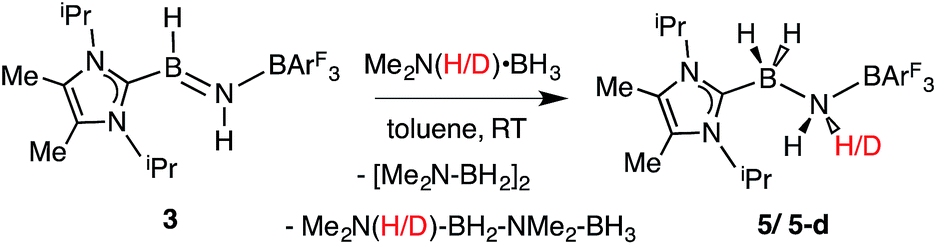DOI:
10.1039/C6SC04893E
(Edge Article)
Chem. Sci., 2017,
8, 2337-2343
Reactivity of a coordinated inorganic acetylene unit, HBNH, and the azidoborane cation [HB(N3)]+†
Received
4th November 2016
, Accepted 17th December 2016
First published on 19th December 2016
Abstract
A donor–acceptor complex of HBNH was prepared via thermolysis of a carbene-stabilized azidoborane. The reactivity of the fundamentally important HBNH unit (inorganic alkyne analogue) was explored in detail, including attempts to convert this species and related hydrido(azido)borane cations into molecular complexes of BN. This work provides added impetus for the development of molecular precursors that can release bulk boron nitride (a desirable insulator and thermal conductor) under mild conditions, and from solution.
Introduction
Iminoboranes (RB![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NR′) are inorganic isoelectronic counterparts to alkynes however their isolation is challenging due to the highly polar nature of their core B–N triple bonds, making these species vulnerable to cyclooligomerization.1,2 In seminal studies, Paetzold and coworkers used steric protection to obtain iminoboranes (e.g.tBuBNtBu) as stable entities, and demonstrated initial coordination chemistry.2d More recently, the Braunschweig, Bertrand and Stephan teams employed carbene-based donors to intercept reactive iminoboranes,3 including the halosilyl analogue ClBNSiMe3.3a Despite these excellent studies, the parent iminoborane, HBNH, remained only identifiable in cryogenic matrices (40 K) or as a fleeting species in the gas phase,4,5 yet HBNH is of interest as a possible intermediate in the laser-induced dehydrogenative synthesis of boron nitride (BN) from H3N·BH3.6
NR′) are inorganic isoelectronic counterparts to alkynes however their isolation is challenging due to the highly polar nature of their core B–N triple bonds, making these species vulnerable to cyclooligomerization.1,2 In seminal studies, Paetzold and coworkers used steric protection to obtain iminoboranes (e.g.tBuBNtBu) as stable entities, and demonstrated initial coordination chemistry.2d More recently, the Braunschweig, Bertrand and Stephan teams employed carbene-based donors to intercept reactive iminoboranes,3 including the halosilyl analogue ClBNSiMe3.3a Despite these excellent studies, the parent iminoborane, HBNH, remained only identifiable in cryogenic matrices (40 K) or as a fleeting species in the gas phase,4,5 yet HBNH is of interest as a possible intermediate in the laser-induced dehydrogenative synthesis of boron nitride (BN) from H3N·BH3.6
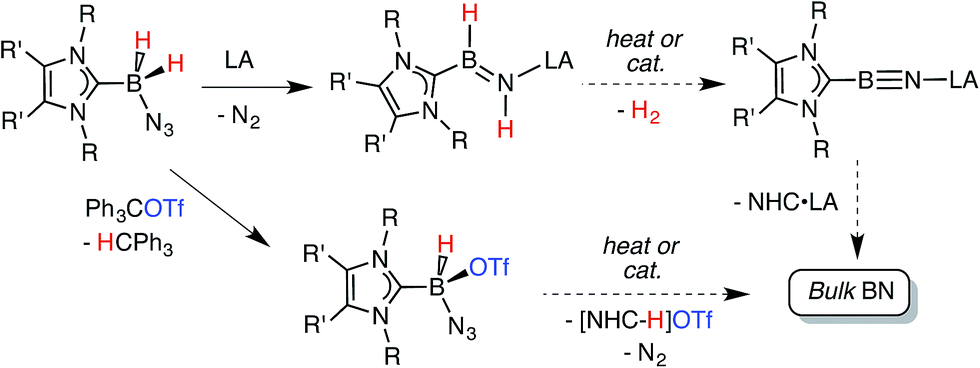
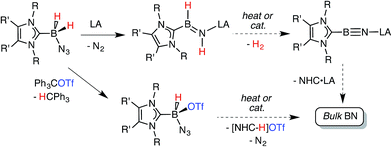
Recently our group was successful in intercepting the first example of a stable complex of HBNH by placing this unsaturated unit in between a sterically encumbered N-heterocyclic carbene (NHC) donor and a large triarylfluoroborane acceptor.7,8 Unfortunately the use of these bulky substituents restricted access to the HBNH array by potential reagents/catalysts. In this Edge Article we introduce a more reactive HBNH adduct and describe our attempts to convert this species into LB·BN·LA complexes (LA = Lewis acid; LB = Lewis base; Scheme 1); in addition we investigate the reactivity of the donor-stabilized azidohydride boronium cation [BH(N3)]+.9 The ultimate goal of our program would be to use these newly developed B–N species for the mild solution-based preparation of bulk boron nitride (Scheme 1). BN and its nanodimensional analogues are highly coveted in the context of advancing modern electronics due to their refractory nature, and desirable electronically insulating and heat dissipating properties.10,11
 |
| | Scheme 1 Synthetic routes explored in this paper are each connected by a common goal of obtaining bulk BN under mild conditions. | |
Results and discussion
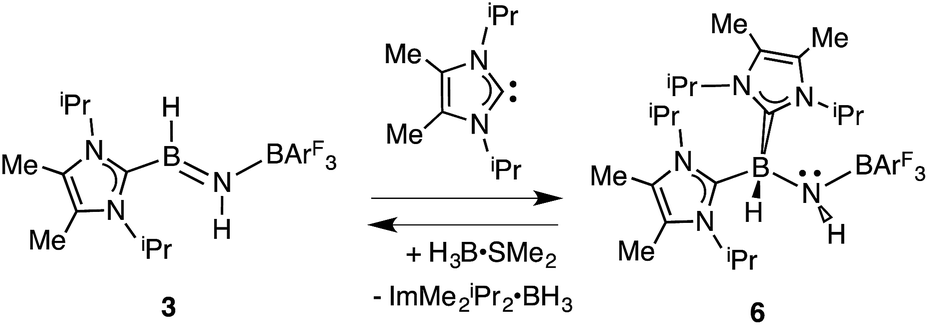
Our initial donor–acceptor HBNH complex IPr·HB![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH·BArF3 [IPr = [(HCNDipp)2C:]; Dipp = 2,6-iPr2C6H3; ArF = 3,5-(F3C)2C6H3]7a was generated by the Lewis acid (BArF3) promoted loss of N2 from the known boron azide IPr·BH2N3,12 followed by an intramolecular 1,2 hydride shift from B to N (Scheme 1). The presence of both hydridic (B–Hδ−) and acidic (N–Hδ+) residues in the HBNH unit prompted us to explore the dehydrogenation of this iminoborane species as a possible route to a molecular adduct of boron nitride, IPr·BN·BArF3. However IPr·HBNH·BArF3 was found to be unreactive in the presence of common dehydrogenation pre-catalysts13 such as [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene).7a The inertness of the iminoborane array was initially attributed to the presence of an extremely congested coordination environment. Thus we decided to generate an HBNH complex supported by the less hindered NHC, ImMe2iPr2 [ImMe2iPr2 = (MeCNiPr)2C:].14
NH·BArF3 [IPr = [(HCNDipp)2C:]; Dipp = 2,6-iPr2C6H3; ArF = 3,5-(F3C)2C6H3]7a was generated by the Lewis acid (BArF3) promoted loss of N2 from the known boron azide IPr·BH2N3,12 followed by an intramolecular 1,2 hydride shift from B to N (Scheme 1). The presence of both hydridic (B–Hδ−) and acidic (N–Hδ+) residues in the HBNH unit prompted us to explore the dehydrogenation of this iminoborane species as a possible route to a molecular adduct of boron nitride, IPr·BN·BArF3. However IPr·HBNH·BArF3 was found to be unreactive in the presence of common dehydrogenation pre-catalysts13 such as [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene).7a The inertness of the iminoborane array was initially attributed to the presence of an extremely congested coordination environment. Thus we decided to generate an HBNH complex supported by the less hindered NHC, ImMe2iPr2 [ImMe2iPr2 = (MeCNiPr)2C:].14
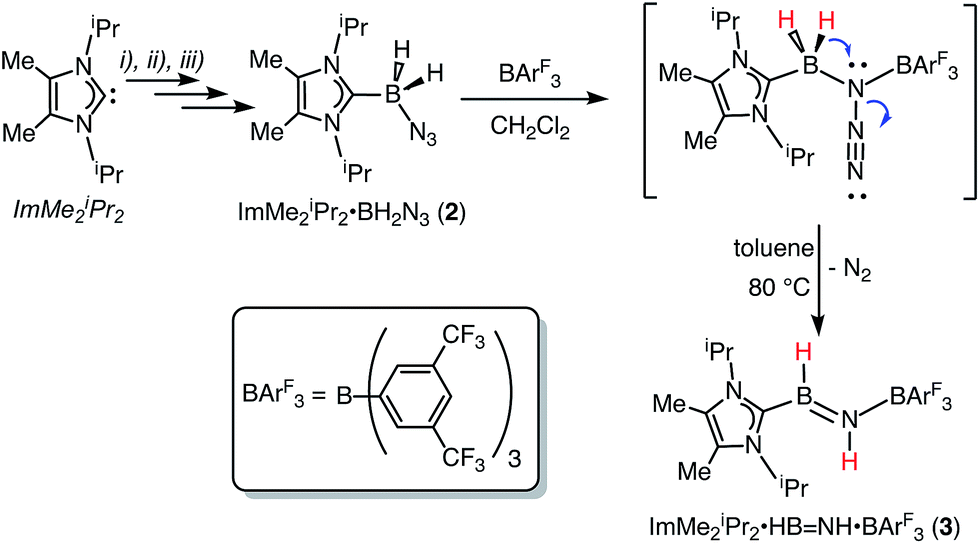
The required azidoborane for our HBNH adduct synthesis, ImMe2iPr2·BH2N3 (2), was prepared from ImMe2iPr2·BH315 in two high yielding steps (Scheme 2). ImMe2iPr2·BH2N3 (2) was then combined with a stoichiometric amount of the fluoroarylborane, BArF3, followed by heating to 80 °C for 12 h in toluene to afford the target iminoborane adduct ImMe2iPr2·HBNH·BArF3 (3) as a colorless solid in a 64% yield (mp = 142–146 °C). Based on prior studies7a this reaction is believed to proceed via initial N2 elimination and trapping of the resulting nitrene adduct, ImMe2iPr2·H2B–N·BArF3 by a 1,2-hydride migration from B to N (Scheme 2). It is salient to mention that the generation of transient nitrenes from boron azides is known in the literature.1a,16
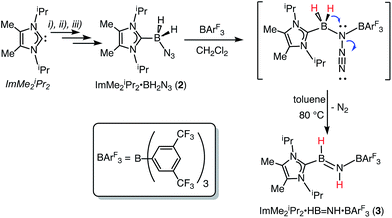 |
| | Scheme 2 Synthesis of ImMe2iPr2·HBNH·BArF3 (3) starting from the azidoborane adduct ImMe2iPr2·BH2N3 (2). Reagents: (i) THF·BH3, THF, rt (95% yield); (ii) 0.5 equiv. I2, benzene, rt (90% yield); (iii) NaN3, DMSO, rt (68% yield). | |
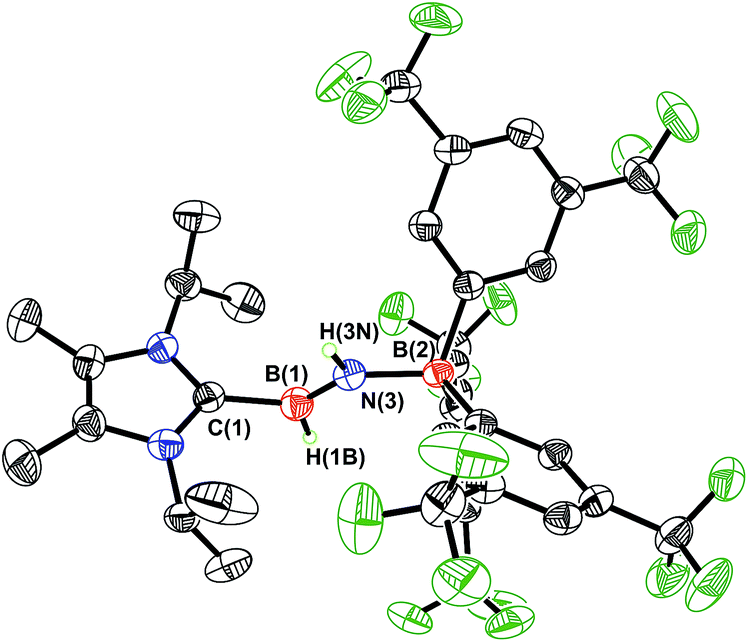
As expected, the 1H{11B} NMR spectrum of ImMe2iPr2·HBNH·BArF3 (3) gave discernable N–H and B–H resonances at 5.42 and 4.62 ppm, respectively (in C6D6), which are similar to the corresponding resonances found in IPr·HBNH·BArF3.7a X-ray crystallography later conclusively identified the presence of an HBNH moiety in 3 (Fig. 1). The core iminoborane unit in 3 adopts a trans arrangement [C–B–N–B dihedral angle = 178.1(2)°] thereby minimizing intramolecular repulsion between the ImMe2iPr2 and BArF3 groups. The central BN and C(NHC)–B bond distances in 3 are 1.369(3) Å and 1.596(4) Å, which are the same within experimental error as in IPr·HBNH·BArF3.7a A slightly elongated B–N distance was reported in the iminoborane (HCC)2B–NiPr2 (1.385(3) Å).17
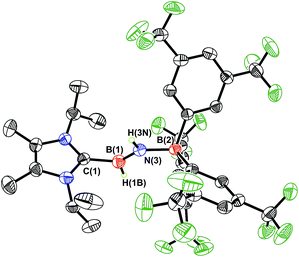 |
| | Fig. 1 Molecular structure of ImMe2iPr2·HBNH·BArF3 (3) with thermal ellipsoids presented at a 30% probability level. All carbon-bound hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): C(1)–B(1) 1.596(2), B(1)–N(3) 1.369(3), N(3)–B(2) 1.572(2); C(1)–B(1)–N(3) 121.8(2), B(1)–N(3)–B(2) 130.5(2), N(3)–B(1)–H(1B) 125.2(16), B(1)–N(3)–H(3N) 115.8(19). | |
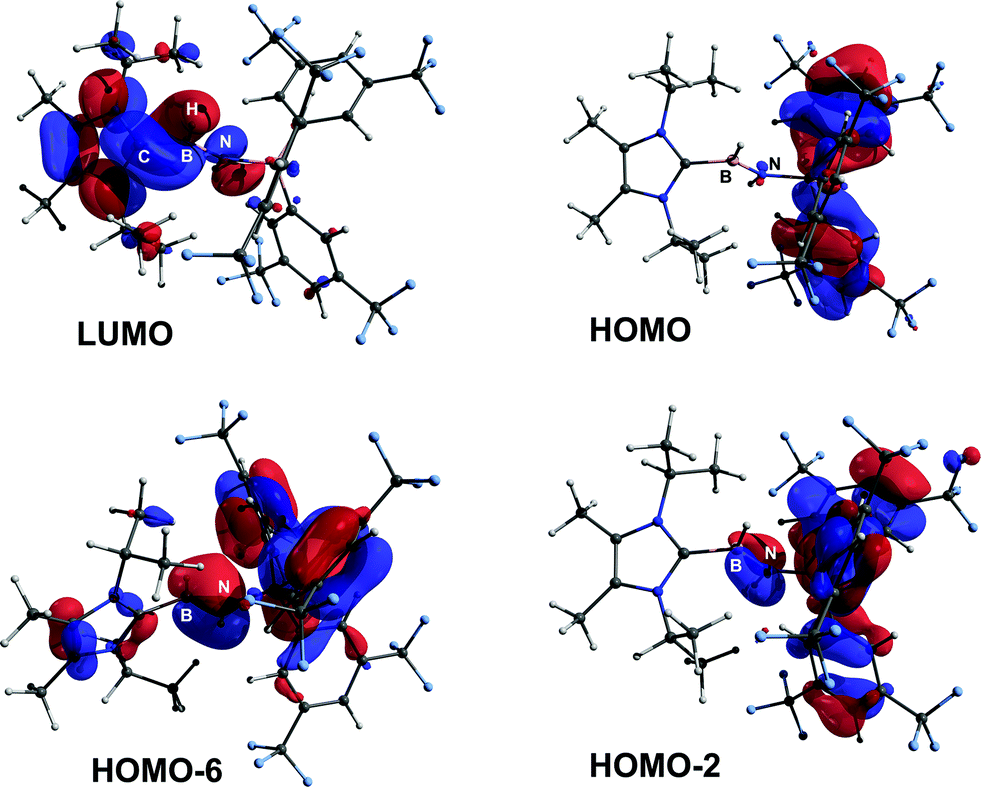
ImMe2iPr2·HBNH·BArF3 (3) was examined by computational methods and an overall charge of −0.13e was found for the central HBNH moiety. As anticipated, the BN linkage (Wiberg bond index, WBI = 1.33) has considerable polarization of the σ- and π-components towards N (ca. 80% located on N), according to NBO analysis. The LUMO shows B–N π* and B–C π-character, while contributions to the B–N π-manifold appear in HOMO−2 and HOMO−6 (Fig. 2).18 The computed HOMO–LUMO gap is 173 kcal mol−1 and is in agreement with the observed inertness of 3 (vide infra).
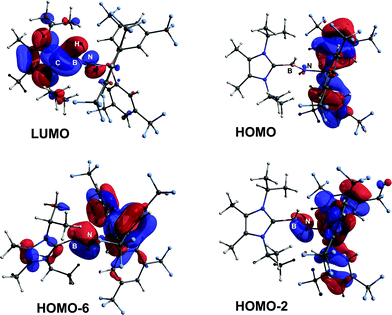 |
| | Fig. 2 POV-ray depiction of selected Kohn–Sham orbitals of 3. | |
With the less hindered HBNH complex 3 in hand, we attempted to promote its dehydrogenation to afford the BN adduct ImMe2iPr2·BN·BArF3. When compound 3 was treated with the well-known dehydrogenation pre-catalyst [Rh(COD)Cl]2 (2–5 mol%) in toluene, no reaction occurred at room temperature. When the same dehydrogenation reaction was attempted at 90 °C for 7 days, only partial decomposition of 3 (<10%; [ImMe2iPr2–H]+ salt) was noted. Moreover, compound 3 was also combined with the potential dehydrogenation catalyst CpFe(CO)2OTf and the Frustrated Lewis Pair (FLP), tBu3P and BArF3, (both known to promote H2 loss from amine-boranes) however in each case no reaction with 3 transpired. Likewise attempted H2 release from 3 by photolysis (300 W Hg lamp in Et2O) gave no reaction.
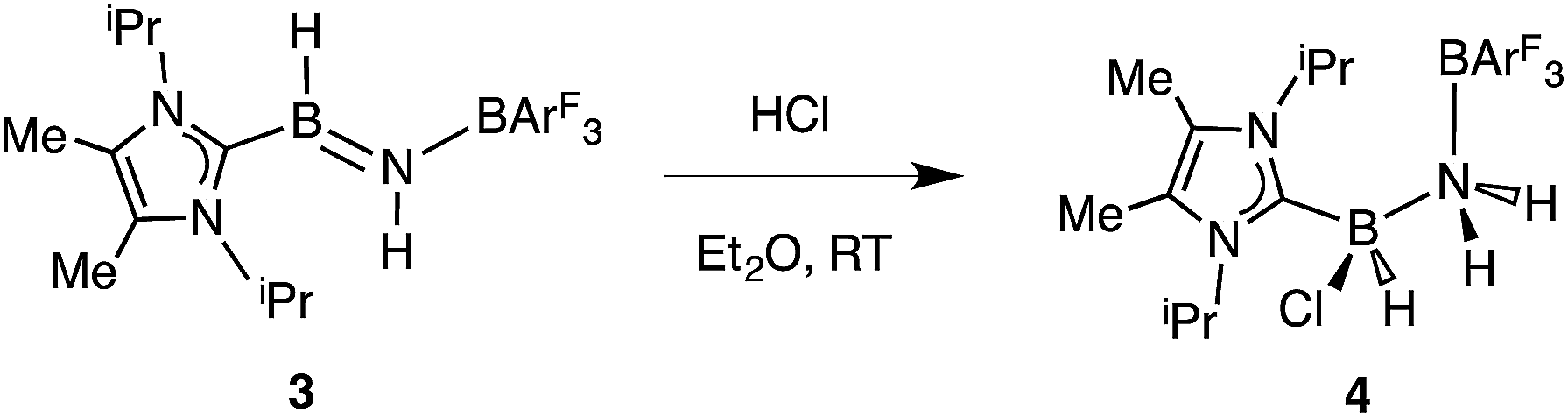
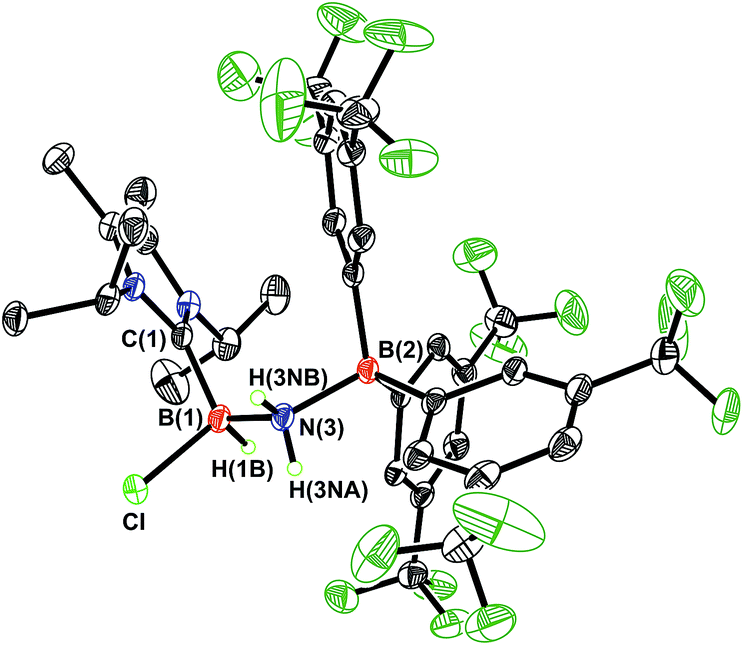
Undaunted by the lack of thermally- or catalytically-instigated H2 release from 3, we decided to see if the core HBNH unit underwent chemical transformations one would expect for a polarized BN linkage.19 When ImMe2iPr2·HBNH·BArF3 (3) was combined with one equivalent of HCl in Et2O, the resulting 11B NMR spectrum was consistent with the presence of two four-coordinate boron centers (δ = −3.7 and −9.5 ppm in C6D6). X-ray crystallography confirmed the successful addition of HCl across the BN bond to form ImMe2iPr2·H(Cl)B–NH2·BArF3 (4) as a racemic mixture due to the presence of a chiral boron atom (Fig. 3; eqn (1)). The addition of chloride at the boron center in 4 illustrates the Lewis acidic nature of the boron atom in coordinated HBNH in 3. The central B–N bond distance in 4 is 1.585(4) Å and is comparable to the B–N bond lengths found in structurally related amine-boranes, such as IPr·BH2NH2BH3.20 The C(NHC)–B bond distance in 4 is 1.616(5) Å which, somewhat to our surprise, is similar in length as the corresponding C(NHC)–B bond distance of 1.596(4) Å in 3, despite the change in hybridization at boron to sp3 in 4; however, the capping N–BArF3 interaction in 4 (1.632(4) Å) is longer than in the HBNH adduct 3 (1.572(2) Å). Addition of HCl also leads to a substantial canting of the relative arrangement of the capping NHC and borane groups (vs. in 3), as evidenced by the C–B–N–B dihedral angle of 65.3(3)°.
| | 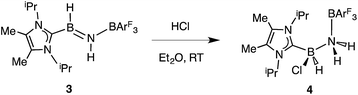 | (1) |
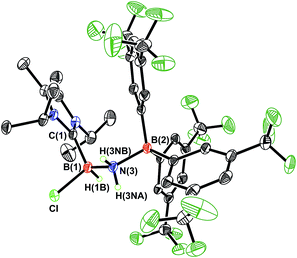 |
| | Fig. 3 Molecular structure of ImMe2iPr2·H(Cl)B–NH2·BArF3 (4) with thermal ellipsoids presented at a 30% probability level. All carbon-bound hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): C(1)–B(1) 1.616(5), B(1)–N(3) 1.585(4), N(3)–B(2) 1.632(4), B(1)–Cl 1.906(4); C(1)–B(1)–N(3) 115.7(3), B(1)–N(3)–B(2) 124.4(2), N(3)–B(1)–Cl 107.2(2), B(1)–N(3)–H(3NA) 105(2). | |
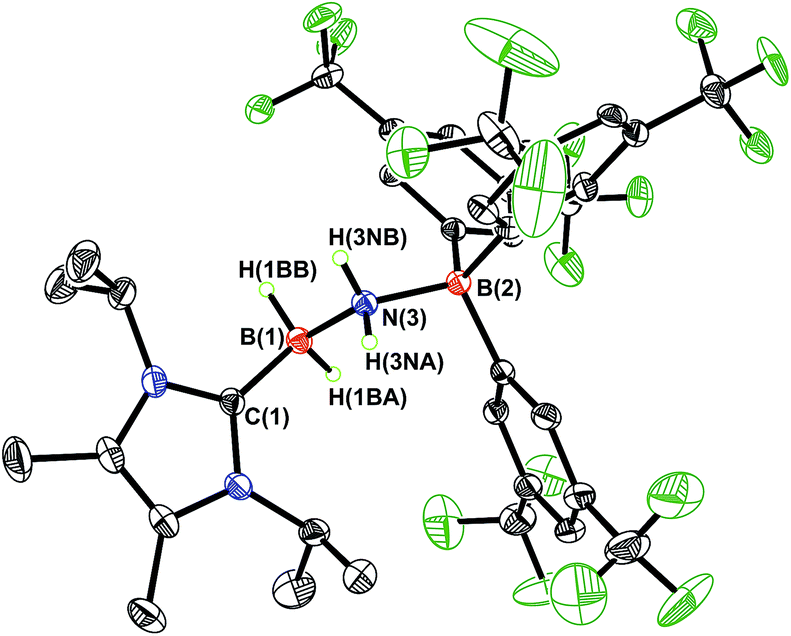
While the polarized BN linkage in ImMe2iPr2·HBNH·BArF3 (3) did not exhibit Frustrated Lewis Pair (FLP) type reactivity with H2, CO or CO2,21 effective transfer hydrogenation22 occurred between the amine-borane Me2NH·BH3 and 3 (eqn (2)). The resulting hydrogenated product ImMe2iPr2·H2B–NH2·BArF3 (5) formed after 12 h at room temperature; the expected dehydrogenated by-products [Me2N–BH2]2 and Me2NH–BH2–NMe2–BH3 were also detected by NMR spectroscopy. To probe the mechanism of this transformation in more detail, compound 3 was combined with Me2ND·BH3; the resulting product ImMe2iPr2·H2B–N(H)D·BArF3 (5-d)18 suggested direct H/D atom transfer from B to B and N to N.22a The molecular structure of 5 (Fig. 4) has similar overall structural features as the HCl addition product ImMe2iPr2·H(Cl)B–NH2·BArF3 (4) with an elongated CNHC–B distance of 1.627(3) Å in accordance with the decreased electrophilicity of the BH2–NH2–BArF3 unit in 5.
| | 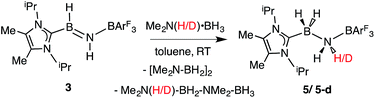 | (2) |
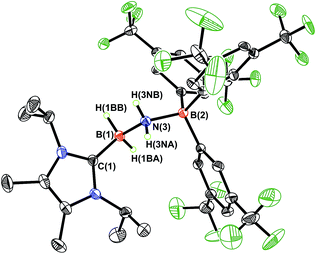 |
| | Fig. 4 Molecular structure of ImMe2iPr2·H2B–NH2·BArF3 (5) with thermal ellipsoids presented at a 30% probability level. All carbon-bound hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): C(1)–B(1) 1.627(3), B(1)–N(3) 1.613(3), N(3)–B(2) 1.622(2); C(1)–B(1)–N(3) 110.23(15), B(1)–N(3)–B(2) 120.11(14), N(3)–B(1)–H(1BB) 109.0(12), B(1)–N(3)–H(3NA) 106.8(15). | |
Despite the presence of both hydridic and acidic H atoms in ImMe2iPr2·H2B–NH2·BArF3 (5), our efforts to induce dehydrogenation (and reform the HBNH adduct 3) by heating up to 100 °C in the presence of known dehydrogenation pre-catalysts [Rh(COD)Cl]2 or CpFe(CO)2OTf led to no discernable reaction. Furthermore, 5 remained unreactive towards the possible H2 acceptors, PhNNPh and the FLP (tBu3P/BArF3), and did not yield 3 upon attempted photolysis (300 W Hg lamp). Accordingly, the calculated NPA charges for 5 show less hydridic character for the B–H array (−0.009 and −0.020e) compared to the reactive amine-borane MeNH2·BH3 (B–H charges of −0.030 to −0.034e), thus partially explaining the higher reactivity for the latter species. The computed positive charges for N-bound hydrogen atoms in 5 (0.429 and 0.437e) are similar to those in MeNH2·BH3.18
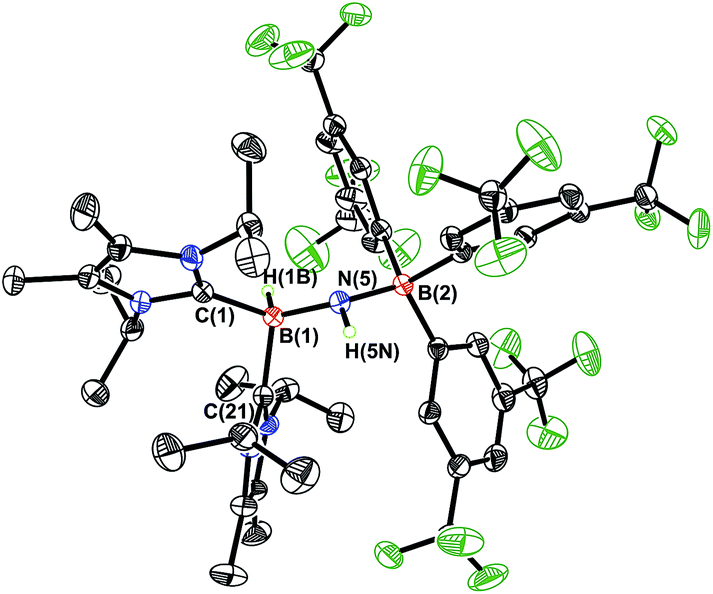
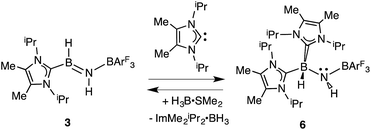
In order to directly probe the Lewis acidity of the HBNH unit in 3,23 an additional equivalent of the carbene donor ImMe2iPr2 was combined with ImMe2iPr2·HBNH·BArF3 (3). While the expected bis adduct (ImMe2iPr2)2HBNH·BArF3 (6) could be isolated in the solid state as a yellow solid (88% yield) and characterized by X-ray crystallography (Fig. 5, vide infra), the NMR spectra of this product in solution exhibited dynamic behavior, consistent with partial dissociation of one NHC ligand. Addition of the Lewis acid acceptor BH3 (delivered in the form of Me2S·BH3) led to the quantitative removal of one equiv. of ImMe2iPr2 from 6 to reform 3 (eqn (3)). Consistent with weaker overall CNHC–B interactions in 6 relative to in the HBNH adduct 3, elongated distances of 1.684(3) and 1.660(2) Å were found in 6 (by ca. 0.06–0.08 Å). For comparison, the C–B distances in Bertrand's mixed NHC/CAAC complex [CAAC·B(L)H(OTf)]BPh4 [CAAC = cyclic alkyl(amino) carbene; L = benzimidazolylidene] were slightly shorter (1.645(2) and 1.627(2) Å).24 Coordination of two NHCs at boron in 6 resulted in substantial lengthening of the core B–N distance from a value of 1.369(3) in 3 to 1.512(2) Å, suggesting a lack of a B–N π-bond interaction in 6. Our computational studies on 6 support this postulate with a computed B–N Wiberg bond index (WBI) of 0.85 (vs. 1.33 in 3). Moreover, interaction of the Lewis base ImMe2iPr2 with the LUMO in 3 populates an orbital with B–N π*-character (Fig. 2).18
| |  | (3) |
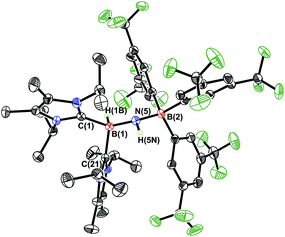 |
| | Fig. 5 Molecular structure of [ImMe2iPr2]2·HB-NH·BArF3 (6) with thermal ellipsoids presented at a 30% probability level. All carbon-bound hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): C(1)–B(1) 1.684(3), C(21)–B(1) 1.660(2), B(1)–N(5) 1.512(2), N(5)–B(2) 1.539(2); C(1)–B(1)–N(5) 117.28(14), B(1)–N(5)–B(2) 125.03(14), N(5)–B(1)–C(21) 112.26(14), N(5)–B(1)–H(1B) 113.4(11), B(1)–N(5)–H(5N) 112.5(14). | |
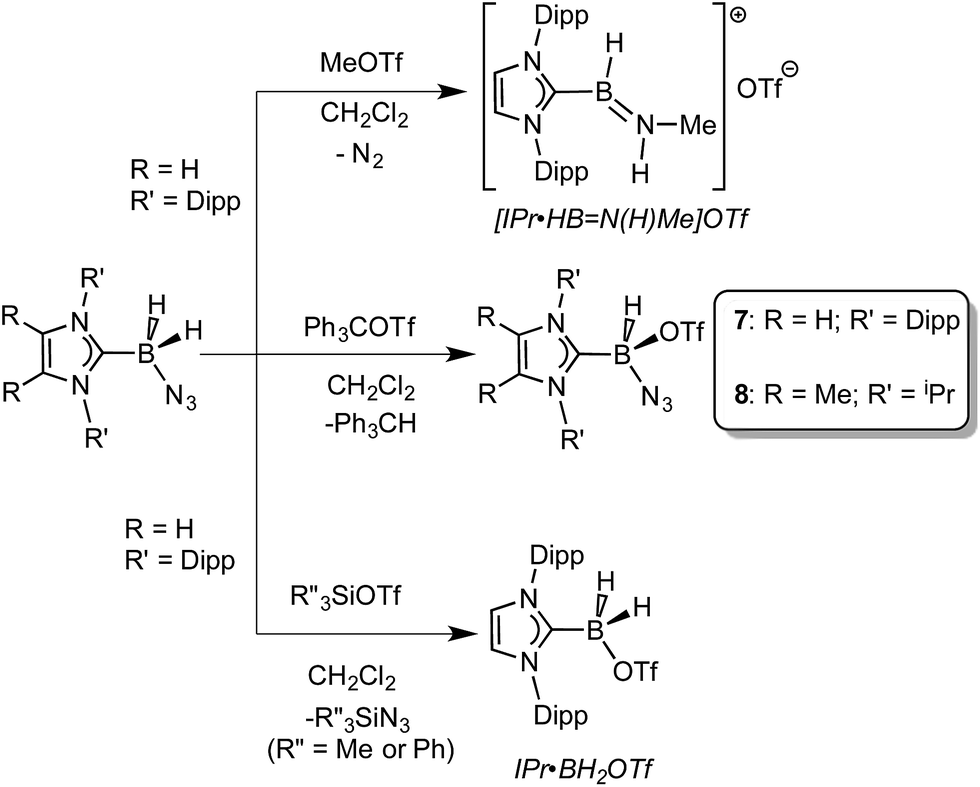
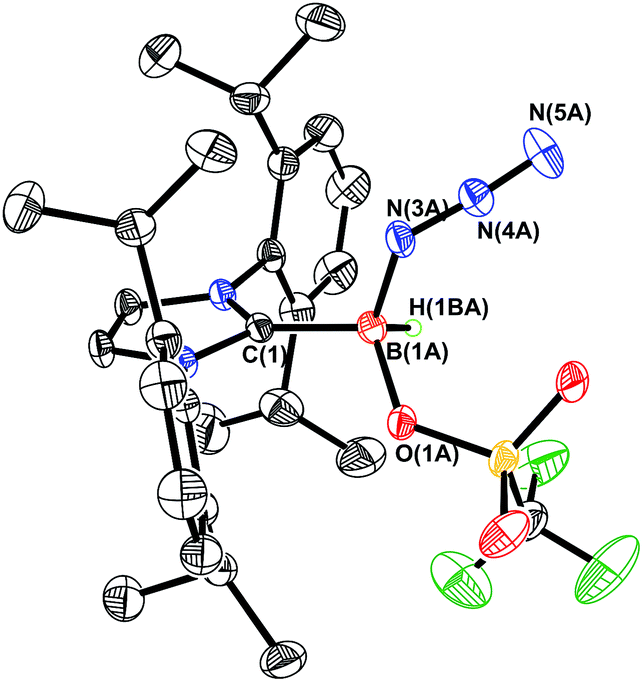
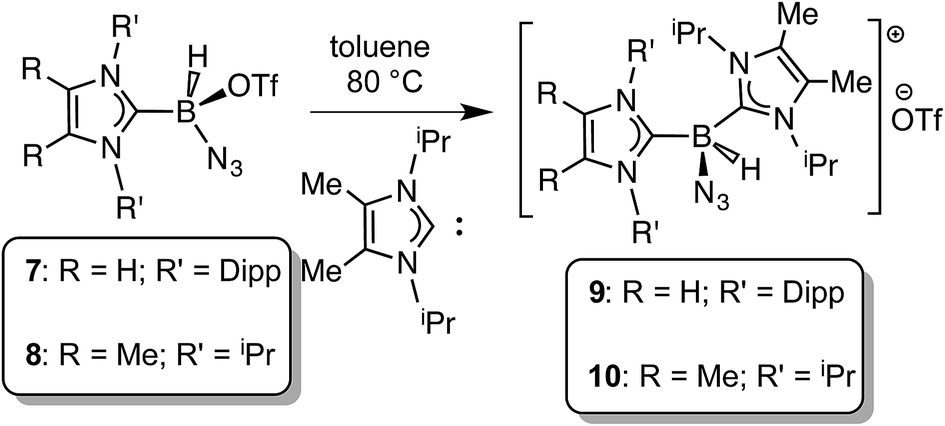
Prior work in our group showed that N2 loss/1,2-hydride migration in IPr·BH2N3 could also be instigated with the methylating agent MeOTf (Scheme 3), eventually leading to the formation of [IPr·HBN(Me)H]OTf.7a Accordingly we wanted to expand the range of known electrophiles that could trigger this potentially general transformation. However, with Ph3COTf and R3SiOTf (R = Me and Ph), divergent reactivity was uncovered (Scheme 3). Specifically, when IPr·BH2N3 or the less hindered analogue ImMe2iPr2·BH2N3 (2) was combined with Ph3COTf in CH2Cl2, hydride abstraction occurred to yield triphenylmethane (Ph3CH) and the new azido(hydrido)borane adducts IPr·BH(OTf)N3 (7) and ImMe2iPr2·BH(OTf)N3 (8) in isolated yields of 95 and 66%, respectively (see Fig. 6 and S1† for the corresponding X-ray structures).18 The 19F NMR spectra of 7 and 8 show the retention of strong B-OTf contacts in solution (e.g. δ = −76.9 ppm for 7 in C6D6), while intense azide IR stretches were present at 2117 and 2116 cm−1 for compounds 7 and 8, respectively; these values compare well with the ν(N3) of 2117 cm−1 reported for Cummins' azido borate salt [nBu4N][(N3)B(C6F5)3].25 Thus by simply replacing MeOTf with Ph3COTf, H/OTf exchange chemistry can transpire in place of N2 loss.
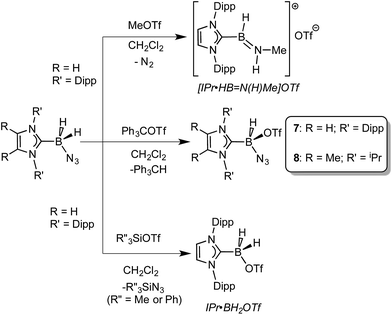 |
| | Scheme 3 Divergent reactivity of NHC·BH2N3 adducts with MeOTf, R′′3SiOTf (R′′ = Me or Ph), and Ph3COTf. | |
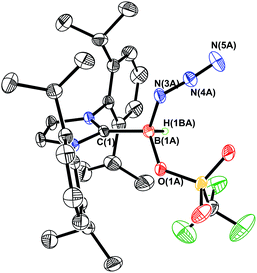 |
| | Fig. 6 Molecular structure of IPr·BHN3(OTf) (7) with thermal ellipsoids presented at a 30% probability level. All carbon-bound hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg) with parameters associated with a second molecule in the asymmetric unit listed in square brackets: C(1)–B(1A) 1.590(11) [1.652(10)], B(1A)–N(3A) 1.542(8) [1.482(12)], N(3A)–N(4A) 1.223(7) [1.211(8)], N(4A)–N(5A) 1.168(9) [1.145(11)], B(1A)–O(1A) 1.552(11) [1.562(11)]; N(3A)–N(4A)–N(5A) 175.0(11) [178.2(11)]. | |
Yet another reaction pathway occurred when IPr·BH2N3 was combined with the silyltriflates Me3SiOTf and Ph3SiOTf (Scheme 3). In each case, complete OTf/azide exchange transpired to form the corresponding silylazides (Me3SiN3 and Ph3SiN3; identified by NMR spectroscopy) and the known borane adduct IPr·BH2OTf.12 It appears that N3/OTf exchange is driven by the relatively strong Si–N bonds (ca. 355 kJ mol−1)26 in relation to the C–N linkages (ca. 305 kJ mol−1), thus azide abstraction by Ph3C+ sources is not as favorable. To recap, NHC·BH2N3 shows three distinct possible reactivity pathways in the presence of electrophiles: (a) HBNH formation via N2 loss/1,2-H shift; (b) hydride abstraction; (c) azide abstraction.
The accidentally uncovered high yield syntheses of the NHC·BH(OTf)N3 adducts 7 and 8 (Scheme 3) opened another possible path to boron nitride (BN). Motivated by the balanced equation (NHC·BH(OTf)N3 → BN + N2 + [NHC–H]OTf; Scheme 1) we decided to investigate the reactivity of both 7 and 8 in more detail. Initially we explored the direct thermolysis of 7 and 8 in solution at temperatures approaching 100 °C (Caution!) but these adducts proved to be stable under these conditions. Treatment of 8 with potassium as a reducing agent (in order to promote the possible reaction: 8 + K → ½H2 + N2 + KOTf + BN + NHC) produced the free carbene ImMe2iPr2 as the only soluble product by NMR spectroscopy. Whereas the reaction of 8 with KC8 produced three different carbene containing products: free carbene ImMe2iPr2, ImMe2iPr2·BH2N3 and ImMe2iPr2·BH3.27 Analysis of the insoluble fractions from both of the reactions by IR identified the presence of K[N3] and K[OTf], indicating that B–N(azide) bond scission transpired in place of H2 loss and boron nitride formation; in support of this reaction path, no IR bands for bulk BN could be found in the product mixture. Furthermore, the LUMO computed for the model species ImMe2·B(H)N3(OTf) (ImMe2 = (HCNMe)2C:) revealed B–N σ*-character, thus explaining the preferential B–N bond scission noted upon reduction.18
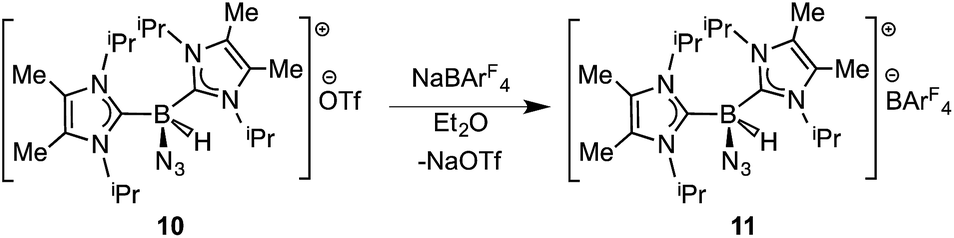
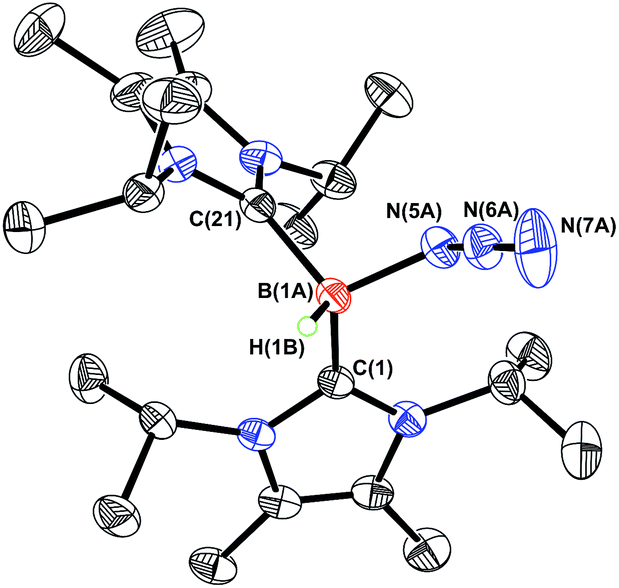
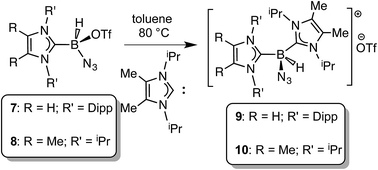
In order to induce 1,2-H transfer in the NHC·BHN3(OTf) species 7 and 8 the donor ImMe2iPr2 (ref. 28) was added to form the respective bis(carbene) boronium salts [IPr(ImMe2iPr2)·BH(N3)]OTf (9) and [(ImMe2iPr2)2·BH(N3)]OTf (10) (eqn (4)). The spectral parameters of these salts were consistent with free OTf− counteranions (e.g.19F resonance at −78.1 ppm for 10 in CDCl3) and the retention of boron-bound azide and hydride substituents (e.g. IR stretches at ca. 2107 and 2400 cm−1 for 9). Structural confirmation of the proposed bonding environment was provided by an X-ray structure of the tetraarylfluoroborate salt [(ImMe2iPr2)2·BH(N3)]BArF4 (11) (eqn (5); Fig. 7). With the goal of taking advantage of possibly higher nucleophilic character of the azide group in 11 in relation to the mono-carbene congener 8, we combined 11 with one equivalent of BArF3. In place of observing Lewis acid-assisted N2 elimination/H-migration to give the “trapped” BNH adduct [(ImMe2iPr2)2·BNH·BArF3]OTf, no reaction transpired. Likewise no conversion of 11 was noted upon heating this species with BArF3 at 90–100 °C or under UV irradiation.
| |  | (4) |
| | 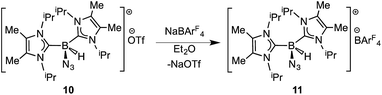 | (5) |
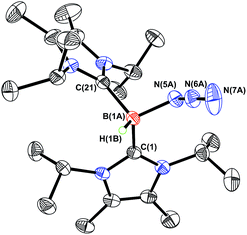 |
| | Fig. 7 Molecular structure of [(ImMe2iPr2)2·BHN3][B{C6H3(m-CF3)2}4] (11) with thermal ellipsoids presented at a 30% probability level. All carbon-bound hydrogen atoms and BArF4 anion have been omitted for clarity. Selected bond lengths (Å) and angles (deg) with parameters associated with a second molecule in the asymmetric unit listed in square brackets: C(1)–B(1A) 1.642(9) [1.71(3)], C(21)–B(1A) 1.650(9) [1.59(3)], B(1A)–N(5A) 1.553(7) [1.514(13)], N(5A)–N(6A) 1.202(6) [1.206(11)], N(6A)–N(7A) 1.147(10) [1.159(14)]; N(5A)–N(6A)–N(7A) 173.7(6) [158(2)]. | |
Conclusion
In this article we present efficient methods to prepare complexes of HBNH and [HB(N3)]+, starting from readily available carbene–azidoborane adducts. In addition, this study provides key insights into the reactivity of the fundamentally important HBNH unit, an inorganic analogue of acetylene. While our detailed investigations aimed at forming bulk boron nitride (BN) from these species under mild conditions were not directly successful, we hope that this work inspires others to seek low temperature (<200 °C) routes to this inorganic wide band gap material. By suitable modification of the capping stabilizing groups, related B–N sources could be potentially used as building blocks for the rational construction of boron nitride materials and π-extended structures.29
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant and CREATE grants for E. R.; CREATE fellowship for A. K. S.), and the Canada Foundation for Innovation (CFI). C. H.-J. acknowledges the Alexander von Humboldt Foundation for a Feodor-Lynen Postdoctoral Fellowship. The authors also acknowledge Mark Miskolzie and Nupur Dabral for experimental assistance. The authors also thank Matthew M. D. Roy and Dr Urmibhusan Bhakta for helpful discussions.
References
-
(a) P. Paetzold, Adv. Inorg. Chem., 1987, 31, 123 CrossRef CAS;
(b) H. Nöth, Angew. Chem., Int. Ed. Engl., 1988, 27, 1603 CrossRef;
(c) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877 CrossRef CAS PubMed;
(d) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924 CrossRef CAS PubMed;
(e) O. Ayhan, T. Eckert, F. A. Plamper and H. Helten, Angew. Chem., Int. Ed., 2016, 55, 13321 CrossRef CAS PubMed.
-
(a) P. Paetzold and C. von Plotho, Chem. Ber., 1982, 115, 2819 CrossRef CAS;
(b) H.-U. Meier, P. Paetzold and E. Schröder, Chem. Ber., 1984, 117, 1954 CrossRef CAS;
(c) J. Kiesgen, J. Münster and P. Paetzold, Chem. Ber., 1993, 126, 1559 CrossRef CAS;
(d) E. Bulak, G. E. Herberich, I. Manners, H. Mayer and P. Paetzold, Angew. Chem., Int. Ed. Engl., 1988, 27, 958 CrossRef.
-
(a) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159 CrossRef CAS PubMed;
(b) F. Dahcheh, D. W. Stephan and G. Bertrand, Chem.–Eur. J., 2015, 21, 199 CrossRef CAS PubMed;
(c) H. Braunschweig, W. C. Ewing, K. Geetharani and M. Schäfer, Angew. Chem., Int. Ed., 2015, 54, 1662 CrossRef CAS PubMed.
-
(a) E. R. Lory and R. F. Porter, J. Am. Chem. Soc., 1973, 95, 1766 CrossRef CAS;
(b) Y. Kawashima, K. Kawaguchi and E. Hirota, J. Chem. Phys., 1987, 87, 6331 CrossRef CAS;
(c) C. A. Thompson and L. Andrews, J. Am. Chem. Soc., 1995, 117, 10125 CrossRef CAS;
(d) F. Zhang, P. Maksyutenko, R. I. Kaiser, A. M. Mebel, A. Gregusova, S. A. Perera and R. J. Bartlett, J. Phys. Chem. A, 2010, 114, 12148 CrossRef CAS PubMed.
- For selected computational studies on HBNH, see:
(a) N. C. Baird and R. K. Datta, Inorg. Chem., 1972, 11, 17 CrossRef CAS;
(b) M. H. Matus, D. J. Grant, M. T. Nguyen and D. A. Dixon, J. Phys. Chem. C, 2009, 113, 16553 CrossRef CAS;
(c) R. Sundaram, S. Scheiner, A. K. Roy and T. Kar, J. Phys. Chem. C, 2015, 119, 3253 CrossRef CAS.
- H. Liu, P. Jin, Y.-M. Xue, C. Dong, X. Li, C.-C. Tang and X.-W. Du, Angew. Chem., Int. Ed., 2015, 54, 7051 CrossRef CAS PubMed.
-
(a) A. K. Swarnakar, C. Hering-Junghans, K. Nagata, M. J. Ferguson, R. McDonald, N. Tokitoh and E. Rivard, Angew. Chem., Int. Ed., 2015, 54, 10666 CrossRef CAS PubMed;
(b) Our HBNH adducts can also be considered as analogues of known Frustrated Lewis Pair (FLP) complexes of alkynes, see: M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef CAS PubMed.
- For related examples of donor–acceptor stabilization of main group species, see:
(a) U. Vogel, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2001, 40, 4409 CrossRef CAS;
(b) P. A. Rupar, M. C. Jennings, P. J. Ragogna and K. M. Baines, Organometallics, 2007, 26, 4109 CrossRef CAS;
(c) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Angew. Chem., Int. Ed., 2011, 50, 8354 CrossRef CAS PubMed;
(d) T. Yamaguchi, A. Sekiguchi and M. Driess, J. Am. Chem. Soc., 2010, 132, 14061 CrossRef CAS PubMed;
(e) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565 CrossRef CAS PubMed;
(f) Y.-P. Zhou, M. Karni, S. Yao, Y. Apeloig and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 15096 CrossRef CAS PubMed.
- For a computational study on LB·BN·LA species, see: M. R. Momeni, L. Shulman, E. Rivard and A. Brown, Phys. Chem. Chem. Phys., 2015, 17, 16525 RSC.
-
(a) Y. Tian, B. Xu, D. Yu, Y. Ma, Y. Wang, Y. Jiang, W. Hu, C. Tang, Y. Gao, K. Luo, Z. Zhao, L.-M. Wang, B. Wen, J. He and Z. Liu, Nature, 2013, 493, 385 CrossRef CAS PubMed;
(b) V. L. Solozhenko, O. O. Kurakevych and Y. Le Godec, Adv. Mater., 2012, 24, 1540 CrossRef CAS PubMed;
(c) H. Sumiya, S. Uesaka and S. Satoh, J. Mater. Sci., 2000, 35, 1181 CrossRef CAS;
(d) Y. Kubota, K. Watanabe, O. Tsuda and T. Taniguchi, Science, 2007, 317, 932 CrossRef CAS PubMed;
(e) K. Watanabe, T. Taniguchi, T. Niiyama, K. Miya and M. Taniguchi, Nat. Photonics, 2009, 3, 591 CrossRef CAS;
(f) S. Bernard, C. Salameh and P. Miele, Dalton Trans., 2016, 45, 861 RSC.
- Our group has used LB·GeH2·LA complexes as precursors to both bulk germanium and luminescent nanoparticles:
(a) E. Rivard, Dalton Trans., 2014, 43, 8577 RSC;
(b) T. K. Purkait, A. K. Swarnakar, G. B. De Los Reyes, F. A. Hegmann, E. Rivard and J. G. C. Veinot, Nanoscale, 2015, 7, 2241 RSC.
- A. Solovyev, Q. Chu, S. J. Geib, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 15072 CrossRef CAS PubMed.
-
(a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS PubMed;
(b) E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817 CrossRef CAS PubMed.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561 CrossRef CAS.
- N. Kuhn, G. Henkel, T. Kratz, J. Kreutzberg, R. Boese and A. H. Maulitz, Chem. Ber., 1993, 126, 2041 CrossRef CAS.
-
(a) M. Müller, C. Maichle-Mössmer and H. F. Bettinger, Chem. Commun., 2013, 49, 11773 RSC;
(b) M. Müller, C. Maichle-Mössmer and H. F. Bettinger, Angew. Chem., Int. Ed., 2014, 53, 9380 CrossRef PubMed.
- F. Ge, G. Kehr, C. G. Daniliuc, C. Mück-Lichtenfeld and G. Erker, Organometallics, 2015, 34, 4205 CrossRef CAS.
- For complete synthetic, crystallographic and computational details, see the ESI.†.
-
(a) A. W. Laubengayer, O. T. Beachley Jr and R. F. Porter, Inorg. Chem., 1965, 4, 578 CrossRef CAS;
(b) O. T. Beachley Jr and B. Washburn, Inorg. Chem., 1975, 14, 120 CrossRef.
- A. C. Malcolm, K. J. Sabourin, R. McDonald, M. J. Ferguson and E. Rivard, Inorg. Chem., 2012, 51, 12905 CrossRef CAS PubMed.
-
(a) V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskelä, T. Repo, P. Pyykkö and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117 CrossRef CAS PubMed;
(b) M.-A. Legaré, M.-A. Courtemanche, E. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef PubMed;
(c) T. Wang, G. Kehr, L. Liu, S. Grimme, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2016, 138, 4302 CrossRef CAS PubMed;
(d) Z. Mo, A. Rit, J. Campos, E. L. Kolychev and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 3306 CrossRef CAS PubMed;
(e) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306 CrossRef CAS PubMed.
- For related examples of transfer hydrogenation between amine-boranes and unsaturated B–N compounds, see:
(a) A. P. M. Robertson, E. M. Leitao and I. Manners, J. Am. Chem. Soc., 2011, 133, 19322 CrossRef CAS PubMed;
(b) M. W. Lui, N. R. Paisley, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2016, 22, 2134 CrossRef CAS PubMed;
(c) E. M. Leitao, N. E. Stubbs, A. P. M. Robertson, H. Helten, R. J. Cox, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2012, 134, 16805 CrossRef CAS PubMed.
- We also explored the possible Lewis basic nature of 3. When 3 was combined with CuI and BArF3, no reaction was found; treatment of 3 with Me2S·BH3 (as a source of BH3) gave a complicated product mixture.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837 RSC.
- A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716 CrossRef CAS PubMed.
-
(a) S. W. Benson, J. Chem. Educ., 1965, 42, 502 CrossRef CAS;
(b) R. Walsh, Acc. Chem. Res., 1981, 14, 246 CrossRef CAS.
- For the synthesis of BN by treating [XBNH]3 (X = Cl or Br) with molten alkali metals, see:
(a) E. J. M. Hamilton, S. E. Dolan, C. M. Mann, H. O. Colijn, C. A. McDonald and S. G. Shore, Science, 1993, 260, 659 CAS;
(b) E. J. M. Hamilton, S. E. Dolan, C. M. Mann, H. O. Colijn and S. G. Shore, Chem. Mater., 1995, 7, 111 CrossRef CAS.
- We have computed the charge of the boron-bound hydrogen atom in the model species [(ImMe2)2BH(N3)]+ (see the ESI†) and noted slightly acidic character (NPA = +0.010); attempts to promote BN formation from 9 by treatment with sodium metal in Et2O yielded free ImMe2iPr2 with no sign of bulk BN formation by IR spectroscopy.
-
(a) M. J. S. Dewar, V. P. Kubba and R. Pettit, J. Chem. Soc., 1958, 3073 RSC;
(b) D. J. H. Emslie, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2003, 42, 1252 CrossRef CAS PubMed;
(c) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074 CrossRef CAS PubMed;
(d) K. Edel, S. A. Brough, A. N. Lamm, S.-Y. Liu and H. F. Bettinger, Angew. Chem., Int. Ed., 2015, 54, 7819 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and tables of crystallographic data for compounds 3–8 and 11. CCDC 1514190–1514196. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04893e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Robert
McDonald
,
Robert
McDonald