
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative self-powered electrochromic biosensors†
Miguel Aller
Pellitero
a,
Anton
Guimerà
 ab,
Maria
Kitsara
a,
Rosa
Villa
ab,
Camille
Rubio
c,
Boris
Lakard
c,
Marie-Laure
Doche
c,
Jean-Yves
Hihn
c and
F.
Javier del Campo
*a
ab,
Maria
Kitsara
a,
Rosa
Villa
ab,
Camille
Rubio
c,
Boris
Lakard
c,
Marie-Laure
Doche
c,
Jean-Yves
Hihn
c and
F.
Javier del Campo
*a
aInstituto de Microelectrónica de Barcelona, IMB-CNM (CSIC), Campus de la Universidad Autónoma, Esfera UAB, 08193-Bellaterra, Barcelona, Spain. E-mail: Javier.delcampo@csic.es; Tel: +34 935947700
bCIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, Barcelona, Spain
cInstitut UTINAM, UMR 6214 CNRS/Université de Franche-Comté, 16 route de Gray, 25030 Besançon, France
First published on 28th November 2016
Abstract
Self-powered sensors are analytical devices able to generate their own energy, either from the sample itself or from their surroundings. The conventional approaches rely heavily on silicon-based electronics, which results in increased complexity and cost, and prevents the broader use of these smart systems. Here we show that electrochromic materials can overcome the existing limitations by simplifying device construction and avoiding the need for silicon-based electronics entirely. Electrochromic displays can be built into compact self-powered electrochemical sensors that give quantitative information readable by the naked eye, simply controlling the current path inside them through a combination of specially arranged materials. The concept is validated by a glucose biosensor coupled horizontally to a Prussian blue display designed as a distance-meter proportional to (glucose) concentration. This approach represents a breakthrough for self-powered sensors, and extends the application of electrochromic materials beyond smart windows and displays, into sensing and quantification.
Introduction
Self-powered biosensors, first introduced by Katz and Willner in 2001 (ref. 1) are analytical tools in which energy output is proportional to the concentration of a target analyte. Some advantages of this kind of systems are that they are highly specific, and that the sensor consists only of two electrodes without external voltage being applied to them. Their main drawback, though, is that due to their typically low energy production, these systems require some form of control instrumentation to quantify the sensor output. This increases their cost and complexity, and explains why after over a decade their development level remains so low.The commonest approach to developing self-powered and self-contained analytical devices consists in the miniaturization through integration of different components into the same package.2–4 A recent trend is hybrid integration through printed electronics, which enables the convergence of technologies, materials and components aimed at the mass, low-cost production of advanced devices.5,6 Beni, Turner et al. recently described the first fully integrated, screen-printed electrochemical glucose biosensor.7 This system represents a key milestone in the path of point of care devices, as it is the first self-contained and fully disposable demonstrator platform that can be used to develop virtually any kind of biosensor, from enzymatic to affinity-based biosensors. However, its battery and integrated circuits alone cost several euros. This represents too heavy a cost burden, and limits the scope of its potential applications.
Unfortunately, whether integrated along with the sensor or in the form of external instrumentation, silicon-based electronics are seemingly crucial to all known self-powered devices.8 In contrast, we believe that electrochromic materials9–11 can overcome this dependence on silicon-based electronics and their inherent limitations in self-powered chemical sensors and biosensors. The use of electrochromic materials in the construction of self-powered displays is hardly new,12–14 but their application in stand-alone quantitative analytical devices is.3,15,16
Electrochromic materials such as Prussian blue17–19 have been recently introduced in the construction of self-powered devices with minimal instrumentation needs, so that detection reactions can be monitored directly by eye.20 Liu and Crooks presented a paper-based glucose sensor powered by a metal–air battery,21 which enabled the detection of glucose above a reported detection limit of 0.1 mM. This device provides a ‘yes/no’ type of answer, and quantification only seems possible by means of an optical reader, as the concentration of glucose may be estimated from the measurement of the Prussian blue spot colour intensity. This is also the principle of operation behind the device reported more recently by Zloczewska et al.,15 who presented a self-powered sensor for the determination of ascorbic acid in fruit juice and other foodstuffs. The authors claim that by using a biofuel cell (BFC) to reduce or oxidize an electrochromic display they can create a reusable, truly self-powered biosensor.
During the detection phase, the presence of ascorbic acid results in the reduction of Prussian blue into colourless Prussian white, and the concentration of ascorbic acid can be inferred from the rate of discolouration of the electrochromic pigment using a camera and image analysis software. Once the measurement is complete, the Prussian blue can be recovered connecting it to an air-breathing oxygen cathode. Similar systems can be found which rely on other enzymes22 or which use other electrochromes, such as methylene green.16
Despite the advantages of this approach, none of the analytical devices described so far allow the direct quantification of an analyte, as they rely on colour readers to translate the Prussian blue cathode hue into analyte concentration values. This may be just a minor inconvenience, given the widespread availability of smartphones with massive processing capabilities and increasingly better cameras.15,23 However, in line with other recent studies using externally powered electrochromic redouts,24 our results demonstrate that no reader other than the user's eyes may be needed to obtain a quantitative result directly.
The approach used here also couples an electrochromic display to a biosensor so that they form a galvanic system. The novelty of this system is in the geometric combination of its components, which enables the system to work without the involvement of silicon-based electronics. We have used a horizontal configuration, as shown in Fig. 1, instead of the vertical construction of conventional electrochromic displays.25–28 Vertical displays, where anode and cathode face each other, minimize internal resistance and allow for quicker responses and larger number of on–off cycles. However, as previous reports demonstrate, quantification in systems built as vertical displays requires the aid of a spectrophotometer. This is because the electrochromic material switches colour simultaneously across the entire surface area, so one can at best hope to read a colour intensity. However, changing the configuration to avoid overlap between anode and cathode enables the display to act as a visual coulometer readable by the naked eye. This is schematized in Fig. 1, which shows images of the device used in this work next to a diagram showing the principle of operation. The construction exploited here relies on electrochromic materials to control the current path through the device, and uses internal resistance to its advantage, as the display first changes its colour in the region closer to the anode through the path of least electric resistance. Because electrochromic materials change colour with oxidation state, they can be used as self-indicating coulometers. Internal resistance can be used to prevent current leaks in the absence of glucose, and to ensure that the system responds only above a certain analyte threshold concentration. Our results demonstrate that controlling the current path in electrochromic devices enables the construction of simple and yet very powerful electroanalytical devices. The charge generated by the biosensor is balanced at the display following the path of least resistance, which results in a progressive discolouration of the display in the horizontal plane that is easy to read by sight.
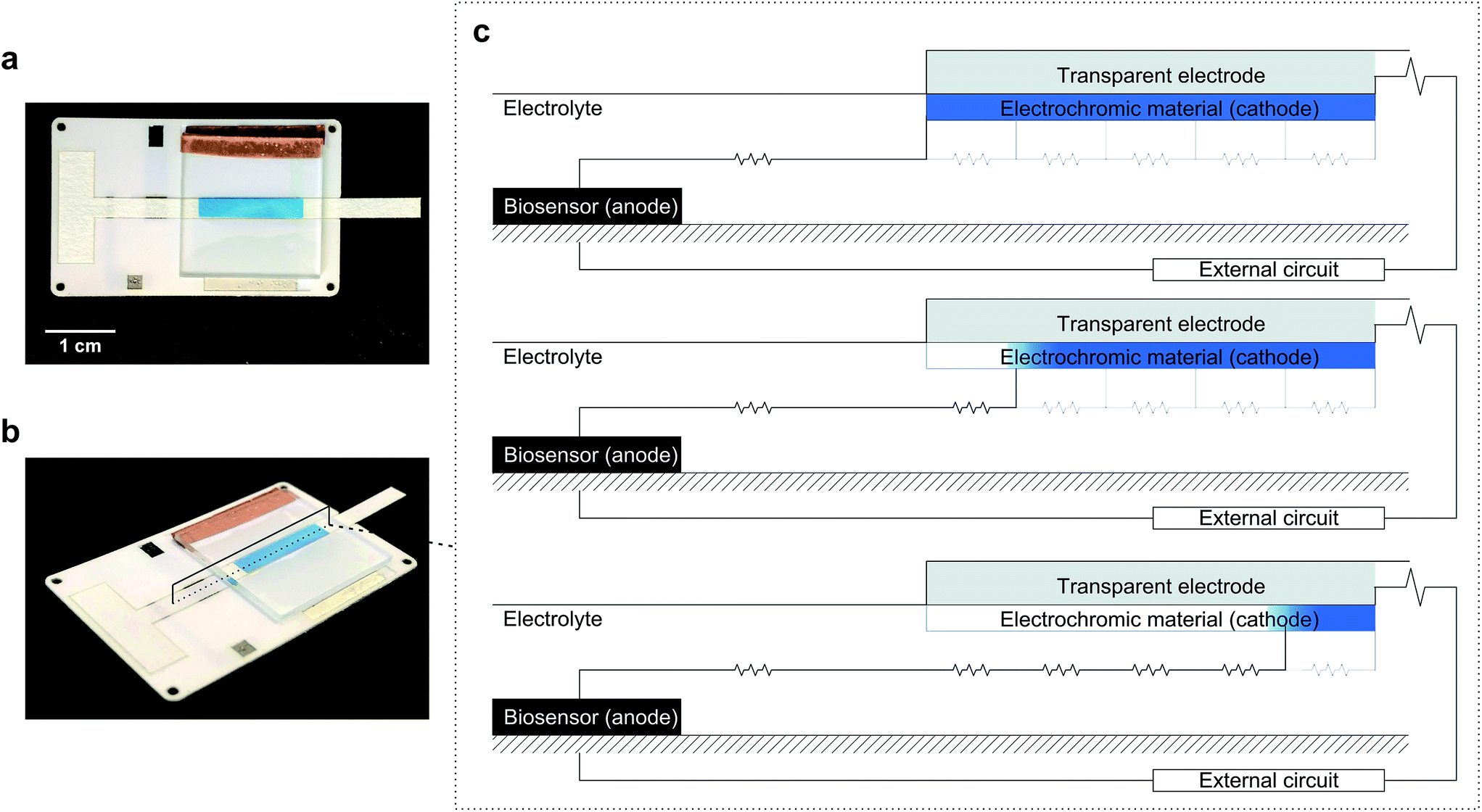 | ||
| Fig. 1 Electrochromic biosensor and its operating principle. (a and b) Photographs of the actual electrochromic biosensor device. (c) Schematic representation of the device side view, with the biosensor (anode) placed to the left of the cathode (electrochromic display). As the analyte concentration or reaction time increases, the charge consumed at the cathode increases, resulting in a gradual color change along the electrode length, following the path of least resistance. | ||
Results and discussion
Horizontal electrochromic displays
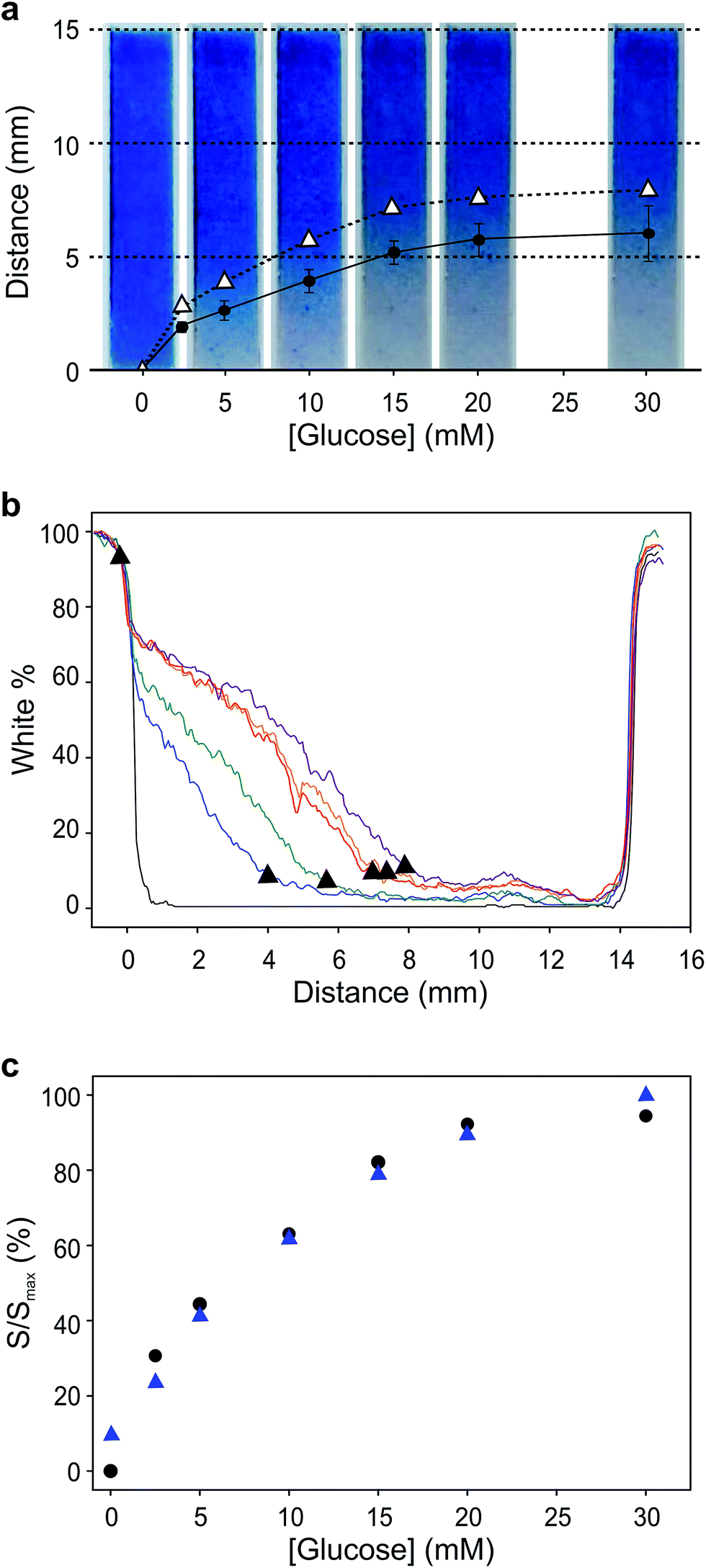
We first demonstrate the concept of visual coulometry using a Prussian blue horizontal display. This Prussian blue display consists of a 3 × 15 mm transparent indium tin oxide (ITO) electrode coated by a thin layer of electrodeposited Prussian blue, 100 ± 15 nm thick. This electrochromic electrode is 5 mm upstream from a stencilled graphite paste anode using a lateral flow membrane that enables flowing glucose and electrolyte solutions as needed.29 The thickness of this membrane, combined with electrolyte concentration, can be used to adjust the electrical resistance between anode and cathode, and therefore, the device performance, i.e. detection thresholds and response linearity.To demonstrate the operation of this visual coulometer, we assembled the device shown in Fig. 1a and b (see ESI† for details). It features stencilled electrodes separated by a lateral flow membrane and a Prussian blue coated ITO electrode on the other. The lateral flow membrane is used to contain the electrolyte, which fills the system by capillary force, and also facilitates the introduction of electrolyte solutions of different composition. We carried out a series of experiments in which a known charge was passed through the system under different conditions, as follows. The anode is a 3 × 3 mm graphite electrode, and the current was forced by a potentiostat. First, the charge contained in the Prussian blue cathode was determined by integration of the current peaks in a stable cyclic voltammogram. Typically, Prussian blue coatings deposited had a charge of 1.2 ± 0.2 mC (details can be found in ESI†). This charge was forced through the system at a constant current of 40 μA, in line with the maximum current output of our biosensor (see below), and the display behaviour was recorded. Fig. 2a shows images of the Prussian blue electrode taken at different times. As charge is passed, the colour front moves away from the anode. The figure shows a comparison in the position chosen as the end of the colour bar subjectively by a person (circles), or by numerical analysis of the colour profiles obtained using ImageJ30 (triangles). Details of the mathematical analysis are given in ESI.†Fig. 2b shows the colour intensity profiles determined by ImageJ at different times during the experiment. Clear colours are represented by high % values on the y-axis, whilst darker colours (blue) take lower % values. As Fig. 2a shows, the length covered by the colour change is directly proportional to the charge drawn from the electrochromic layer, as expected. Fig. 2c, on the other hand, shows images taken after 30 seconds passing different current levels through the system, which again represents different total charges. This experiment aimed to study the behaviour of the horizontal display as a visual coulometer in the form of a metering bar in the presence of different glucose concentrations. As seen in Fig. 2d, the system continues to respond well, and the distance travelled by the colour front remains proportional to the total charge. Note that this experiment is only an approximation, because during a biosensor experiment both cell potential and current change over time. Nevertheless, this experiment demonstrates proof of principle, and strongly suggests that a clear visual readout is possible if a biosensor with the right oxidation potential is combined with a suitable electrochromic counter electrode.
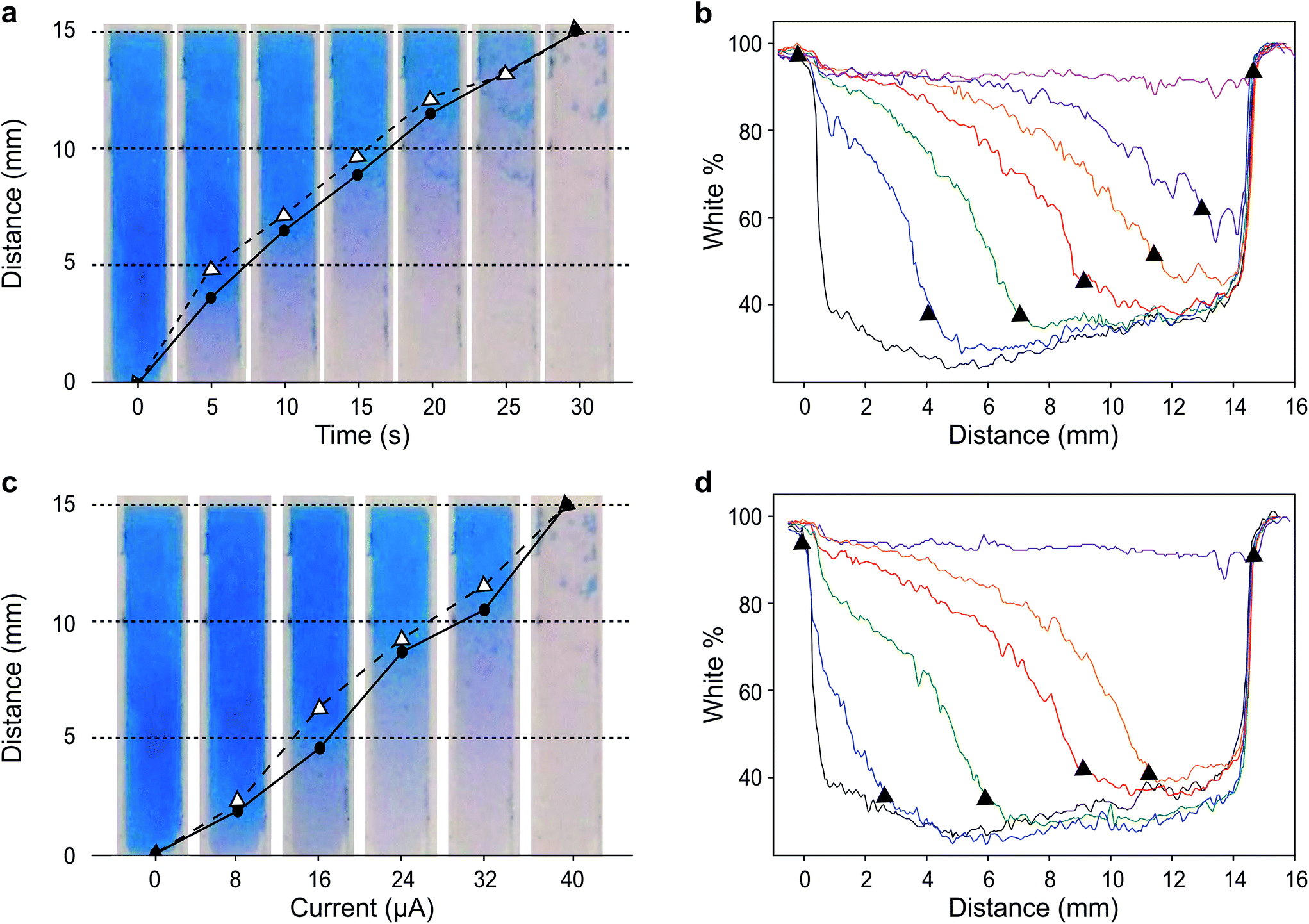 | ||
| Fig. 2 Behaviour of the horizontal electrochromic display. (a) Captures of the display at different times under a constant applied current of 40 μA. The points signal the position of the color front chosen arbitrarily by a user (circles) and determined by analysis of the corresponding color profiles (triangles). (b) Color profiles of the electrochromic display obtained with ImageJ software. (c) Captures of the display recorded 30 s after application of different current levels. (d) Color profiles of the electrochromic display obtained with ImageJ software. The triangles in (a and b) represent the calculated distance obtained with ImageJ software. Circles represent the position estimated subjectively by eye. | ||
Enzymatic biosensors enable self-powered electrochromic devices
Next, we connected the Prussian blue electrode to a glucose biosensor using N,N,N′,N′-tetramethyl-p-phenylenediamine, TMPD, as electron mediator between the enzyme glucose oxidase and the graphite electrode (see ESI† for details).29 The electrode reactions taking place, and which allow the system to work as a galvanic cell, are:At the biosensors:
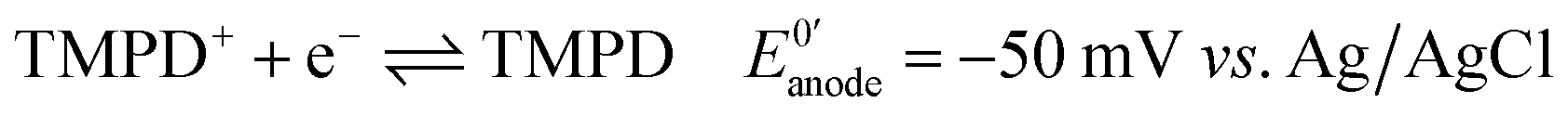 | (1) |
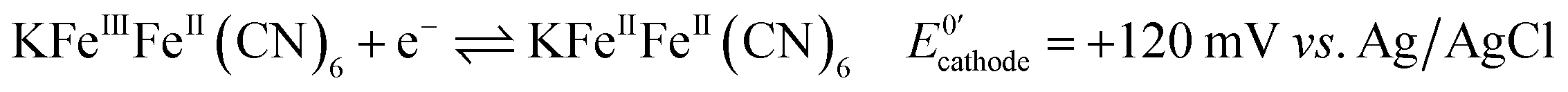 | (2) |
For each electron taken from TMPD at the anode (biosensor), an electron is transferred to the Fe(III) sites in the Prussian blue cathode (display) while ions carry the charge inside the cell. Amongst these ions, potassium facilitates a rapid cathode response and ensures the reversibility of the electrochromic process.31 As seen in Fig. 3a, under constant polarization at 0.2 V vs. Ag/AgCl, the biosensor displays relatively high currents and a wide dynamic range. Fig. 3b shows typical cyclic voltammograms of both the glucose oxidase/TMPD anode and the Prussian blue cathode. Note that the onset of the Prussian blue reduction at the cathode is observed at 0.3 V vs. Ag/AgCl, while the oxidation current of TMPD, resulting from the activity of the enzyme at the anode begins at approximately −0.15 V vs. Ag/AgCl, leading to cell potentials around 0.45 V. This is in line with the open circuit potential (OCP) observed in the polarization curves recorded at various glucose concentrations (Fig. 3c). In the absence of glucose, the open circuit potential is around 100 mV, but the internal resistance prevents the Prussian blue display from bleaching. The behaviour of the self-powered device was studied using glucose solutions of different concentrations. The electrochromic cathode was connected to the biosensing anode, thus initiating the enzymatic reaction. Fig. 3c and d show the corresponding polarization and power curves, where the ‘limited’ amount of charge available at the Prussian blue electrode is apparent at high glucose concentrations. As the Prussian blue is depleted, the current passing through the system drops. Fig. 3c and d also show a progressive increase in the current and power density maxima produced by the cell as the fuel concentration, i.e. glucose, is increased, reaching a maximum current of 40 μA for a glucose concentration of 20 mM, in agreement with the biosensor Michaelis–Menten curve (Fig. 3a). As other self-powered devices, the system power output is proportional to analyte concentration. In this case a maximum power density around 13 μW cm−2 was obtained.
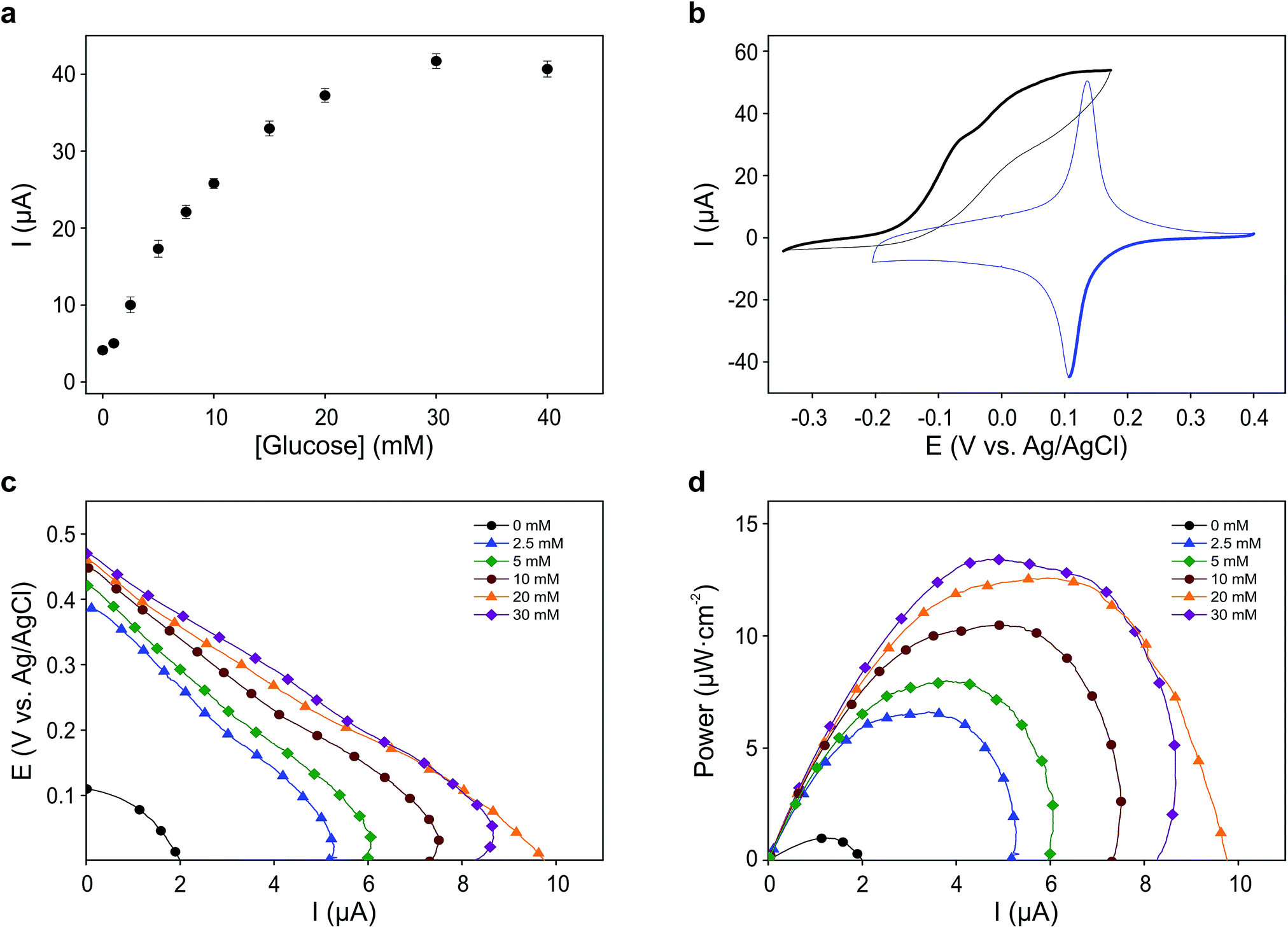 | ||
| Fig. 3 Electrochemical characterization of the self-powered glucose sensor. (a) Biosensor current response to different glucose concentrations. Error bars represent the standard deviation of the measurements from four different devices. (b) Cyclic voltammograms showing the reduction and oxidation of Prussian blue on an ITO electrode (blue) and the response of a stencilled biosensor in contact with a saturating concentration of glucose (black). Measurements were carried out at 5 mV s−1 against a stencilled Ag/AgCl pseudo-reference electrode. (c) Polarization curves taken from the OCP to 0 V at a scan rate of 1 mV s−1. (d) Power curves obtained from the polarization curves. | ||
Fig. 4a shows images taken 30 seconds after the connection of the electrodes. The points represent the position of the colour front determined subjectively (circles) and by ImageJ colour profile analysis (triangles; see also Fig. 4b). The use of analysis software improves the accuracy of the measurements, while the sensitivity of both methods is comparable. Another interesting feature of these experiments is that the behaviour of the biosensor differs slightly from that observed during the controlled charge experiments described above. In the controlled charge experiment the display colour was switched completely. In contrast, the biosensor was only able to turn the colour on roughly half the cathode area, even though the biosensor was able to draw currents of similar magnitude as those used in the controlled current experiments. Also, the colour front is blurred to different extents in both experiments. These two effects are brought about by the device internal resistance, which changes during the experiment. The blurriness in the colour front is mostly caused by the potential drop inside the cell arising from the resistivity of the electrolyte medium between anode and cathode. At low currents the extent of this iR drop decreases and a wider region of the cathode is able to observe the same potential. In other words, when the current passing is sufficiently low, the potential distribution at the electrode is more homogenous, causing a spread or blur of the colour front. The current goes from zero initially, then increases as the enzyme reaction equilibrates, and then decreases again once the internal resistance increases as a result of the colour front moving away from the biosensor. Thus, the colour front is blurrier at short and long times, and is sharper when the biosensor current is at its maximum.
 | ||
| Fig. 4 Self-powered electrochromic biosensor response. (a) Captures of the display 30 seconds after the addition of different glucose solutions. The points signal the position of the color front determined subjectively (circles) and from the analysis of color profiles (open triangles). The error bars correspond to the standard deviation of the measurements obtained from four different devices. (b) Color profiles of the electrochromic display obtained with ImageJ software. The triangles represent the position of the color front for each glucose concentration. (c) Plot of the normalized to the maximum biosensor current (blue triangles) and color front position (black circles) as a function of glucose concentration. | ||
Another effect of the increasing internal resistance is that the colour front only advances 6–7 mm into the electrode (9–10 mm away from the biosensor edge) instead of consuming all the Prussian blue available at the cathode. As the colour front moves farther away from the biosensor, the length of the current path increases, and with it the internal resistance of the device. As internal resistance increases, the system operation point moves along the polarization curve towards lower potential values. At some point the iR drop overcomes the cell electromotive force, emf, causing the colour front to stop. However, before it does, the low current results in a blurred front.
The variability shown by the self-powered biosensor is thought to be due to differences in construction of the Prussian blue/ITO display. Indeed, since the display is pressure-mounted on the lateral flow membrane holding the electrolyte, changes in geometry arising from different assemblies affect sample flow conditions and internal electrical resistance. These problems did not appear in the controlled current experiments because the potentiostat was able to adjust the potential of the electrodes in order to supply the required current levels. When a biosensor is connected to the display, the potential and the current depend on a number of factors, among which analyte concentration, mass transport conditions, and internal resistance are the most important. On the other hand, Fig. 4c shows that, in spite of all this, the self-powered device follows the Michaelis–Menten curve of the biosensor. The current data from Fig. 3a and the distances measured in Fig. 4a have been normalized and plotted together. As the figure shows, the two normalized datasets are in excellent agreement. Last, the device was tested with a diluted sample of a commercial fruit juice as described in ESI.† The amount of glucose present in this sample was previously determined using a commercial glucometer (CardioCheck, Novalab ES) leading to a value of 7.6 ± 0.2 mM. The interpolation of the length of the colour change in the strip when the sample was let to flow yielded a glucose concentration of 7.3 ± 0.4 mM, demonstrating the value of this instrumentation-less device to quantify the concentration of analyte in a sample easily and without additional instrumentation.
Experimental
Chemicals and materials
Carbon (C2030519P4) and silver/silver chloride (C61003P7) screen printing pastes (Gwent Electronic Materials Ltd., UK) were used for the fabrication of the electrodes. Pressure sensitive adhesives ARcare® 8259 and ARcare® 8939 (Adhesive Research Ltd., IE) were used as a substrate for the printed electrochemical cell and as structural material outlining the electrode areas and contacts respectively. Whatman 1 (GE Healthcare, FR) lateral flow membrane was used as a carrier for the solutions. Indium tin oxide on 1.1 mm float glass wafers (CEC020S, Präzisions Glas & Optik, GmbH) was etched through a vinyl mask in a hydrochloric acid and nitric acid (Sigma-Aldrich, 37% and 69% w/w respectively). Isopropanol (Honeywell, 99.5%), acetone (Honeywell, 99.5%) and ammonia (Panreac, 30%) were used to clean the ITO electrodes. Potassium hexacyanoferrate(III) trihydrate (Sigma-Aldrich), iron(III) nitrate nonahydrate, hydrochloric acid and potassium chloride (Sigma-Aldrich) were used to electrodeposit the Prussian blue cathode on the indium tin oxide (ITO) electrode. A 0.05 M phosphate buffer (Fluka) and 0.1 M potassium chloride (Sigma-Aldrich) solution at pH = 7 was used as supporting electrolyte for all the measurements. Glucose oxidase, GOx (Sekisui Diagnostics, EC 1.1.3.4, 236 U mg−1), glutaraldehyde solution, GA (Sigma-Aldrich, 50% w/w), bovine serum albumin, BSA (Sigma-Aldrich), alcoholic Nafion solution (Sigma-Aldrich, 20% w/w), N,N,N′,N′-tetramethyl-p-phenylenediamine, TMPD (Sigma-Aldrich) and ethanol (Panreac, 96%) were used to fabricate the glucose biosensor.Instrumentation
A 30 W Epilog Mini 24 CO2 laser engraver (Laser Project, ES) was used to cut pressure sensitive adhesive and lateral flow membrane parts. Adhesive vinyl sheets and copper tape were cut using a CAMM1-GX24 Servo cutter plotter (Roland DG Ibérica, ES). Scanning electron microscopy (SEM) images of the ITO and Prussian blue layers were recorded in an Auriga (Carl Zeiss) system. The measurements were performed by applying a beam voltage of 5 kV. Images at different magnifications were obtained on at least five different areas in the same sample using the InLens detector (Fig. S4 and S5†). Thickness measurements of the Prussian blue layer were performed by a mechanical profilometer (Alpha-Step 200 Profiler from Tencor Instruments). A μ-Autolab III potentiostat (Metrohm) controlled by a GPES 4.1 software was used for the electrochemical measurements.Fabrication and characterization of the fully integrated device
The electrochemical cells were fabricated as described in ESI† and were activated applying 10 potential steps from 0 to −1.5 V using a 0.5 M KNO3 solution. The glucose biosensors were fabricated as follows: 10 μL of a 25 mM TMPD solution prepared in ethanol were spread on the surface of the working electrode and let to dry. Then, 5 μL of a solution containing 15 mg mL−1 GOx, 0.15% (w/w) GA, 0.6 mg mL−1 BSA and 0.5% (w/w) alcoholic Nafion solution were cast on the surface of the TMPD film and let to dry overnight at 4 °C in darkness. The electrode was rinsed with deionized water and then polarized at 0.2 V vs. Ag/AgCl for 15 minutes in buffer solution. ITO electrodes were shaped by using an etching protocol. For this, an adhesive vinyl cut to a desired shape was fixed on the surface of the ITO substrate, leaving exposed the surface of the oxide to be removed. The electrodes were immersed in a HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3:H2O solution (4:1:1) for 1 hour. ITO electrodes were cleaned before and after the etching using 10% ammonia solution, isopropanol and acetone, and employing ultrasonication for 10 minutes in each cleaning step. The synthesis of the cathode/electrochromic display was done electrochemically. Briefly, 10 mL of 0.02 M aqueous solutions of potassium hexacyanoferrate(III) and iron(III) nitrate nonahydrate were mixed with 4 mL of 1 M hydrochloric acid and 0.1 M potassium chloride to a final volume of 40 mL. A homogeneous Prussian blue film was achieved by applying a potential of 0.4 V vs. Ag/AgCl (KCl 3 M) for 20 seconds under constant stirring. The ITO electrode was then rinsed with deionized water and dried in air. Electrochemical activation of the Prussian blue coating was required in order to obtain electrochemically reversible films (Fig. S2†). Highly reproducible and reversible films were obtained using this method (Fig. S3†). Furthermore the electrodeposited Prussian blue layer on top of the ITO was characterized physically using scanning electron microscopy (SEM) and profilometry. SEM images show the characteristic dense granular structure of the sputtered ITO layer on top of glass, and the overlying uniform layer of Prussian blue after its electrochemical activation (Fig. S4 and S5†). The ITO/Prussian blue electrode was assembled on the surface of the electrochemical cell as seen in Fig. S1† so the device was ready to be used.
:HNO3:H2O solution (4:1:1) for 1 hour. ITO electrodes were cleaned before and after the etching using 10% ammonia solution, isopropanol and acetone, and employing ultrasonication for 10 minutes in each cleaning step. The synthesis of the cathode/electrochromic display was done electrochemically. Briefly, 10 mL of 0.02 M aqueous solutions of potassium hexacyanoferrate(III) and iron(III) nitrate nonahydrate were mixed with 4 mL of 1 M hydrochloric acid and 0.1 M potassium chloride to a final volume of 40 mL. A homogeneous Prussian blue film was achieved by applying a potential of 0.4 V vs. Ag/AgCl (KCl 3 M) for 20 seconds under constant stirring. The ITO electrode was then rinsed with deionized water and dried in air. Electrochemical activation of the Prussian blue coating was required in order to obtain electrochemically reversible films (Fig. S2†). Highly reproducible and reversible films were obtained using this method (Fig. S3†). Furthermore the electrodeposited Prussian blue layer on top of the ITO was characterized physically using scanning electron microscopy (SEM) and profilometry. SEM images show the characteristic dense granular structure of the sputtered ITO layer on top of glass, and the overlying uniform layer of Prussian blue after its electrochemical activation (Fig. S4 and S5†). The ITO/Prussian blue electrode was assembled on the surface of the electrochemical cell as seen in Fig. S1† so the device was ready to be used.
Conclusions
Herein we have shown proof of concept of an unconventional use of electrochromic materials in sensing, through a functional prototype able to quantify glucose concentration in aqueous solutions and commercial fruit juice. The versatility and broad applicability of the approach used here stem from the fact that the principles described can be further extended to the analysis of other analytes beyond glucose. This is because the sensor may be either an anode or a cathode, provided that a complementary electrochromic process is used as counter electrode. The only pre-requisite for the system to work is that the cell ΔV > 0 over a desired analyte concentration range, so that the electrochromic display can be driven.Our results demonstrate the feasibility of developing quantitative and self-powered electroanalytical devices without the need for silicon-based electronics. While coupling an optical reader to the device might improve reliability and facilitate automation, the information can already be interpreted by sight.
The horizontal configuration demonstrated here could foster the development of novel analytical devices, particularly non-invasive ones, based on electrochromic materials, introducing new opportunities for electrochromic materials and devices beyond smart glasses, displays and mirrors.
Regarding their industrial impact, because these systems only require two electrodes and an electrolyte to work, they can be entirely produced using large area printing processes. This can lead to extremely cost effective devices including skin patches, contact lenses and other non-invasive and easy to use wearables.13,14,32–36 Moreover, the use of reversible electrochromic materials may lead to the development of silicon-electronics-free sensors for continuous measurements.
Acknowledgements
This work was partly funded by the Spanish Ministry of Economy through the DADDi2 project (TEC2013-48506). MK acknowledges funding through the Beatriu de Pinós program (BP-DGR-2013), supported by the Secretary for Universities and Research of the Ministry of Economy and Knowledge of the Government of Catalonia and the Cofund programme of the Marie Curie Actions of the 7th R&D Framework Programme of the European Union.References
- E. Katz, A. F. Bückmann and I. Willner, J. Am. Chem. Soc., 2001, 123, 10752–10753 CrossRef CAS PubMed
.
- J. P. Esquivel, J. Colomer-Farrarons, M. Castellarnau, M. Salleras, F. J. del Campo, J. Samitier, P. Miribel-Catala and N. Sabate, Lab Chip, 2012, 12, 4232–4235 RSC
- A. P. Turner, Chem. Soc. Rev., 2013, 42, 3184–3196 RSC
- W. Gao, S. Emaminejad, H. Y. Nyein, S. Challa, K. Chen, A. Peck, H. M. Fahad, H. Ota, H. Shiraki, D. Kiriya, D. H. Lien, G. A. Brooks, R. W. Davis and A. Javey, Nature, 2016, 529, 509–514 CrossRef CAS PubMed
- D. H. Kim, N. Lu, R. Ma, Y. S. Kim, R. H. Kim, S. Wang, J. Wu, S. M. Won, H. Tao, A. Islam, K. J. Yu, T. I. Kim, R. Chowdhury, M. Ying, L. Xu, M. Li, H. J. Chung, H. Keum, M. McCormick, P. Liu, Y. W. Zhang, F. G. Omenetto, Y. Huang, T. Coleman and J. A. Rogers, Science, 2011, 333, 838–843 CrossRef CAS PubMed
- K. Fukuda, Y. Takeda, Y. Yoshimura, R. Shiwaku, L. T. Tran, T. Sekine, M. Mizukami, D. Kumaki and S. Tokito, Nat. Commun., 2014, 5, 4147 CAS
- V. Beni, D. Nilsson, P. Arven, P. Norberg, G. Gustafsson and A. P. F. Turner, ECS J. Solid State Sci. Technol., 2015, 4, S3001–S3005 CrossRef CAS
- R. L. Arechederra and S. D. Minteer, Anal. Bioanal. Chem., 2011, 400, 1605–1611 CrossRef CAS PubMed
-
P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, 2007 Search PubMed
- R. J. Mortimer, Annu. Rev. Mater. Res., 2011, 41, 241–268 CrossRef CAS
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed
- M. Möller, N. Leyland, G. Copeland and M. Cassidy, Eur. Phys. J.: Appl. Phys., 2010, 51, 33205 CrossRef
- L.-M. Huang, C.-W. Hu, H.-C. Liu, C.-Y. Hsu, C.-H. Chen and K.-C. Ho, Sol. Energy Mater. Sol. Cells, 2012, 99, 154–159 CrossRef CAS
- J. Wang, L. Zhang, L. Yu, Z. Jiao, H. Xie, X. W. Lou and X. W. Sun, Nat. Commun., 2014, 5, 4921 CrossRef CAS PubMed
- A. Zloczewska, A. Celebanska, K. Szot, D. Tomaszewska, M. Opallo and M. Jonsson-Niedziolka, Biosens. Bioelectron., 2014, 54, 455–461 CrossRef CAS PubMed
- P. Pinyou, F. Conzuelo, K. Sliozberg, J. Vivekananthan, A. Contin, S. Poller, N. Plumere and W. Schuhmann, Bioelectrochemistry, 2015, 106, 22–27 CrossRef CAS PubMed
- W. P. Griffith, Q. Rev. Chem. Soc., 1962, 16, 188 RSC
- V. D. Neff, J. Electrochem. Soc., 1978, 125, 886–887 CrossRef CAS
-
C. G. Granqvist, Handbook of Inorganic Electrochromic Materials, Elsevier, 1995 Search PubMed
- B. Kong, C. Selomulya, G. Zheng and D. Zhao, Chem. Soc. Rev., 2015, 44, 7997–8018 RSC
- H. Liu and R. M. Crooks, Anal. Chem., 2012, 84, 2528–2532 CrossRef CAS PubMed
- A. N. Sekretaryova, V. Beni, M. Eriksson, A. A. Karyakin, A. P. Turner and M. Y. Vagin, Anal. Chem., 2014, 86, 9540–9547 CrossRef CAS PubMed
- L. Shen, J. A. Hagen and I. Papautsky, Lab Chip, 2012, 12, 4240–4243 RSC
- D. D. Liana, B. Raguse, J. J. Gooding and E. Chow, ACS Appl. Mater. Interfaces, 2015, 7, 19201–19209 CAS
- P. Andersson Ersman, J. Kawahara and M. Berggren, Org. Electron., 2013, 14, 3371–3378 CrossRef CAS
- J. Kawahara, P. A. Ersman, I. Engquist and M. Berggren, Org. Electron., 2012, 13, 469–474 CrossRef CAS
- J. Kawahara, P. Andersson Ersman, D. Nilsson, K. Katoh, Y. Nakata, M. Sandberg, M. Nilsson, G. Gustafsson and M. Berggren, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 265–271 CrossRef CAS
- D. Eric Shen, A. M. Österholm and J. R. Reynolds, J. Mater. Chem. C, 2015, 3, 9715–9725 RSC
- M. Aller Pellitero, M. Kitsara, F. Eibensteiner and F. J. Del Campo, Analyst, 2016, 141, 2515–2522 RSC
-
W. S. Rasband, ImageJ Search PubMed
- A. A. Karyakin, Electroanalysis, 2001, 13, 813–819 CrossRef CAS
- H. Yao, A. J. Shum, M. Cowan, I. Lahdesmaki and B. A. Parviz, Biosens. Bioelectron., 2011, 26, 3290–3296 CrossRef CAS PubMed
- J. R. Windmiller and J. Wang, Electroanalysis, 2013, 25, 29–46 CrossRef CAS
- W. Jia, G. Valdes-Ramirez, A. J. Bandodkar, J. R. Windmiller and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 7233–7236 CrossRef CAS PubMed
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS PubMed
- A. J. Bandodkar, W. Jia, C. Yardimci, X. Wang, J. Ramirez and J. Wang, Anal. Chem., 2015, 87, 394–398 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04469g |
| This journal is © The Royal Society of Chemistry 2017 |