thermal decomposition. Interestingly, an evident difference in the decomposition temperature of the second weight-loss step of Au(100)-1 and Au(150)-1 is observed, despite the virtually identical polylysine mass content. This points to stronger interchain interactions in Au(150)-1 and, more generally, to a delicate balance between chain length and number.
Information about the formation of the polylysine layer was obtained by using FT-IR spectroscopy, a technique particularly useful for peptide MPCs.57 For peptides, proteins and poly(amino acids), the absorption bands in the amide A (N–H stretching, 3300–3500 cm−1), amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, 1700–1600 cm−1), and amide II (N–H bending and C–N stretching, 1480–1575 cm−1) regions are strongly related to the peptide secondary structure and spatial orientation.58Fig. 3 shows these bands for the polylysine grafted Au nanoclusters, in the protonated state.
O stretching, 1700–1600 cm−1), and amide II (N–H bending and C–N stretching, 1480–1575 cm−1) regions are strongly related to the peptide secondary structure and spatial orientation.58Fig. 3 shows these bands for the polylysine grafted Au nanoclusters, in the protonated state.
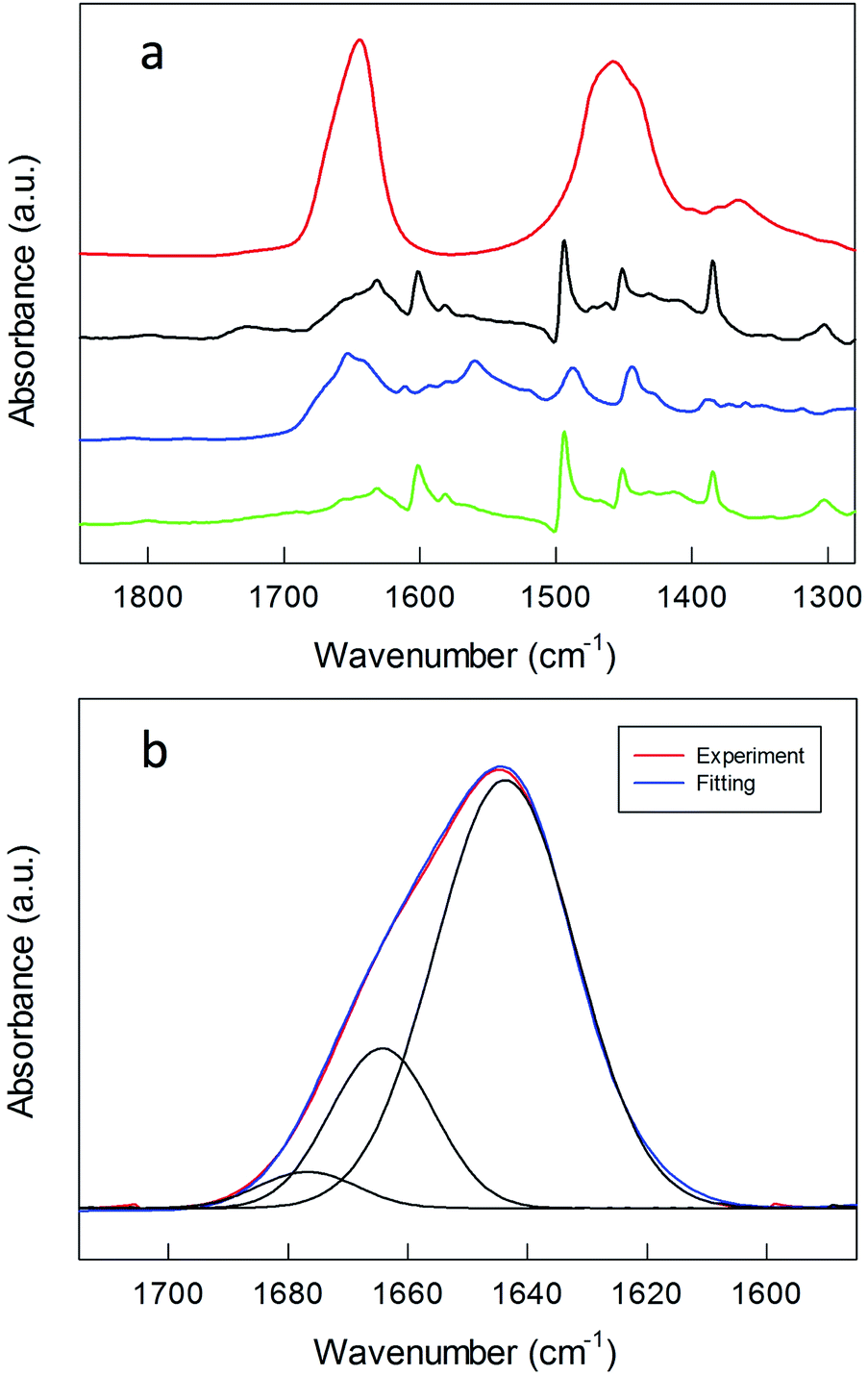 | ||
| Fig. 3 (a) FT-IR spectra of (green) Au144(SCH2CH2Ph)60, (blue) TrtS–CH2CH2CO–NH–CH2CH2–NH2, (black) Au144-NH2, all obtained in KBr, and (red) Au(150)-1 in D2O. The spectra are shifted for the sake of comparison. (b) Gaussian curve fit of the amide I region of the FT-IR spectrum of Au(150)-1 in D2O. | ||
For comparison, the FT-IR spectra of Au144-NH2 and TrtS–CH2CH2–CO–NH–CH2CH2–NH2, i.e., 1 where S is protected by a trityl group, are included (Fig. 3a). The spectrum of Au144-NH2 is quite similar to that of native Au144(SCH2CH2Ph)60, but for the presence in the latter of a band at 1654 cm−1 due to the stretching vibrations of the carbonyl group, which is also present in the spectrum of the protected ligand. After polymerization, the FT-IR spectra are dominated by the broad amide A, amide I, and amide II bands of the polylysine coating, with maxima at 3402, 1644 and 1457 cm−1 (Fig. S7 and S8†). These frequency values have been observed previously for polylysine hydrochloride in D2O.59 Different concentrations of monomer and initiator do not affect the FT-IR pattern. Both the broad amide A and amide I point to the formation of a large inter- and intramolecular H bond network. Curve-fitting analysis of the amide I band (Fig. 3b) allows the distinguishing of three vibration modes at 1644, 1664 and 1677 cm−1 that are assigned to random coils, β-turns, and β-sheets, respectively.58,60,61 These results are in agreement with the work of Guo et al.32 in which it is shown that, at low pH, random coils and β-turns constitute the dominant conformations of polylysine bonded via complexation to large gold NPs.
STEM images of the core–shell MPCs were taken to verify that the gold size was maintained after NCA polymerization. In TEM it is often difficult to gather information on polymer coatings, especially for polymers composed of poly(amino acids), because of their low contrast and sensitivity to high energy electron beam impact.62 Therefore, only the gold part can be seen in the STEM images. Fig. 4 shows typical images obtained for Au(50)-1, Au(100)-1, and Au(150)-1, whereas the histograms are in Fig. S9.†
 | ||
| Fig. 4 STEM images of series 1. | ||
According to STEM, the diameters of Au(50)-1, Au(100)-1, Au(150)-1 are 1.9 ± 0.2, 1.9 ± 0.2, and 1.8 ± 0.5 nm, respectively. The data thus show that polymerization does not cause any evident change in size, which remains the same, within error, as that of the pristine Au144(SCH2CH2Ph)60 cluster, 2.0 ± 0.5 nm. Also, no aggregation of clusters is detected, which indicates that even in the conditions for sample preparation for acquiring the STEM images the clusters remain well dispersed. Finally, there is no indication of gold nanoclusters being incorporated in the polylysine network during the polymerization followed by formation of a 3D matrix with encapsulated Au MPCs.
To examine the overall size of the core–shell nanosystems obtained, including both the nanoparticle core and polylysine layer, we carried out dynamic light scattering (DLS) measurements (Table 2 and Fig. S10†). The measurements were carried out using the Fmoc-deprotected core–shell NPs, as the positively charged polylysine chains were expected to prevent NP coagulation through coulombic repulsion. All measurements were carried out in water, where the Fmoc-deprotected core–shell NP systems are fully soluble. We found that polylysine grafted NPs obtained using different ratios of monomer and initiator have diameters ranging from 22 to 35 nm with polydispersity indices (PDIs) ranging from 0.22 to 0.46. Within each of the two sets of NPs labeled as 1 or 2 (Table 2, entries 1–3 and 4–6, respectively), the average NP diameter depends on the NCA monomer/initiator ratio, in agreement with the TGA results. In particular, as the monomer content in the reaction mixture decreases, thinner polylysine layers form. It is also worth noting that, despite the lower polymer content, the core–shell NPs of series 2 have similar hydrodynamic diameters to those of series 1; this indicates that in series 2 the polylysine chains grow particularly long or are more mobile, and therefore that the polylysine shell is less dense. To exclude the possibility of aggregation artifacts, the results were also compared to those obtained after the dilution of each sample, but no differences were observed.
Small angle X-ray scattering (SAXS) is very sensitive to the electron density distributions in the structures of nanoassemblies averaged over time,63 and thus is suitable for providing a detailed picture of the core–shell architecture of the NPs. SAXS data on 1.0 wt% Au(50)-1, Au(100)-1 and Au(150)-1 solutions (Table 2) were expressed as the scattering intensity as a function of the scattering vector q, which is defined as (4π/λ)sin(θ/2) where θ and λ are the scattering angle and wavelength, respectively. Fig. 5 shows monotonic decays in all three SAXS patterns. The Au(150)-1 curve also shows an extra oscillation-like decay at 0.04 Å−1 < q < 0.08 Å−1. On the basis of the TEM and DLS data, we performed data fitting using the polydisperse-core-multi-shell spherical model; details are provided in the ESI.† For all clusters, the radius of the Au cluster was kept constant at 1 nm (according to the TEM results) and a minimal number of shells was used to fit the SAXS in order to minimize the number of fitting parameters.
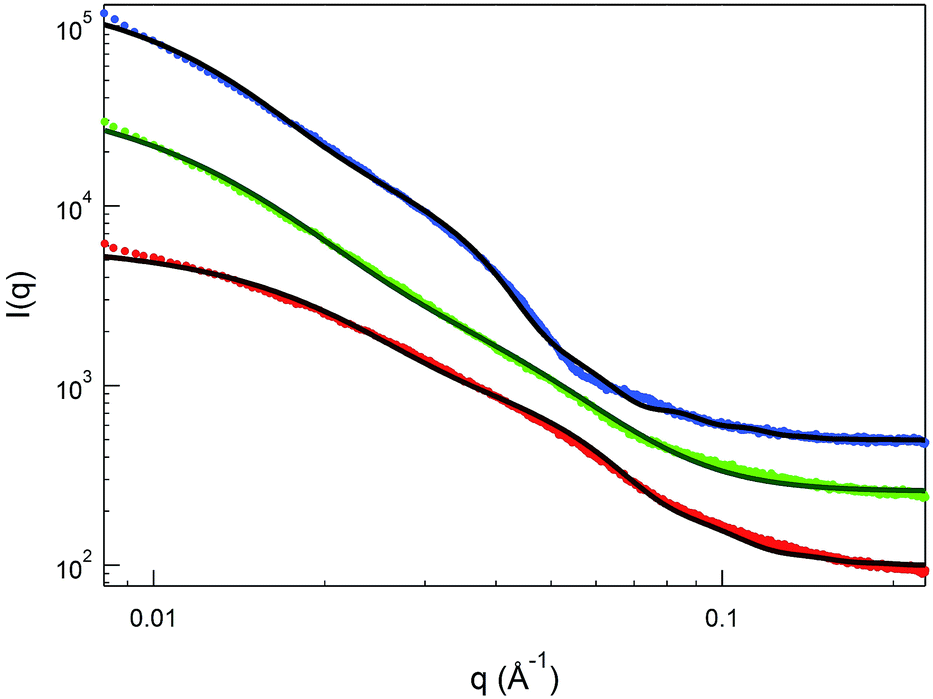 | ||
| Fig. 5 SAXS data on 1 wt% Au(50)-1 (red circles), Au(100)-1 (green circles), and Au(150)-1 (blue circles) in 10% acetic acid solution. The solid lines are the best fits to the SAXS data: a polydisperse-core-one-shell model was used for Au(50)-1 and Au(100)-1, whereas a two-shell model was used for Au(150)-1. | ||
The overall best-fit diameters from the SAXS data (cluster diameter plus twice the thickness value) are larger than the hydrodynamic diameters obtained from the DLS measurements by 25–50%. The discrepancy may originate from applying a constant electron density to the shells, to simplify the model, whereas in reality the density is expected to decrease from the inside out. This simplified analysis, however, can still provide important insights into the shell thickness and distribution. First, the thickness increases from Au(50)-1 to Au(150)-1 (Table 2, entries 1–3) in keeping with the longer polylysine chains for the core–shell NPs obtained with a greater feed of NCA during polymerization. Second, the relative shell-to-solvent contrast increases by more than 50% as one goes from Au(50)-1 to Au(100)-1, and this indicates that the polymer density also increases. In fact, for Au(150)-1, the one-shell polydisperse-core model can no longer provide a reasonable fit to the SAXS data and, therefore, a second inner shell (Table 2) was introduced in the analysis. The thickness of this inner shell is around 6 nm, with a much higher electron density than for the outer shell. This is indeed in good agreement with the data obtained by TGA analysis, where, despite the similar content of polylysine in Au(100)-1 and Au(150)-1, for the latter the decomposition temperature is higher.
The combined STEM, DLS and SAXS analysis thus points to the so-prepared MPCs having a cluster size as small as in the pristine clusters and a capping layer consisting of a high-density inner polylysine layer, whose thickness increases upon increasing the monomer/initiator ratio, and a less dense outer layer composed of long polylysine chains. No aggregation phenomena are detectable, whether in the solid state or solution.
To gain insights into the formation of the polylysine capped MPCs, the polymerization was followed by optical absorption spectroscopy by periodically collecting samples that were then diluted and studied either as such (Fig. S11†) or after deprotection (Fig. 6). At the initial stages of the reaction, no changes in the spectra are observed, whereas after 4 days a weak but clearly perceivable SPR-like band appears at ca. 530 nm. The same band is also seen for Au(50)-2, for which the concentrations of both Au144-NH2 and NCA are very low, which excludes the possibility that the appearance of such a band is caused by aggregation of the NPs during polymerization. This SPR-like band is more evident when the samples are Fmoc-deprotected (cf.Fig. 6 and S11†). Its intensity also increases when the amount of monomer in the mixture is larger (Fig. S12†). However, even when particularly evident, as for Au(150)-1, this band is weak (the maximum value of the derivative is 0, at 509 nm) and the color of the NP solution remains brown, as observed for Au144-NH2. That the gold cluster is still Au144 is supported by the very nice overlap between the traces of the derivative absorption, shown in comparison with Au144-NH2. It is noteworthy that, even when the SPR-like band is at its maximum, fine fingerprint features for Au144 are still present at 380–415 and 660–730 nm.
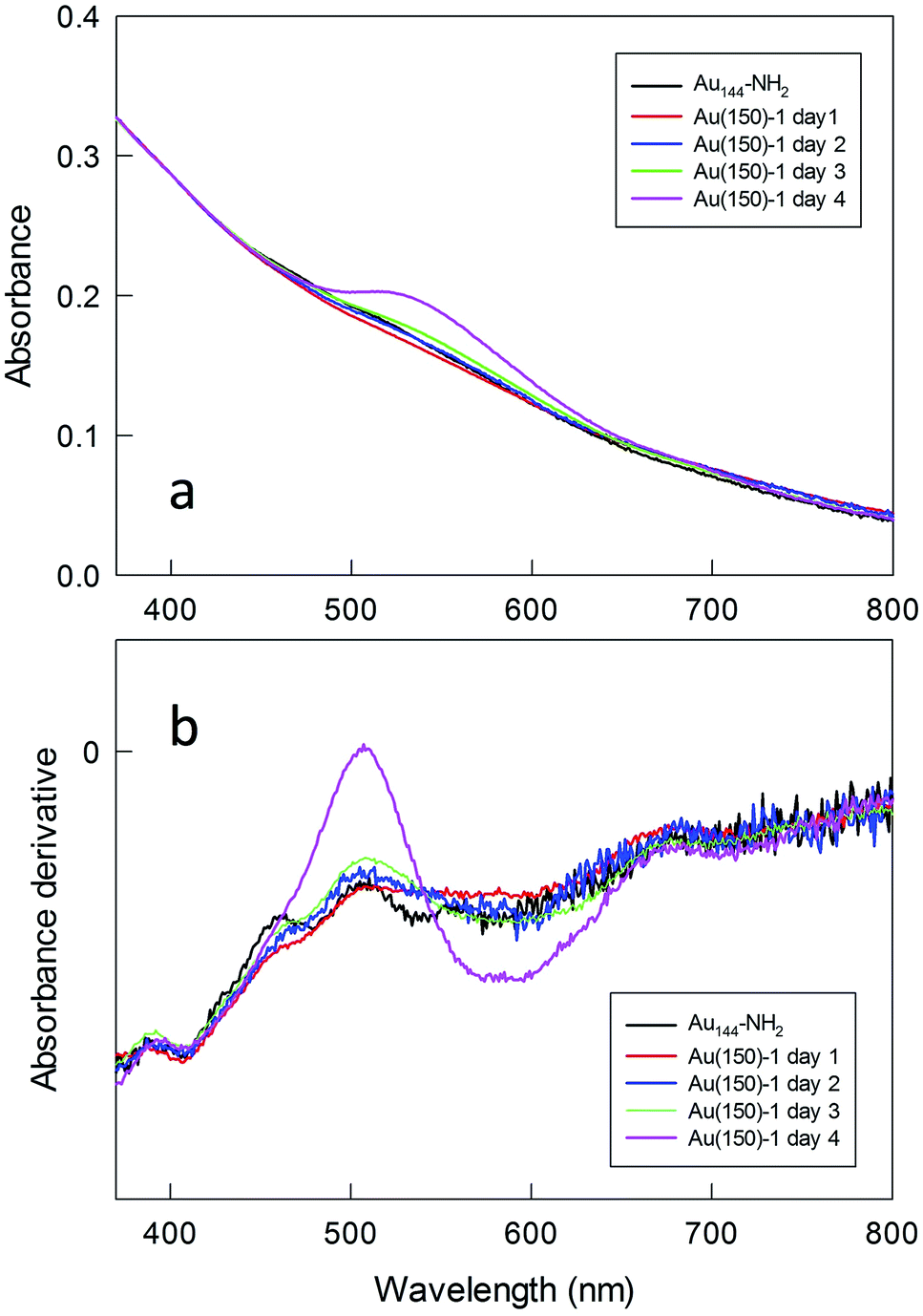 | ||
| Fig. 6 Evolution of the (a) optical absorption spectrum and (b) its derivative for Au(150)-1. Samples were taken from the reaction mixture, Fmoc-deprotected, and dissolved in water + 1% HCl. For the sake of comparison, the spectra have been normalized at 400 nm. | ||
UV-vis spectra were also taken at the end of the polymerization, after extensive purification of the Fmoc-deprotected polylysine capped MPCs. In this state, the NPs are only soluble in water. We found that the spectra undergo a drastic time-dependent change, accompanied by a clearly detectable color shift from brown to pink. Fig. 7 illustrates a typical example (for a sample of Au(100)-1) of the time evolution of the spectral profile showing the very significant growth of an intense SPR-like band at 531 nm that reaches its maximum after 3–4 days. This SPR-like band also strongly increases for NPs obtained by polymerization using a lower amount of NCA and Au144-NH2 (series 2) or other ratios thereof, with only limited differences in the ratio of the absorbance value at the peak to that at the minimum at ca. 470 nm. Its intensity and position do not depend on the sample dilution (Fig. S13†), which excludes the possibility that the growth of the band is caused by NP aggregation caused by interaction of the polylysine chains. Importantly, the formation of the band is irreversible. As a matter of fact, after reaching a fully developed SPR-like band the samples can be dried and then redissolved in water to immediately yield the same spectrum. As all the spectra were acquired under aerobic conditions, we also checked whether the oxygen content could affect the spectra. However, we found that the SPR-like band is unaffected when collecting the spectrum under strictly anaerobic conditions (argon) or after prolonged bubbling with pure oxygen. To verify whether the appearance of the SPR-like band is associated with the presence of free amino groups in the polylysine shell, the NPs were acetylated after Fmoc deprotection. In this case, however, no band appears even upon keeping the NPs in solution for one week (Fig. S14†). This experiment suggests that the presence of free amino groups is relevant to the observation of a fully developed band.
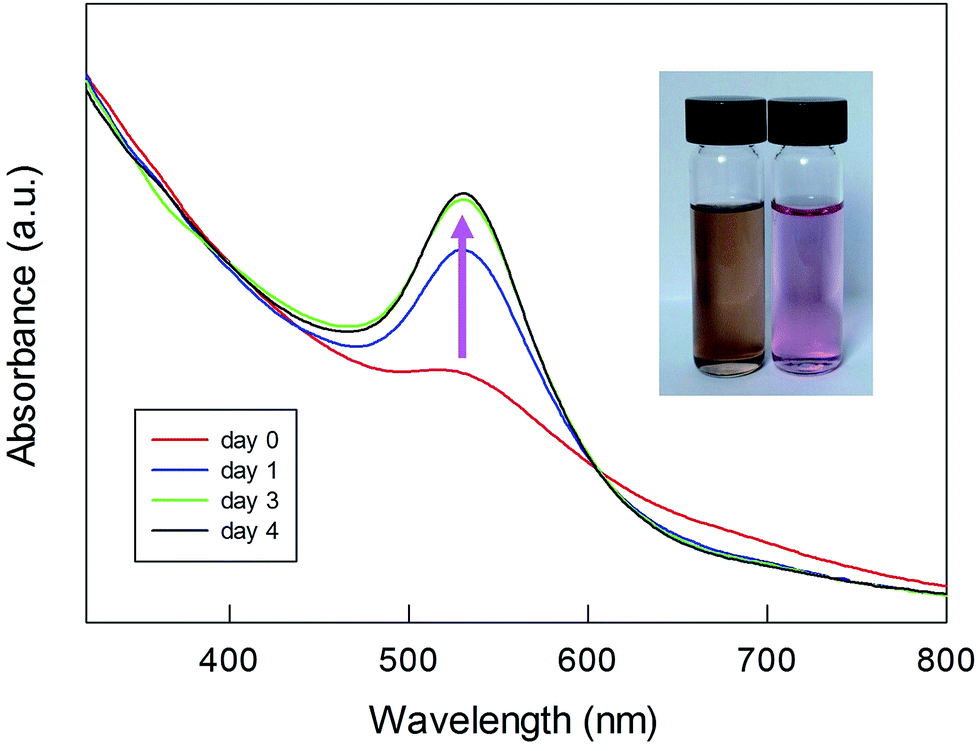 | ||
| Fig. 7 Time evolution of the SPR-like band for Fmoc-deprotected Au(100)-1. The inset shows the overall change in the color of the sample. | ||
We checked the response of these NP systems to large changes in pH. It is known60 that pH affects the conformation of polylysine; in particular, a large amount of β-sheet structure is expected to form in basic conditions, possibly causing NP aggregation. As a matter of fact, a broadening and red shift of the SPR band has been observed for polylysine-capped NPs.32Fig. 8 shows the UV-vis spectra recorded for Au(150)-1 at different pH values. Upon increasing the pH, however, only a slight increase in the intensity and no shift of the SPR-like band is observed. These results thus indicate that the systems investigated are highly stable with respect to possible conformational changes and aggregation between chains. We attribute this to tight packing and crowding of the poly(amino acid) chains in the shell formed during polymerization, as well as steric hindrance to conformational changes.
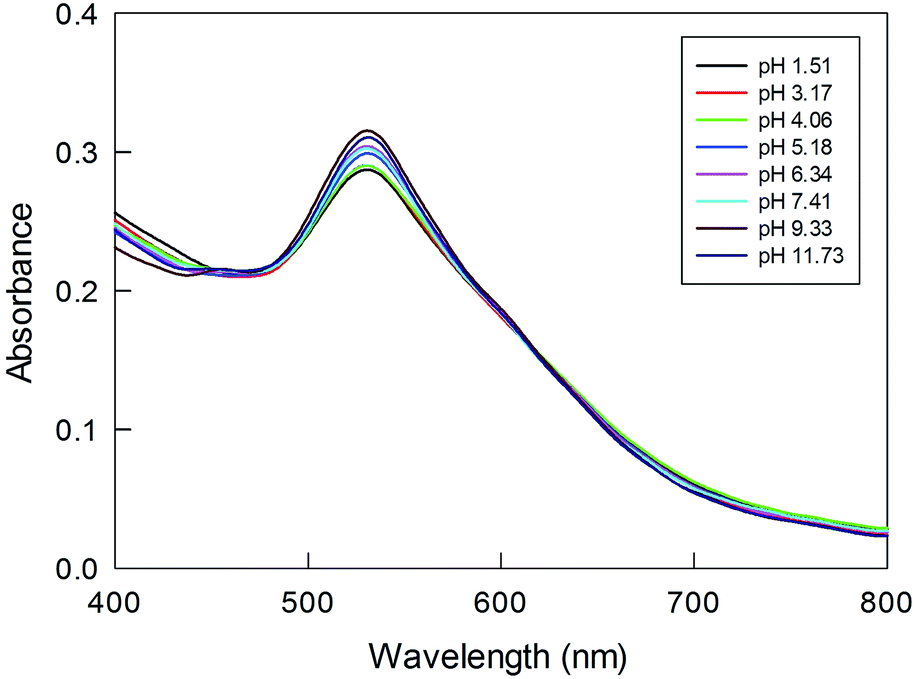 | ||
| Fig. 8 Effect of pH, as shown in the legend, on the SPR-like band for Au(150)-1. The sample was preliminarily kept for one day in solution. | ||
As further stability tests, and also in view of the possible applications of these NP systems in biomedicine, we checked their reactivity toward ligand place-exchange. Glutathione-mediated desorption of the protective polymer layer may indeed present a problem in the design of core–shell gold NPs for use in biological media, particularly in the intracellular environment where the concentration of glutathione can be as high as 10 mM. We thus checked the stability of our systems in the presence of 10 mM glutathione, but found that the UV-vis spectrum does not change even after one week (Fig. S15†). We also checked that the core–shell NPs are stable in the presence of the very short ethanethiol, even when present at a very high concentration; only a small blueshift of the SPR-like band and a small decrease in its intensity were observed (Fig. S16†). Finally, the remarkable high stability of the so-prepared core–shell nanosystem and, therefore, protective function of the polymer layer are also demonstrated by the lack of reactivity in the presence of excess iodine, whereas under the same conditions clusters protected by short thiolates react very quickly by releasing gold metal and disulfides, as mentioned previously.
These results and control experiments clearly show that the observed SPR-like band is stable with respect to environment changes and is not caused by artifacts, such as NP aggregation. It is known that the SPR band is caused by collective oscillation of the conduction electrons under incident light.45 It is influenced by several parameters, mainly the metal NP size and shape6,8,64 and the dielectric properties of the surrounding environment.65,66 The first factor is particularly intriguing because SPR bands are observed for large Au NP systems, whereas the polylysine systems that we prepared and characterized are based on grafting from Au144. When capped with common thiolates, the UV-vis absorption spectrum of Au144(SR)60 only shows a trace of what for larger Au clusters and NPs becomes a true SPR band. Au144(SR)60 and slightly smaller MPCs only possess what has been defined as a core-localized plasmon46 and, according to electronic-energy-relaxation measurements, Au144(SR)60 is the smallest-known nanocluster to exhibit optical properties characteristic of metals.67 However, clear optical features obtained at low temperature indicate that Au144(SR)60 still shows quantum features.68 Only slightly larger clusters start displaying a more bulk-like, plasmonic behavior.47 Ligands affect the optical behavior of Au144(SR)60 to a very limited extent. In this context, it is worth mentioning that clusters of similar sizes (average TEM-determined diameter 1.2 nm) generated by the reaction of HAuCl4 with a polymer, poly(N-vinyl-2-pyrrolidone), have been recently found to show the appearance of a small SPR-type shoulder when exposed to sodium borohydride; however, the shoulder quickly disappears (in hours) particularly under aerobic conditions.69 A similar observation has also been reported, again using an analogous (average diameter 1.4 nm) poly(N-vinyl-2-pyrrolidone) stabilized gold cluster.70 In both cases, however, the core size could not be controlled with atomic precision, and the shoulder was very sensitive to the oxygen content of the medium.
Au144(SR)60 is one of the most studied and stable clusters, with a structure that is still elusive and so far has only been estimated, by DFT calculations. According to theoretical models and different TEM measurements,46–48,52,68,71 the gold structure in Au144(SR)60 has a diameter in the range from 1.6 to 2.0 nm and the cluster does not display any evident SPR behavior. Our present results (with an average 1.9 nm diameter) show that although Au144 does not change its size during polymerization, a well-developed and stable SPR-like band forms. If one makes a simple calculation of the size by using the intensity of the observed SPR-like band,64,72 the striking result is that the Au144 cluster capped by polylysine behaves as if it is as large as 7.3 ± 1.8 nm (an average of the values observed after 10 days for the six samples), i.e., nearly 4 times larger than is evident from the optical and TEM analyses. In connection, it is worth mentioning that larger nanoclusters, including ∼Au940, which is a particularly large nanocrystal characterized in detail and which has a TEM diameter of 2.9 nm, only show very weak plasmonic bands.73 The birth of SPR bands of various intensities was observed upon doping Au144 and Au349 with copper or silver atoms.74–76 The fact that the band that we observe is fully stable with respect to oxygen, pH, and thiols indicates that these MPC systems must evidently obtain some unique characteristics related to the capping polylysine chains; according to the outcome of the acetylation experiments, the chains should keep their amino groups free. Regardless, our results show that the medium provided by the specific ligands and solvent may have an unexpectedly large effect on the optical behavior of this cluster. Very recent results obtained for Au25(SR)18 indicate that for alkanethiolate ligands the chemically relevant orbitals expand by ca. 6 Å from the actual gold core.77 On the other hand, time-dependent density-functional-theory simulations indicate that, e.g., tuning the electronic conjugation in thiophenolate-type ligands protecting smaller clusters can expand electronic resonance from the core to the ligands.78 For polylysine-grafted Au144 clusters, the conjugation might be related to a rearrangement of the inner polymer shell and, perhaps, the surface Au-SR motifs. The effect of ligands on the optical properties of intrinsically nonplasmonic clusters is indeed still largely unexplored and underestimated.79 In this context, our results thus provide a new scenario and call for the in-depth investigation of this phenomenon by targeted experimental and theoretical studies.
Conclusions
We have described a new method for the preparation of robust nanosystems formed of a gold core coated by linear polylysine chains. Direct N-carboxyanhydride polymerization is directly performed on a Au144 nanocluster bearing free amino groups in the protective monolayer. Polymerization, tested under different experimental conditions, proceeds smoothly, and the density and thickness of the polylysine shell depends on the initial monomer/initiator ratio and the rate of polymerization. The resulting systems are robust and are not affected by the pH or oxygen content of the medium, or the presence of other thiols. Equally important is that the resulting MPCs show an SPR-like band that grows until it reaches a maximum after a few days. This band is very stable with respect to different environmental changes and ligand exchange reactions. The results exclude the possibility that this effect is caused by either a change in the gold cluster size or the aggregation of NPs in solution. In fact, the final system displays a band that would be expected for a particle with a diameter ca. 4 times larger than that of Au144. These results could be of particular interest for the preparation of very stable, functionalized nanosystems suitable for biomedicine applications, such as thermotherapy and bioimaging. Regarding the method itself, it is simple, easily controllable, and could be conceivably extended to the preparation of core–shell nanosystems based on other mono- or co-poly(amino acids).Experimental
Thermogravimetric analysis
TGA was performed with a Q5000 IR Thermogravimetric Analyzer (TA Instruments). The TGA curves were recorded under a nitrogen-flow atmosphere with the samples (0.5–1 mg) accurately weighed and placed on an open platinum pan. To eliminate traces of water, an isothermal stage at 45 °C was carried out for 30 min before starting the measurements. In the Hi-Res Ramp mode (ramp 1.5–50 °C min−1, depending on the rate of mass loss with temperature) in the 45–900 °C range, the organic volatilization and ensuing mass loss occurred for all samples at temperatures above 200 °C, leaving a gold metal residue in the pan. For Au(50)-2, Au(100)-2, and Au(150)-2, at the end of each run an isothermal stage in the presence of oxygen was carried out to ensure complete decomposition of the samples.FT-IR absorption spectroscopy
FT-IR spectra were recorded on a nitrogen flushed Nicolet 770 FT-IR spectrophotometer at a 2 cm−1 nominal resolution, using cells with an optical path length of 0.05 mm and CaF2 windows. For the samples in D2O solution (10–60 mg ml−1) and KBr, we used an average of 25 and 20 scans, respectively. Gaussian curve fitting was performed in the amide I band region with the Fityk software.UV-vis absorption spectroscopy
Spectra were taken with an Evolution 60S UV-Vis spectrophotometer (Thermo Scientific), using standard cuvettes with a path length of 1 or 0.2 cm. The spectral resolution was 1 nm.MALDI-TOF mass spectrometry
Experiments were carried out with an Applied Biosystems 4800 MALDI-TOF/TOF spectrometer equipped with a Nd:YAG laser operating at 355 nm, with a laser firing rate of 200 Hz and an accelerating voltage of 25 kV. DCTB (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile, Sigma-Aldrich, ≥98%) was used as the matrix. The instrument was calibrated with Au25(SCH2CH2Ph)25.80 A 0.1 mM solution of Au144(SCH2CH2Ph)60 in dichloromethane, containing DCTB in a 1:400 MPC/matrix ratio, was drop casted onto the sample plate and air-dried. Spectra were recorded using the linear positive mode.
Dynamic light scattering
DLS analysis was performed with a Zetasizer Nano S90 (Malvern) with a 633 nm laser beam. All studies were carried out at 25 °C. The samples were dissolved in ultrapure water, aged for 5 days, and filtered before each measurement.Scanning transmission electron microscopy
STEM was carried out on a Zeiss Libra 200FE microscope with an X-Max energy dispersive X-ray detector. TEM samples were prepared by drop casting 5 μL of 1–5 mg ml−1 aqueous solution of the samples onto carbon-film-coated copper grids. Histograms of the core sizes and the corresponding Gaussian fit to the data were obtained from digitized images.Small angle X-ray scattering
For the SAXS experiment, liquid samples (each 80 μL) were loaded into a standard Bruker quartz liquid holder. For each sample, the scattering and transmittance were measured separately. SAXS measurements were conducted on a Bruker NanoSTAR instrument. X-rays were generated by a Turbo (rotating anode) X-ray source (TXS). A wavelength of 1.542 Å was chosen from the Cu-Kα emission using a Göbel mirror. A pair of “scatterless” pinholes with diameters of 500 and 350 μm were used for beam collimation. The 2-D intensity data was collected by a MikroGap VÅNTEC-2000 detector with a sample-to-detector distance of 108 cm to cover a scattering vector q ranging from 0.007 to 0.25 Å−1. The raw SAXS data was corrected for the sample transmission and the empty-cell scattering. The 1-D data (I vs. q) was obtained by circularly averaging the 2-D data with respect to the beam center.Polymerization
To a solution of N-carboxyanhydride in anhydrous N,N-dimethylformamide (DMF), a solution of Au-NH2 (5 mg, 1.36 × 10−4 mmol) in anhydrous DMF was added to obtain the sought concentrations and ratios of NCA/initiator. The reaction was left for 4 days at 4 °C or 25 °C, without stirring. The product was precipitated in diethyl ether (or in a diethyl ether/methanol 1/1 mixture in the case of the polymerization carried out at 4 °C), separated by centrifugation, washed with diethyl ether or a diethyl ether/methanol mixture and dried in vacuo over P2O5. The obtained core–shell nanoparticles were further Fmoc-deprotected by using a 10% piperidine solution in DMF, purified after precipitation in diethyl ether by repetitive washing with diethyl ether, water and methanol, and dried in vacuo over P2O5. The polymerization yields ranged from 70 to 90% for both series 1 and 2, whereas they decreased to 10% for polymerization at 4 °C.Acknowledgements
This work was financially supported by the Italian Association for Cancer Research, AIRC (FM, Grant 12214: Innovative tools for cancer risk assessment and early diagnosis – 5 per mille), the Russian Science Foundation (TT, Grant #145000069), and the National Science Foundation (MPN, Grant NSF-CBET 1605971). The SAXS instrument at the University of Connecticut is also partially funded through an NSF major research instrument grant (NSF-DMR 1228817). TEM data were obtained by using the facilities of the Interdisciplinary Resource Centre for Nanotechnology of St. Petersburg State University, Russia.References
- K. T. Nguyen and Y. Zhao, Acc. Chem. Res., 2015, 48, 3016–3025 CrossRef CAS PubMed.
- J. A. Kemp, M. S. Shim, C. Y. Heo and Y. J. Kwon, Adv. Drug Delivery Rev., 2016, 98, 3–18 CrossRef CAS PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- A. Sasidharan and N. A. Monteiro-Riviere, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 779–796 CrossRef CAS PubMed.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed.
- N. S. Abadeer and C. J. Murphy, J. Phys. Chem. C, 2016, 120, 4691–4716 CAS.
- Protected Metal Clusters: From Fundamentals to Applications, Frontiers of Nanoscience, ed. T. Tsukuda and H. Häkkinen, Elsevier, Amsterdam, 2015, vol. 9 Search PubMed.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 4134–4142 CrossRef CAS PubMed.
- C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef PubMed.
- H. Holm, M. Ceccato, R. L. Donkers, L. Fabris, G. Pace and F. Maran, Langmuir, 2006, 22, 10584–10589 CrossRef PubMed.
- P. S. Ghosh, C.-K. Kim, G. Han, N. S. Forbes and V. M. Rotello, ACS Nano, 2008, 2, 2213–2218 CrossRef CAS PubMed.
- Y. Kang and T. A. Taton, Angew. Chem., 2005, 117, 413–416 CrossRef.
- Y. Chen, J. Cho, A. Young and T. A. Taton, Langmuir, 2007, 23, 7491–7497 CrossRef CAS PubMed.
- H. Y. Chen, S. Abraham, J. Mendenhall, S. C. Delamarre, K. Smith, I. Kim and C. A. Batt, ChemPhysChem, 2008, 9, 388–392 CrossRef CAS PubMed.
- M. Rai, A. P. Inglea, I. Gupta and A. Brandelli, Int. J. Pharm., 2015, 496, 159–172 CrossRef CAS PubMed.
- A. Lalatsa, A. G. Schätzlein, M. Mazza, T. B. H. Le and I. F. Uchegbu, J. Controlled Release, 2012, 161, 523–536 CrossRef CAS PubMed.
- A. Sulistio, J. Lowenthal, A. Blencowe, M. N. Bongiovanni, L. Ong, S. L. Gras, X. Zhang and G. G. Qiao, Biomacromolecules, 2011, 12, 3469–3477 CrossRef CAS PubMed.
- H. Xiao, R. Qi, S. Lui, X. Hu, T. Duan, Y. Zheng, Y. Huang and X. Jing, Biomaterials, 2011, 32, 7732–7739 CrossRef CAS PubMed.
- S.-I. Park, E.-O. Lee, H.-M. Yang, C. W. Park and J.-D. Kim, Colloids Surf., B, 2013, 110, 333–338 CrossRef CAS PubMed.
- T. Akagi, P. Piyapakorn and M. Akashi, Langmuir, 2012, 28, 5249–5256 CrossRef CAS PubMed.
- T. Suma, K. Miyata, T. Ishii, S. Uchida, H. Uchida, K. Itaka, N. Nishiyama and K. Kataoka, Biomaterials, 2012, 33, 2770–2779 CrossRef CAS PubMed.
- Y. Bao, G. Shen, H. Liu and Y. Li, Polymer, 2013, 54, 652–660 CrossRef CAS.
- A. A. Bogdanov Jr, S. Gupta, N. Koshkina, S. J. Corr, S. Zhang, S. A. Curley and G. Han, Bioconjugate Chem., 2015, 26, 39–50 CrossRef PubMed.
- M. Gkikas, J. Timonen, J. Ruokolainen, P. Alexandridis and H. Iatrou, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1448–1456 CrossRef CAS.
- R. Zhang, S. Xu, J. Luo, D. Shi, C. Liu and X. Liu, RSC Adv., 2014, 4, 25106–25113 RSC.
- D. Perego, N. Masciocchi, A. Guagliardi, J. M. Domìnguez-Vera and N. Gálvez, Nanotechnology, 2013, 24, 075102 CrossRef PubMed.
- C. Wang, F. Tang, X. Wang and L. Li, Colloids Surf., A, 2016, 506, 425–430 CrossRef CAS.
- V. S. Murthy, J. N. Cha, G. D. Stucky and M. S. Wong, J. Am. Chem. Soc., 2004, 126, 5292–5299 CrossRef CAS PubMed.
- Y. Guo, Y. Ma, L. Xu, J. Li and W. Yang, J. Phys. Chem. C, 2007, 111, 9172–9176 CAS.
- H. Chen, P. Zou, J. Connarn, H. Paholak and D. Sun, Nano Res., 2012, 5, 815–825 CrossRef CAS.
- D. Li, Q. He and J. Li, Adv. Colloid Interface Sci., 2009, 149, 28–38 CrossRef CAS PubMed.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 1999, 121, 7409–7410 CrossRef CAS.
- F. Patolsky, Y. Weizmann, O. Lioubashevski and I. Willner, Angew. Chem., 2002, 114, 2429–2433 CrossRef.
- Y. Chen, Z. Xu, D. Zhu, X. Tao, Y. Gao, H. Zhu, Z. Mao and J. Ling, J. Colloid Interface Sci., 2016, 483, 201–210 CrossRef CAS PubMed.
- J. Deng, S. Wu, M. Yao and C. Gao, Sci. Rep., 2016, 6, 31595 CrossRef CAS PubMed.
- S. Nuβ, H. Böttcher, H. Wurm and M. L. Hallensleben, Angew. Chem., 2001, 40, 4016–4018 CrossRef.
- R. Jordan, N. West, A. Ulman, Y.-M. Chou and O. Nuyken, Macromolecules, 2001, 34, 1606–1611 CrossRef CAS.
- K. J. Watson, J. Zhu, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 1999, 121, 462–463 CrossRef CAS.
- L. Wu, U. Glebe and A. Böker, Polym. Chem., 2015, 6, 5143–5184 RSC.
- K. Zawada, W. Tomaszewski and E. A. Megiel, RSC Adv., 2014, 4, 23876–23885 RSC.
- H. H. Park and T. R. Lee, J. Nanopart. Res., 2011, 13, 2909–2918 CrossRef CAS.
- J. A. Scholl, A. L. Koh and J. A. Dionne, Nature, 2012, 483, 421–427 CrossRef CAS PubMed.
- S. Malola, L. Lehtovaara, J. Enkovaara and H. Häkkinen, ACS Nano, 2013, 7, 10263–10270 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS PubMed.
- A. Fields-Zinna, R. Sardar, C. A. Beasley and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 16266–16271 CrossRef PubMed.
- O. Lopez-Acevedo, J. Akola, R. L. Whetten, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2009, 113, 5035–5038 CAS.
- H. Qian and R. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS PubMed.
- S. Das, A. Goswami, M. Hesari, J. F. Al-Sharab, E. Mikmeková, F. Maran and T. Asefa, Small, 2014, 10, 1473–1478 CrossRef CAS PubMed.
- L. Becucci, R. Guidelli, F. Polo and F. Maran, Langmuir, 2014, 30, 8141–8151 CrossRef CAS PubMed.
- E. Heikkilä, H. Martinez-Seara, A. A. Gurtovenko, I. Vattulainen and J. Akola, Biochim. Biophys. Acta, 2014, 1838, 2852–2860 CrossRef PubMed.
- H.-C. Weissker, O. Lopez-Acevedo, R. L. Whetten and X. López-Lozano, J. Phys. Chem. C, 2015, 119, 11250–11259 CAS.
- K. M. Ø. Jensen, P. Juhas, M. A. Tofanelli, C. L. Heinecke, G. Vaughan, C. J. Ackerson and S. J. L. Billinge, Nat. Commun., 2016, 7, 11859 CrossRef CAS PubMed.
- O. Baseggio, M. De Vetta, G. Fronzoni, M. Stener and A. Fortunelli, Int. J. Quantum Chem., 2016, 116, 1603–1611 CrossRef CAS.
- L. Fabris, S. Antonello, L. Armelao, R. L. Donkers, F. Polo, C. Toniolo and F. Maran, J. Am. Chem. Soc., 2006, 128, 326–336 CrossRef CAS PubMed.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95–120 CrossRef CAS PubMed.
- R. Blout and M. J. Idelson, J. Am. Chem. Soc., 1958, 80, 4909–4913 CrossRef.
- H. Susi, S. N. Timasheff and L. Stevens, J. Biol. Chem., 1967, 242, 5460–5466 CAS.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS PubMed.
- D. T. Grubb, J. Mater. Sci., 1974, 9, 1715–1736 CrossRef CAS.
- W. Wang, L. Li, X. Yu, H. Han and X. Guo, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1681–1688 CrossRef CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- S. Underwood and P. Mulvaney, Langmuir, 1994, 10, 3427–3430 CrossRef CAS.
- M. M. Miller and A. A. Lazarides, J. Phys. Chem. B, 2005, 109, 21556–21565 CrossRef CAS PubMed.
- C. Yi, M. A. Tofanelli, C. J. Ackerson and K. L. Knappenberger Jr, J. Am. Chem. Soc., 2013, 135, 18222–18228 CrossRef CAS PubMed.
- H.-C. Weissker, H. B. Escobar, V. D. Thanthirige, K. Kwack, D. Lee, G. Ramakrishna, R. L. Whetten and X. Lopez-Lozano, Nat. Commun., 2014, 5, 3785 Search PubMed.
- R. Ishida, S. Yamazoe, K. Koyasu and T. Tsukuda, Nanoscale, 2016, 8, 2544–2547 RSC.
- N. Chavda, A. Trivedi, J. Thakarda, Y. K. Agrawal and P. Maity, Catal. Lett., 2016, 146, 1331–1339 CrossRef CAS.
- D. Bahena, N. Bhattarai, U. Santiago, A. Tlahuice, A. Ponce, S. B. H. Bach, B. Yoon, R. L. Whetten, U. Landman and M. Jose-Yacaman, J. Phys. Chem. Lett., 2013, 4, 975–981 CrossRef CAS PubMed.
- E. Oh, K. Susumu, R. Goswami and H. Mattoussi, Langmuir, 2010, 26, 7604–7613 CrossRef CAS PubMed.
- C. Kumara, X. Zuo, D. A. Cullen and A. Dass, ACS Nano, 2014, 8, 6431–6439 CrossRef CAS PubMed.
- C. Dharmaratne and A. Dass, Chem. Commun., 2014, 50, 1722–1724 RSC.
- C. Kumara, X. Zuo, D. A. Cullen and A. Dass, J. Phys. Chem. Lett., 2015, 6, 3320–3326 CrossRef CAS.
- S. Malola, M. J. Hartmann and H. Häkkinen, J. Phys. Chem. Lett., 2015, 6, 515–520 CrossRef CAS PubMed.
- M. Agrachev, S. Antonello, T. Dainese, J. A. Gascón, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, Chem. Sci., 2016, 7, 6910–6918 RSC.
- L. Sementa, G. Barcaro, A. Dass, M. Stener and A. Fortunelli, Chem. Commun., 2015, 51, 7935–7938 RSC.
- P. Hu, L. Chen, X. Kang and S. Chen, Acc. Chem. Res., 2016, 49, 2251–2260 CrossRef CAS PubMed.
- A. Venzo, S. Antonello, J. A. Gascón, I. Guryanov, R. D. Leapman, N. V. Perera, A. Sousa, M. Zamuner, A. Zanella and F. Maran, Anal. Chem., 2011, 83, 6355–6362 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details on methods and syntheses, and further characterization results. See DOI: 10.1039/c6sc05187a |
| This journal is © The Royal Society of Chemistry 2017 |