
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium-catalyzed asymmetric hydrogenation of racemic α-substituted lactones to chiral diols†
Xiao-Hui
Yang
a,
Hai-Tao
Yue
a,
Na
Yu
a,
Yi-Pan
Li
a,
Jian-Hua
Xie
*a and
Qi-Lin
Zhou
*ab
aState Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China. E-mail: jhxie@nankai.edu.cn; qlzhou@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China
First published on 15th November 2016
Abstract
We report a protocol for the highly efficient iridium-catalyzed asymmetric hydrogenation of racemic α-substituted lactones via dynamic kinetic resolution. Using Ir-SpiroPAP (R)-1d as a catalyst, a wide range of chiral diols were prepared in a high yield (80–95%) with a high enantioselectivity (up to 95% ee) under mild reaction conditions. This protocol was used for enantioselective syntheses of (−)-preclamol and a chiral 2,5-disubstituted tetrahydropyran.
Introduction
Transition-metal-catalyzed asymmetric hydrogenation of ketones is an efficient and reliable method for the synthesis of optically active chiral secondary alcohols.1 In contrast, the enantioselective synthesis of chiral primary alcohols by catalytic asymmetric hydrogenation of their corresponding aldehydes or esters is difficult, and work on the development of practical methods is underway. In 2007, we reported the first example of the catalytic asymmetric hydrogenation of racemic α-branched aldehydes, via dynamic kinetic resolution (DKR), for the synthesis of chiral primary alcohols.2 Subsequently, List3 and Lin4et al. also reported the synthesis of chiral primary alcohols, by means of ruthenium-catalyzed asymmetric hydrogenation of racemic α-substituted aldehydes. Although a wide range of catalysts have been developed for the hydrogenation of esters,5 efficient chiral catalysts for the asymmetric hydrogenation of racemic α-substituted esters via DKR are rare. The biggest challenge for the direct asymmetric hydrogenation of racemic esters to form optically active primary alcohols is to find a catalyst that can discriminate between the enantiomers of chiral esters and then hydrogenate them to alcohols selectively. In 2011, Ikariya et al.6 described the enantioselective hydrogenation of a racemic mixture of an α-substituted γ-lactone to a chiral 1,4-diol via DKR using chiral ruthenium catalysts bearing chiral 1,2-diamine ligands (Scheme 1). However, the enantioselectivity of the reaction was low (up to 32% ee). Recently, as part of our work on the asymmetric hydrogenation of ketones, we found that chiral Ru-SDPs/diamine catalysts and chiral Ir-SpiroPAP catalysts can also mediate the hydrogenation of ester groups.7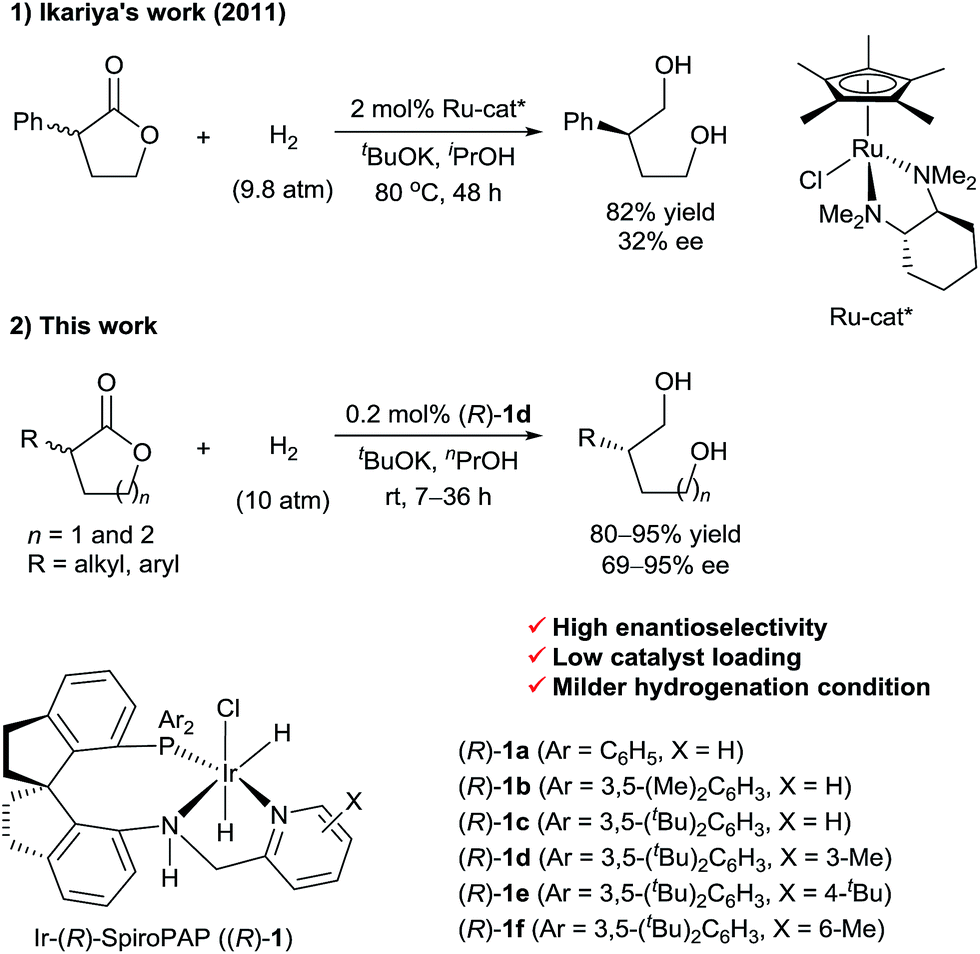 | ||
| Scheme 1 Asymmetric hydrogenation of racemic α-substituted lactones via DKR. | ||
In this communication, we report a protocol for the Ir-SpiroPAP-catalyzed asymmetric hydrogenation of racemic α-substituted lactones to afford chiral diols in a high yield (80–95%) with a high enantioselectivity (up to 95% ee, Scheme 1).
Results and discussion
We initially performed the hydrogenation of racemic α-phenyl δ-valerolactone (2a) to evaluate the activity and enantioselectivity of various catalysts (Table 1). Under the previously reported reaction conditions7b (catalyst loading = 0.2 mol% (S/C = 500), [2a] = 0.25 M, [tBuOK] = 0.04 M, EtOH, 10 atm H2 and 25–30 °C), no reaction occurred in the presence of the catalyst (R)-1d (entry 1). However, when the concentration of tBuOK was increased to 0.06 M, the hydrogenation reaction occurred and provided (R)-3a in 15% yield with 91% ee (entry 2); further increasing the concentration of tBuOK increased the reaction rate and the yield of 3a substantially. For example, when 0.25 M tBuOK (2a/tBuOK/(R)-1d = 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500:1) was used, the reaction was complete within 10 h, providing (R)-3a in 91% yield with 92% ee (entry 3). The absolute configuration of (R)-3a was determined by comparing the sign of its optical rotation with the literature data.8 Evaluation of various chiral Ir-SpiroPAP catalysts (R)-1 revealed that the substituents on the pyridine and phenyl groups of the catalysts had little effect on the yield or enantioselectivity (entries 4–8), with (R)-1d giving the best results. Experiments with various solvents showed that nPrOH was suitable (entries 9–11); the reaction was complete within 10 h, affording (R)-3a in 92% yield with 93% ee. In addition to tBuOK, tBuONa also gave a high yield with a high enantioselectivity, but the use of KOH, NaOH, or K2CO3 resulted in low yields (entries 12–15).
:500:1) was used, the reaction was complete within 10 h, providing (R)-3a in 91% yield with 92% ee (entry 3). The absolute configuration of (R)-3a was determined by comparing the sign of its optical rotation with the literature data.8 Evaluation of various chiral Ir-SpiroPAP catalysts (R)-1 revealed that the substituents on the pyridine and phenyl groups of the catalysts had little effect on the yield or enantioselectivity (entries 4–8), with (R)-1d giving the best results. Experiments with various solvents showed that nPrOH was suitable (entries 9–11); the reaction was complete within 10 h, affording (R)-3a in 92% yield with 93% ee. In addition to tBuOK, tBuONa also gave a high yield with a high enantioselectivity, but the use of KOH, NaOH, or K2CO3 resulted in low yields (entries 12–15).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Base | [Base] | Solvent | Time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 1.0 mmol scale, [2a] = 0.25 M, 0.2 mol% of catalyst, solvent (4.0 mL) and room temperature (25–30 °C). b Isolated yield. c Determined by HPLC using a chiral column. The absolute configuration of 3a is R determined by comparing its optical rotation with the literature data (see ESI). | |||||||
| 1 | (R)-1d | t BuOK | 0.04 | EtOH | 24 | ND | ND |
| 2 | (R)-1d | t BuOK | 0.06 | EtOH | 24 | 15 | 91 |
| 3 | (R)-1d | t BuOK | 0.25 | EtOH | 10 | 91 | 92 |
| 4 | (R)-1a | t BuOK | 0.25 | EtOH | 10 | 93 | 90 |
| 5 | (R)-1b | t BuOK | 0.25 | EtOH | 10 | 92 | 90 |
| 6 | (R)-1c | t BuOK | 0.25 | EtOH | 10 | 92 | 91 |
| 7 | (R)-1e | t BuOK | 0.25 | EtOH | 17 | 91 | 90 |
| 8 | (R)-1f | t BuOK | 0.25 | EtOH | 10 | 92 | 90 |
| 9 | (R)-1d | t BuOK | 0.25 | MeOH | 7 | 10 | 86 |
| 10 | (R)-1d | t BuOK | 0.25 | n PrOH | 10 | 92 | 93 |
| 11 | (R)-1d | t BuOK | 0.25 | i PrOH | 8 | 89 | 74 |
| 12 | (R)-1d | t BuONa | 0.25 | n PrOH | 10 | 91 | 93 |
| 13 | (R)-1d | KOH | 0.25 | n PrOH | 20 | 48 | 93 |
| 14 | (R)-1d | NaOH | 0.25 | n PrOH | 20 | 36 | 93 |
| 15 | (R)-1d | K2CO3 | 0.25 | n PrOH | 20 | 21 | 92 |
To evaluate the substrate scope of the reaction, we investigated a wide range of racemic α-substituted δ-valerolactones under the established reaction conditions (Table 2). For racemic α-aryl-substituted δ-valerolactones 2a–i (entries 1–9), neither electron-donating nor electron-withdrawing groups on the phenyl ring of the substrates had much effect on the enantioselectivity of the reaction, but substrates with an electron-withdrawing group (entries 2, 5, and 8) showed a higher reaction rate than those with an electron-donating group.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | n | 3 | Time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 1.0 mmol scale, [substrate] = 0.25 M, [tBuOK] = 0.25 M, 0.2 mol% of (R)-1d, nPrOH (4.0 mL) and room temperature (25–30 °C). b Isolated yield. c Determined by HPLC using a chiral column. d The absolute configuration of the product is determined by comparing its optical rotation with the literature data (see ESI). | ||||||
| 1d | C6H5 | 2 | 3a | 10 | 92 | 93 (R) |
| 2 | 4-ClC6H4 | 2 | 3b | 7 | 93 | 93 |
| 3 | 4-MeC6H4 | 2 | 3c | 9 | 92 | 93 |
| 4 | 4-MeOC6H4 | 2 | 3d | 10 | 93 | 93 |
| 5 | 3-ClC6H4 | 2 | 3e | 7 | 95 | 92 |
| 6 | 3-MeC6H4 | 2 | 3f | 10 | 91 | 93 |
| 7 | 3-MeOC6H4 | 2 | 3g | 10 | 93 | 92 (R) |
| 8d | 3,4-Cl2C6H3 | 2 | 3h | 7 | 94 | 92 |
| 9 | 3,4-(MeO)2C6H3 | 2 | 3i | 10 | 91 | 91 |
| 10 | 2-ClC6H4 | 2 | 3j | 13 | 89 | 78 |
| 11 | 2-MeC6H4 | 2 | 3k | 36 | 84 | 77 |
| 12 | 2-MeOC6H4 | 2 | 3l | 20 | 88 | 86 |
| 13d | Me | 2 | 3m | 8 | 92 | 91 (S) |
| 14 | Et | 2 | 3n | 12 | 90 | 87 |
| 15d | i Pr | 2 | 3o | 12 | 90 | 95 (R) |
| 16d | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH2 CHCH2CH2 |
2 | 3p | 12 | 91 | 88 (S) |
| 17 | C6H5 | 1 | 3q | 10 | 80 | 80 (R) |
| 18 | Me | 1 | 3r | 10 | 82 | 69 (S) |
In addition, owing to steric effects, substrates with an ortho-substituent on the phenyl ring gave lower reaction rates, yields, and enantioselectivities (entries 10–12). The hydrogenation of α-alkyl-substituted δ-valerolactones 2m–p also worked well, affording the corresponding 1,5-diols 3m–p in high yields and high enantioselectivities (entries 13–16). Catalyst (R)-1d also catalyzed the hydrogenation of racemic α-substituted γ-butyrolactones 2q and 2r, providing the 1,4-diols 3q (80% ee) and 3r (69% ee), respectively (entries 17 and 18).
We investigated the pathway of the hydrogenation of racemic α-substituted lactones 2 by 1H NMR. As shown in Fig. 1, after reaction for 0.5 h under the optimal reaction conditions, rac-2a was converted to the hydroxyl ester 4, propyl 5-hydroxy-2-phenylpentanoate, in 62% yield with no ee and the diol 3a in 38% yield with 93% ee. Over the following 10 h, the amount of the hydroxyl ester 4 gradually decreased, and the amount of the diol 3a increased. Only a trace amount of lactone 2a was detected from 2 min after the reaction started.
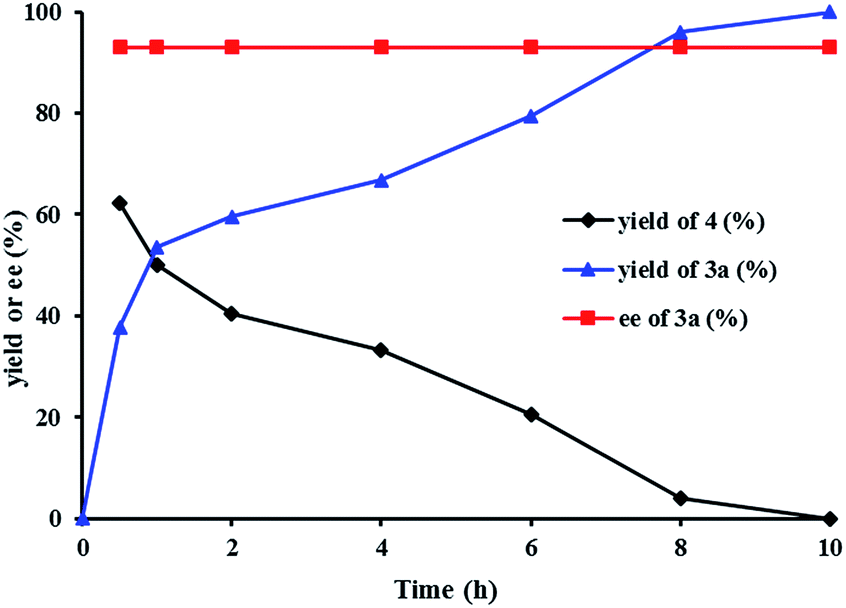 | ||
| Fig. 1 The plots of the hydrogenation of rac-2a with (R)-1d. | ||
Direct hydrogenation of the hydroxyl ester 4 with catalyst (R)-1d provided the diol 3a in 93% yield with 93% ee, which is the same as the result obtained from the hydrogenation of lactone 2a (Scheme 2). We also conducted the hydrogenation of the ester 5, which has a δ-OCH2OMe group instead of a δ-OH group as in the hydroxyl ester 4, and observed no reaction. These results indicated that the hydroxyl group of ester 4 was crucial for the hydrogenation. Thus, although the lactone 2a was readily alcoholized to the hydroxyl ester 4 under the reaction conditions, the hydrogenation of 2a occurred inevitably via its lactone form (Scheme 2).7b,c
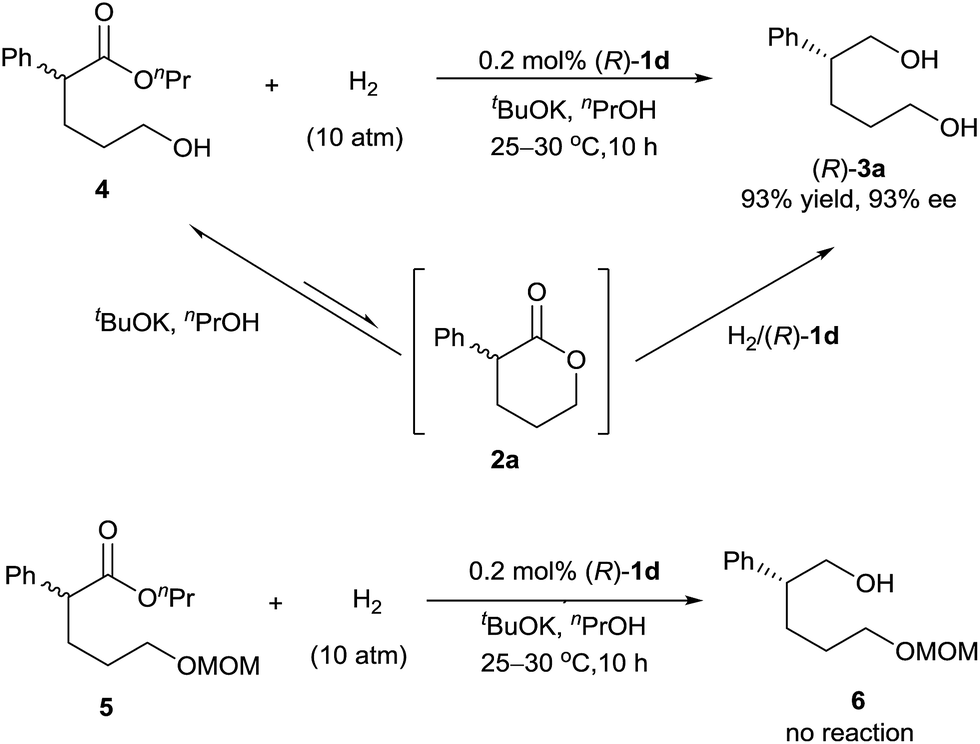 | ||
| Scheme 2 Asymmetric hydrogenation of esters 4 and 5 with (R)-1d. | ||
Chiral 3-aryl/alkyl substituted piperidines are part of an important class of bioactive heterocyclic compounds,9 but they are difficult to synthesize in their optically active forms.10 By using the catalyst (S)-1d, we synthesized (−)-preclamol,11 which is a candidate drug for the treatment of neurological disorders such as Parkinson's disease.12 The hydrogenation of rac-2g (1.65 g) catalyzed by (S)-1d afforded the diol (S)-3g (89% yield and 93% ee), which was subsequently transformed to (−)-preclamol by activation with methanesulfonyl chloride, substitution/cyclization with n-propylamine, and demethylation with hydrobromic acid (84% yield over three steps, Scheme 3).
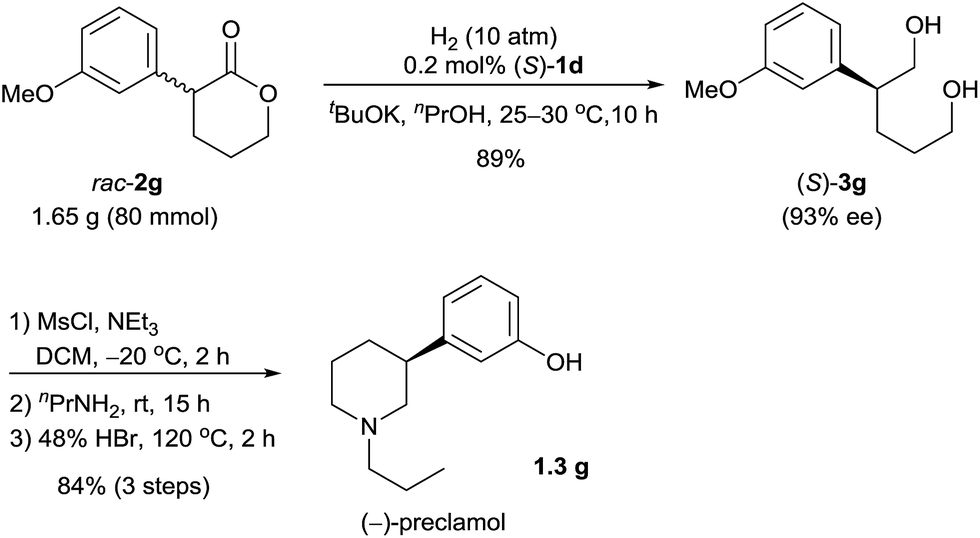 | ||
| Scheme 3 Enantioselective synthesis of (−)-preclamol. | ||
Diol (S)-3p is a useful building block for the synthesis of chiral 2,5-disubstituted tetrahydropyrans, which occur in many biologically active natural products such as the terpenoids rhopaloic acid A13 and barangcadoic acid A14 (Scheme 4), isolated from marine sponges. Iodoetherification of (S)-3p with iodine15 produced the tetrahydropyran 7 in 94% yield as a 2:1 trans/cis mixture. Nucleophilic substitution of tetrahydropyran 7 with NaCN afforded the nitrile 8 (trans/cis = 2:1). Hydrolysis of the nitrile 8 and its subsequent esterification with MeOH afforded the tetrahydropyran 9 in a 72% yield with a higher trans/cis ratio (5:1). Thus, this protocol represents a potential method for the construction of the chiral core structures of rhopaloic acid A and barangcadoic acid A.16
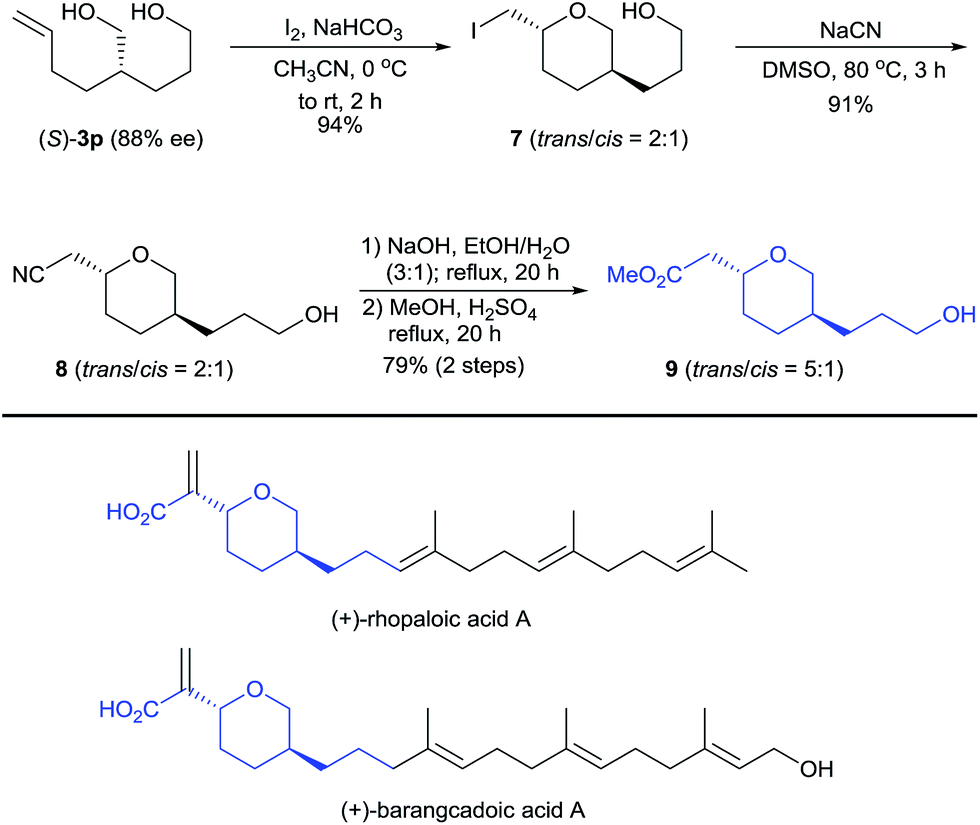 | ||
| Scheme 4 Enantioselective synthesis of a chiral 2,5-disubstituted tetrahydropyran. | ||
Conclusions
In conclusion, we have developed a protocol for the highly efficient iridium-catalyzed asymmetric hydrogenation of racemic α-substituted lactones via DKR. Using an Ir-SpiroPAP catalyst, a series of racemic α-substituted lactones were hydrogenated to chiral diols in high yield with high enantioselectivity under mild reaction conditions. The protocol was used for the enantioselective syntheses of (−)-preclamol and a chiral 2,5-disubstituted tetrahydropyran.Experimental
General procedure for the asymmetric hydrogenation of 2
Into a 20 mL hydrogenation vessel in an autoclave was added racemic α-substituted δ-valerolactone 2 (1.0 mmol), a solution of iridium catalyst (R)-1d in nPrOH (dried with MS 4 Å for 12 h, 0.002 mmol mL−1, 1.0 mL, 0.002 mmol), a solution of tBuOK in nPrOH (0.5 mmol mL−1, 2.0 mL, 1.0 mmol) and nPrOH (1.0 mL). The autoclave was purged with hydrogen by pressurizing to 5 atm and releasing the pressure. This procedure was repeated three times and then the autoclave was pressurized to 10 atm of H2. The reaction mixture was stirred at room temperature (25–30 °C) until no obvious hydrogen pressure drop was observed. The reaction mixture was then quenched with saturated NH4Cl (5 mL) and extracted with EtOAc (5 mL × 3). The combined extracts were washed with brine, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on a silica gel using petroleum ether/ethyl acetate as the eluent to afford the chiral diols 3. The ee values of the chiral diols 3 were determined by HPLC using a chiral column.Acknowledgements
This project was supported by the National Natural Science Foundation of China, the National Basic Research Program of China (973 Program) (No. 2012CB821600) and the “111” project (No. B06005) of the Ministry of Education of China.Notes and references
-
(a)
The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed
; (b) B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS PubMed
-
(a) J.-H. Xie, Z.-T. Zhou, W.-L. Kong and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 1868 CrossRef CAS PubMed
- X. Li and B. List, Chem. Commun., 2007, 1739 RSC
- D. J. Mihalcik and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 6229 CrossRef CAS PubMed
- For reviews on catalytic hydrogenation of esters, see:
(a)
M. L. Clarke and G. J. Roff, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley, New York, 2007, p. 413 Search PubMed
- M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 4240 CrossRef CAS PubMed
-
(a) C. Liu, J.-H. Xie, Y.-L. Li, J.-Q. Chen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 593 CrossRef CAS PubMed
- S. S. M. A. Hakim and T. Sugimura, Org. Lett., 2010, 12, 3626 CrossRef CAS PubMed
-
(a) M. J. Schneider, Alkaloids: Chem. Biol. Perspect., 1996, 10, 155 CAS
- F. Colpaert, S. Mangelinckx and N. De Kimpe, J. Org. Chem., 2011, 76, 234 CrossRef CAS PubMed
- For selected papers on asymmetric synthesis of preclamol, see:
(a) J. Hu, Y. Lu, Y. Li and J. Zhou, Chem. Commun., 2013, 49, 9425 RSC
-
(a) U. Hacksell, L. E. Arvidsson, U. Svensson, J. L. G. Nilsson, D. Sanchez, H. Wikstroem, P. Lindberg, S. Hjorth and A. Carlsson, J. Med. Chem., 1981, 24, 1475 CrossRef CAS PubMed
-
(a) S. Ohta, M. Uno, M. Yoshimura, Y. Hiraga and S. Ikegami, Tetrahedron Lett., 1996, 37, 2265 CrossRef CAS
- K. S. Craig, D. E. Williams, I. Hollander, E. Frommer, R. Mallon, K. Collins, D. Wojciechowicz, A. Tahir, R. Van Soest and R. J. Andersen, Tetrahedron Lett., 2002, 43, 4801 CrossRef CAS
- L. F. Tietze and C. Schneider, J. Org. Chem., 1991, 56, 2476 CrossRef CAS
- For selected papers on the synthesis of rhopaloic acid A and its analogues, see:
(a) K. Kadota and K. Ogasawara, Heterocycles, 2003, 59, 485 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04609f |
| This journal is © The Royal Society of Chemistry 2017 |