DOI:
10.1039/C6SC04367D
(Edge Article)
Chem. Sci., 2017,
8, 1535-1546
A supramolecular Tröger's base derived coordination zinc polymer for fluorescent sensing of phenolic-nitroaromatic explosives in water†
Received
30th September 2016
, Accepted 24th October 2016
First published on 25th October 2016
Abstract
A V-Shaped 4-amino-1,8-napthalimide derived tetracarboxylic acid linker (L; bis-[N-(1,3-benzenedicarboxylic acid)]-9,18-methano-1,8-naphthalimide-[b,f][1,5]diazocine) comprising the Tröger's base (TB) structural motif was rationally designed and synthesised to access a nitrogen-rich fluorescent supramolecular coordination polymer. By adopting the straight forward precipitation method, a new luminescent nanoscale Zn(II) coordination polymer (TB-Zn-CP) was synthesized in quantitative yield using Zn(OAc)2·2H2O and tetraacid linker L (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5) in DMF at room temperature. The phase-purity of as-synthesised TB-Zn-CP was confirmed by X-ray powder diffraction analysis, infra-red spectroscopy, and elemental analysis. Thermogravimetric analysis suggests that TB-Zn-CP is thermally stable up to 330 °C and the morphological features of TB-Zn-CP was analysed by SEM and AFM techniques. The N2 adsorption isotherm of thermally activated TB-Zn-CP at 77 K revealed a type-II reversible adsorption isotherm and the calculated Brunauer–Emmett–Teller (BET) surface area was found to be 72 m2 g−1. Furthermore, TB-Zn-CP displayed an excellent CO2 uptake capacity of 76 mg g−1 at 273 K and good adsorption selectivity for CO2 over N2 and H2. The aqueous suspension of as-synthesized TB-Zn-CP showed strong green fluorescence (λmax = 520 nm) characteristics due to the internal-charge transfer (ICT) transition and was used as a fluorescent sensor for the discriminative sensing of nitroaromatic explosives. The aqueous suspension of TB-Zn-CP showed the largest quenching responses with high selectivity for phenolic-nitroaromatics (4-NP, 2,4-DNP and PA) even in the concurrent presence of other potentially competing nitroaromatic analytes. The fluorescence titration studies also provide evidence that TB-Zn-CP detects picric acid as low as the parts per billion (26.3 ppb) range. Furthermore, the observed fluorescence quenching responses of TB-Zn-CP towards picric acid were highly reversible. The highly selective fluorescence quenching responses including the reversible detection efficiency make the nanoscale coordination polymer TB-Zn-CP a potential material for the discriminative fluorescent sensing of nitroaromatic explosives.
:0.5) in DMF at room temperature. The phase-purity of as-synthesised TB-Zn-CP was confirmed by X-ray powder diffraction analysis, infra-red spectroscopy, and elemental analysis. Thermogravimetric analysis suggests that TB-Zn-CP is thermally stable up to 330 °C and the morphological features of TB-Zn-CP was analysed by SEM and AFM techniques. The N2 adsorption isotherm of thermally activated TB-Zn-CP at 77 K revealed a type-II reversible adsorption isotherm and the calculated Brunauer–Emmett–Teller (BET) surface area was found to be 72 m2 g−1. Furthermore, TB-Zn-CP displayed an excellent CO2 uptake capacity of 76 mg g−1 at 273 K and good adsorption selectivity for CO2 over N2 and H2. The aqueous suspension of as-synthesized TB-Zn-CP showed strong green fluorescence (λmax = 520 nm) characteristics due to the internal-charge transfer (ICT) transition and was used as a fluorescent sensor for the discriminative sensing of nitroaromatic explosives. The aqueous suspension of TB-Zn-CP showed the largest quenching responses with high selectivity for phenolic-nitroaromatics (4-NP, 2,4-DNP and PA) even in the concurrent presence of other potentially competing nitroaromatic analytes. The fluorescence titration studies also provide evidence that TB-Zn-CP detects picric acid as low as the parts per billion (26.3 ppb) range. Furthermore, the observed fluorescence quenching responses of TB-Zn-CP towards picric acid were highly reversible. The highly selective fluorescence quenching responses including the reversible detection efficiency make the nanoscale coordination polymer TB-Zn-CP a potential material for the discriminative fluorescent sensing of nitroaromatic explosives.
Introduction
Supramolecular self-assembly chemistry provides a fascinating platform for developing functional structures and materials that can have applications in various fields.1 Discovery of suitable chemical sensors and sensor materials for the rapid and selective detection of chemical explosives at trace levels has attracted significant attention in recent times.2 The reliable identification of chemical explosives in post-blast detritus is of great importance for forensic analysis and criminal investigations, as well as in an effort to combat terrorism, improve national security and control environmental pollution, etc.2 Among the various known high energetic chemical substances of such nature are the nitroaromatic compounds (NACs). These include picric acid (PA), 2,4,6-trinitrotoluene (TNT), 2,4-dinitrotoluene (2,4-DNT) and 2,6-dinitrotoluene (2,6-DNT); all of which are used as primary components in several known chemical explosives owing to their low cost syntheses and easy transportation.2b,3 Several examples of supramolecular sensory systems for the detection of such explosives have been developed in recent times.2–5 Apart from its explosive nature, PA is also used on a small scale for various medical formulations and as an antiseptic agent and also finds application as yellow pigment in dye/leather industries.6 Because of its multifarious uses and high water solubility (∼14 g L−1 at 20 °C), PA and its derivatives have become a major environmental pollutant and severely affect soil and ground water causing serious health hazards.7 According to the World Health Organization (WHO), the allowed concentration of PA in ground water is 0.001 mg L−1 and above this level it is considered as toxic to living organisms.8 Short time exposure to PA residues induce eye irritation, headache, anemia and skin related diseases while long time exposure may lead to kidney problems and severe liver and respiratory organ damage.6a,9 Therefore, the development of selective and highly sensitive sensors with a fast-response time for trace and discriminative detection of PA from other nitroaromatic explosives is vital for environmental remediation, civilian safety, and for military operations.5b,10
Currently, a wide range of sophisticated techniques like metal detectors, canine teams, ion-mobility spectrometry (IMS), surface-enhanced Raman spectroscopy (SERS), gas chromatography (GC), X-ray diffraction and cyclic voltammetry etc. are available for the detection of nitroaromatic explosives.11 But the routine-field analysis using these techniques is often limited due to their high cost, lack of portability, low selectivity and difficulty to operate on-site. However, in recent years, fluorescence detection techniques have become an effective and alternative detection method for nitroaromatic explosives owing to their high sensitivity, easy visualisation, portability and fast response time for detection.12 In fluorescence-based sensing, the emission characteristic of the fluorophores is perturbed upon binding with analytes.13 The presence of electron withdrawing nitro (–NO2) groups makes the nitroaromatics electron-deficient and thus they form effective charge-transfer complexes with electron-rich fluorophores by subsequent fluorescence quenching.5a,14 In light of this, until recently, a wide variety of fluorescent sensors have been designed and successfully employed for NACs detection.1b,3b,15
One such material that has received immense interest, in recent years, for NACs detection is luminescent metal–organic frameworks (MOFs) or coordination polymers (CPs) on account of their easy synthesis, unique and functional properties, permanent porosity, high thermal/chemical stability, systematically tuneable electronic structures, intriguing structural diversity, good recyclability, and so on.1a,5a,b,7a,13b,16 In contrast to the conventional CPs, the miniaturized coordination polymers can have entirely different size-dependent physico-chemical properties.17 Scaling down the particle size of bulk crystalline CPs to their micro-/nanoscale regime is expected to facilitate their easy dispersion in aqueous media and increases the interaction with analytes and thus enhances the sensing ability.18 To the best of our knowledge, only a limited number of luminescent nanoscale coordination polymers have been developed and exploited as potential fluorescence sensors for the detections of NACs. For example, Maji et al. have synthesised a rod-like Gd(III) coordination polymer from oligo-(p-phenylene-ethynylene)dicarboxylic acid and demonstrated its sensing ability towards NACs in ethanol.19 Zhang et al. reported a fluorescent nanoscale Zn(II)–anthracene coordination polymer with cubic morphology which exhibited efficient sensing characteristics for NACs vapour,20 while Qiu et al. described the nitroaromatic sensing characteristics of Cd(II) based coordination polymer tubular nanotubes in the solid-state.21 Huang et al. synthesized a π-system based fluorescent Tb(III)-coordination polymer hollow nanospheres dispersed in DMF showing good selectivity and sensitivity for sensing NACs.18b,22 However, the nitroaromatic sensing characteristics of these and other already reported nanoscale CPs involve sensing in a pure organic medium or in a vapour phase. For the practical applicability, the sensing of NACs in aqueous media is highly desirable, particularly due to its environmental significance.15e,23 Moreover, and somewhat surprising, there are very few reports on nanoscale CP based sensor materials having discriminative sensing abilities for phenolic-nitroaromatics in aqueous medium.5b,10b,15e,16a One possible way to increase the selectivity of CPs is to functionalise them with Lewis base (amine and pyridine) functional groups that could selectively recognize phenolic-nitroaromatics through intermolecular H-bonding interactions between Lewis bases and the hydroxy group of phenolic-nitroaromatics.5b,15e,24
Based on this consideration, we rationally designed and synthesized a novel fluorescent nanoscale Zn(II)-coordination polymer functionalized with a Tröger's base structural motif, TB-Zn-CP, from the 4-amino-1,8-napthalimide derived tetracarboxylic acid linker (L) as Tröger's bases based on this motif have been shown to be excellent supramolecular building blocks in our laboratory.26 The intrinsic chirality of the Tröger's base moiety favours the unique V-shaped arrangement of two 1,8-naphthalimides (as we have shown by using X-ray crystallography) facilitating the guest binding through multiple supramolecular interactions and subsequently demonstrating superior sensing capability for analyte detection. We foresaw that the Lewis base characteristics of the Tröger's base would increase the selectivity of TB-Zn-CP for the discriminative detection of phenolic nitroaromatics through intermolecular H-bonding interactions.23b As we expected, TB-Zn-CP showed highly sensitive and discriminative sensing ability for the rapid detection of phenolic nitroaromatics such as PA, 2,4-DNT, 4-NP, even in the presence of other potential competing NACs. As it has also recently been shown by McKeown and co-workers, that Tröger's base containing (covalently linked) polymers can be used effectively for gas-uptake and storage of gases,25 we also investigated these properties for TB-Zn-CP. Indeed, TB-Zn-CP displayed an excellent CO2 gas uptake capacity of 76 mg g−1 at 273 K and good adsorption selectivity for CO2 over other gases such as N2 and H2, furthering the application of this type of CPs.
Results and discussion
Synthesis and characterisation of tetracarboxylic acid linker (L)
The 4-amino-1,8-naphthalimide containing Tröger's base tetracarboxylic acid linker (L; bis-[N-(1,3-benzenedicarboxylic acid)]-9,18-methano-1,8-naphthalimide-[b,f][1,5]diazocine) was prepared as a racemic mixture in four steps from the commercially available 4-nitro-1,8-naphthalic anhydride as shown in Scheme 1.
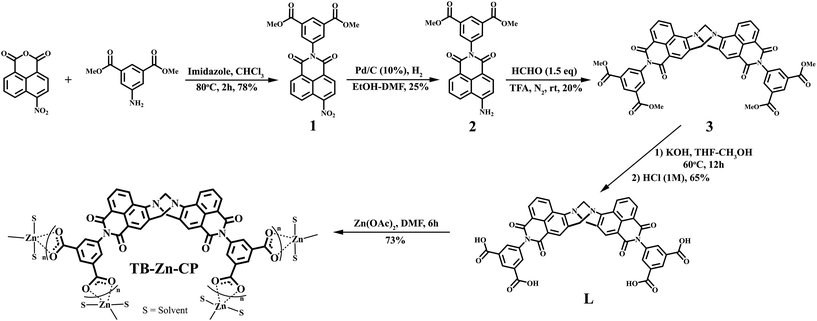 |
| | Scheme 1 Synthesis of the bis-naphthalimide derived Tröger’s base tetracarboxylic acid linker (L) from commercially available 4-nitro-1,8-naphthalic anhydride, that was then used to generate the Zn(II) coordination polymer (TB-Zn-CP). | |
4-Nitro-1,8-naphthalic anhydride and dimethyl-5-aminoisophthalate were refluxed at 80 °C in CHCl3 along with imidazole for 2 hours giving rise to the formation of the 4-nitro-1,8-naphthalimide (1) precursor. The reduction of 1 by catalytic hydrogenation with 10% Pd/C at 3 atm of H2 in ethanol/dimethylformamide gave the corresponding 4-amino-1,8-naphthalimide 2. Compound 2 was next converted into the Tröger's base, using a method developed in our laboratory,26 involving the use of 1.5 equiv. of paraformaldehyde in neat trifluoroacetic acid under an inert atmosphere over 12 hours at room temperature. The resulting reaction mixture was basified (pH > 10) using aq. NH3 and followed by extraction with CH2Cl2 which gave tetra ester derivative 3 in 20% yield after trituration with cold diethyl ether. Subsequent hydrolysis of 3 under basic conditions led to the isolation of target tetracarboxylic acid linker L as a bright orange solid in 65% yield.
The formation of L was fully characterized by various spectroscopic techniques (see Experimental section and ESI†) such as IR, multinuclear (1H and 13C) NMR and HRMS. The 1H NMR spectrum confirmed the identity of L by the presence of a well-separated doublet of doublet peaks in between 5.24 and 4.70 ppm corresponding to the methylene (–CH2N) protons of the diazocine moiety and also reflecting the C2 symmetry of L (Fig. S7, ESI†).26 The high resolution mass spectrometry (HRMS) analysis of L showed two sharp peaks at m/z = 787.1308 and 393.0595 corresponding to the molecular ions [M − H]− and [M − 2H]2−, respectively (Fig. S12, ESI†). The IR spectrum of L showed several intense peaks at ν = 3350 cm−1, 1705 cm−1 and 1239 cm−1 accounting for –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N stretching, respectively which indicated the presence of –COOH and Tröger's base functional groups (Fig. S16, ESI†).27 In view to gain further insight into the molecular structure of L, the energy minimized structure was obtained using density functional theory (DFT) calculation.‡ The structure has been optimized at M062X/6-31G* level and optimized structure along with the HOMO and LUMO orbitals are given in Fig S17 and S18 (see ESI†). The energy minimized structure of L suggests that the methao-1,5-diazocine ring with an angle of 95.40° between the two 1,8-naphthalimide moieties gives rise to the unique V-shaped geometry. It is also evident that the electron density is more localized near the Tröger's base regions and hence the electron-deficient analyte is expected to show preferential binding affinity with Tröger's base moiety. This angle corresponds well with that obtained from crystal structure analysis of a series of 4-amino-1,8-naphthalimide based Tröger's bases developed in our laboratory.26c
O and C–N stretching, respectively which indicated the presence of –COOH and Tröger's base functional groups (Fig. S16, ESI†).27 In view to gain further insight into the molecular structure of L, the energy minimized structure was obtained using density functional theory (DFT) calculation.‡ The structure has been optimized at M062X/6-31G* level and optimized structure along with the HOMO and LUMO orbitals are given in Fig S17 and S18 (see ESI†). The energy minimized structure of L suggests that the methao-1,5-diazocine ring with an angle of 95.40° between the two 1,8-naphthalimide moieties gives rise to the unique V-shaped geometry. It is also evident that the electron density is more localized near the Tröger's base regions and hence the electron-deficient analyte is expected to show preferential binding affinity with Tröger's base moiety. This angle corresponds well with that obtained from crystal structure analysis of a series of 4-amino-1,8-naphthalimide based Tröger's bases developed in our laboratory.26c
Synthesis and characterization of nanoscale Zn(II) coordination polymer TB-Zn-CP
In a typical synthesis, the nanoscale coordination polymer TB-Zn-CP was prepared by stirring Zn(OAc)2·2H2O (1 eq.) and tetracarboxylic acid linker L (0.5 eq.) in DMF solution (4 mL) at room temperature for 6 hours. While several examples of Zn(II) targeted naphthalimide structures have been developed to date, no such naphthalimide Tröger's base ligands have been developed.28 The coordination of Zn(II) ions to the carboxylate groups of the Tröger's base was evidenced from the instantaneous formation of orange colloidal suspensions; which was collected from the reaction mixture by centrifugation, the resulting residue being washed with fresh DMF via three successive dispersion cycles. The chemical composition of TB-Zn-CP was determined through elemental analysis, and it was found to be consistent with a molecular formula of ZnL·2H2O·DMF, in which the deprotonated linker (L–4H) is interconnected by Zn(II) metallic nodes through metal–ligand coordinations. The product was further characterised by using FT-IR spectroscopy, the result showing a strong transitions at 1573 cm−1 (symmetrical) and 1402 cm−1 (asymmetrical), corresponding to the coordinated carboxylate (–COO−) stretching frequencies (Fig. S19, ESI†).29 A strong band at 1657 cm−1 was assigned to CO stretching vibration of entrapped DMF solvent molecules, while a broad band, with the maximum at 3449 cm−1, was assigned to hydrogen bonded H2O molecules (Fig. S19, ESI†).
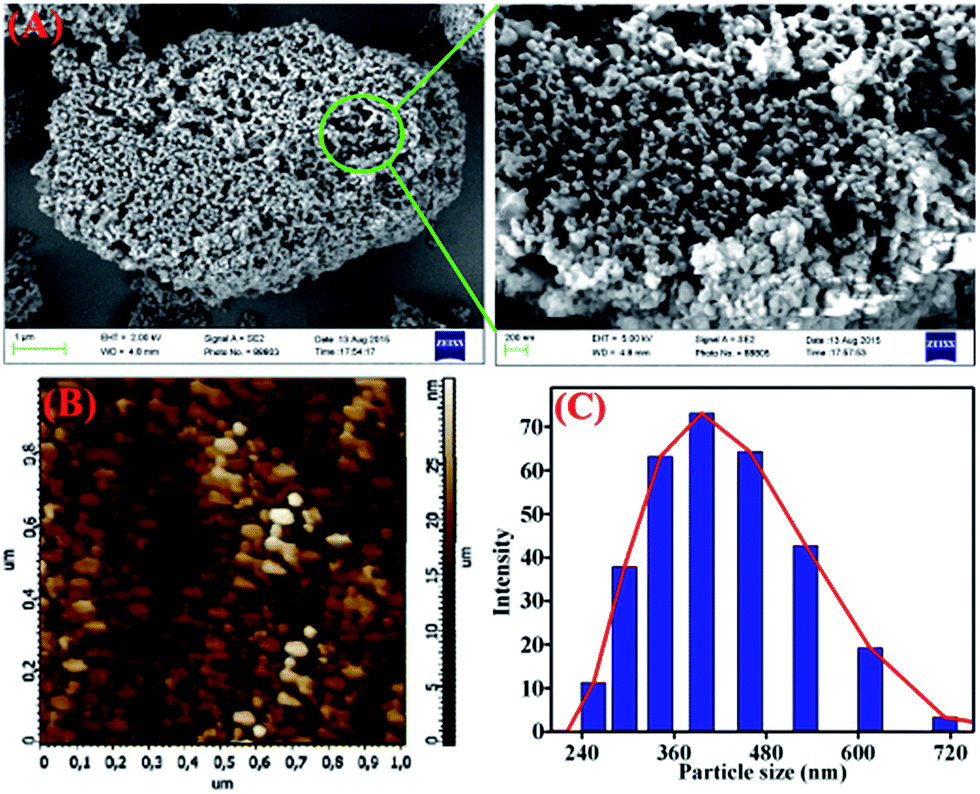
TB-Zn-CP was shown to be amorphous as indicated by X-ray powder diffraction measurement (Fig. S20, ESI†). The TGA analysis of TB-Zn-CP under nitrogen atmosphere showed an initial weight loss of ∼6% at 120 °C due to the loss of coordinated/trapped solvent molecules and the desolvated material was stable up to 330 °C and retains ∼24% of its initial mass at 500 °C (Fig. S21, ESI†). To comprehend the morphology of TB-Zn-CP, we performed Scanning Electron Microscopy (SEM) and atomic-force microscopy (AFM) analysis of the product. As shown in Fig. 1, the SEM (Fig. 1A) and AFM (Fig. 1B) images of TB-Zn-CP showed polydispersed spherical particles that were further aggregated to give a nanoporous structure. The energy-dispersive X-ray spectroscopy (EDX) analysis of TB-Zn-CP exhibited the signature peak for Zn, C, O and N (Fig. S22, ESI†). Quanta area mapping study illustrated the uniform distribution of Zn, C, O and N over the entire sample (Fig. S22, ESI†). Further analysis, using dynamic light scattering (DLS), of aqueous suspension of TB-Zn-CP indicated that the particle sizes are in the nanoscale range, as shown in Fig. 1C.
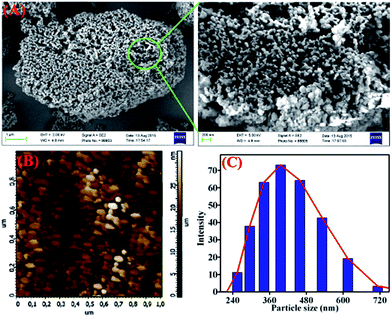 |
| | Fig. 1 (A) Scanning electron microscopy and atomic force microscopy images (B) of as-synthesized TB-Zn-CP. (C) Dynamic light scattering (DLS) measurements of TB-Zn-CP dispersed in water. | |
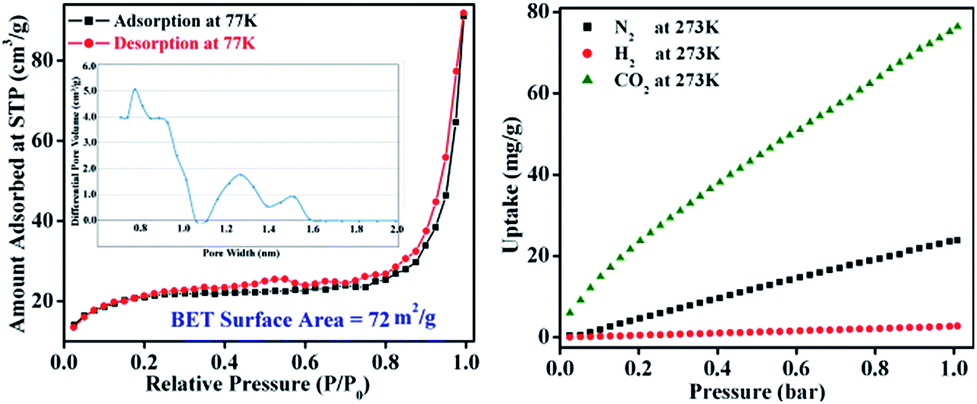
The permanent porosity and surface area of TB-Zn-CP were verified by N2 adsorption isotherms on a Quantachrome AUTOSORP-IQ automated gas sorption analyser. The N2 adsorption isotherm at 77 K exhibits a conventional type-II reversible adsorption isotherm and the calculated Brunauer–Emmett–Teller (BET) surface area was found to be 72 m2 g−1, Fig. 2. The Non-Local Density Functional Theory (NLDFT) pore size distribution of TB-Zn-CP suggests the presence of porosity in the range of 0.7–1.5 nm as shown in Fig. 2. The presence of the CO2-philic nitrogen-rich Tröger's base unit encouraged us to also evaluate the CO2 capture properties. Hence, the TB-Zn-CP was activated under vacuum at 100 °C for 24 hours. The CO2 adsorption measurements of activated TB-Zn-CP up to 1 bar at 273 K showed an uptake of 76 mg g−1 (Fig. 2). The total uptake capacity of TB-Zn-CP for N2 and H2 at 273 K was lower which can be partially attributed to weak interactions with the nitrogen-rich Tröger's base moieties (e.g.Fig. 2). Selectivity for CO2 over N2 was calculated at 273 K by taking the linear part of each uptake slope from 0.025 to 0.178 bar and dividing the slopes which yielded a value of 2.6. The selectivity of TB-Zn-CP for CO2 over N2 can be ascribed to enhanced dipole–quadrupole interactions.25a,30
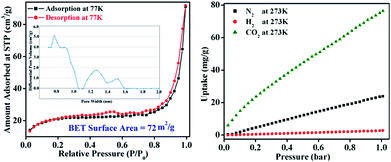 |
| | Fig. 2 The N2 adsorption–desorption isotherm measured at 77 K (inset: corresponding pore-size distribution curve) of TB-Zn-CP (left). The uptake capacitates of TB-Zn-CP for CO2, N2 and H2 at 273 K (right). | |
Photophysical characteristics of TB-Zn-CP and L
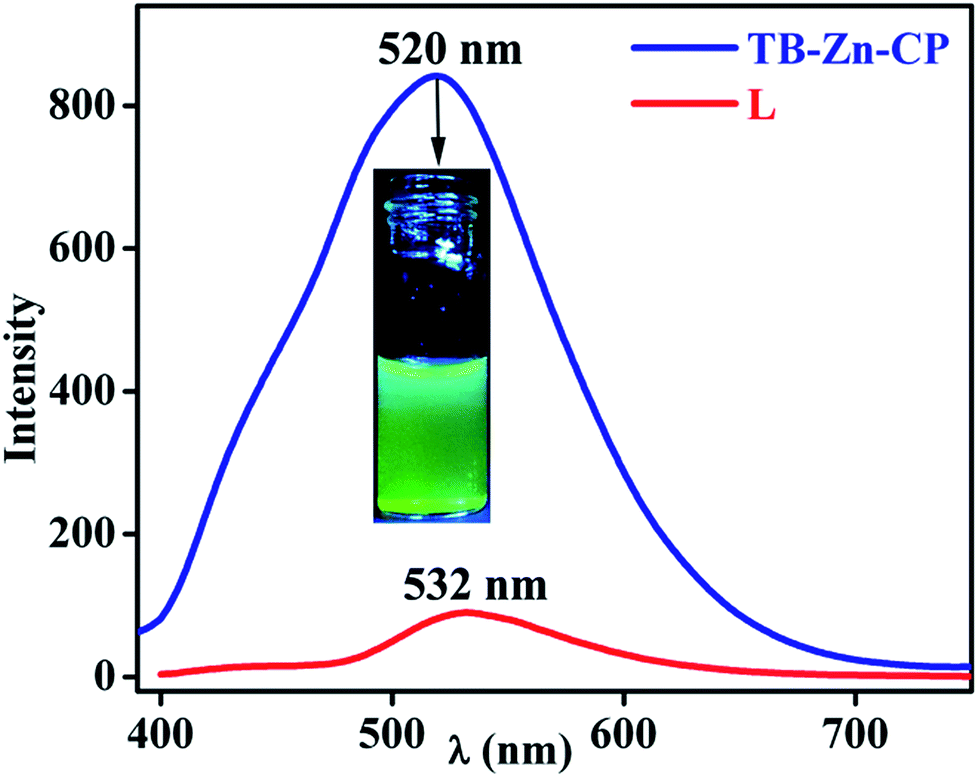
Coordination polymers based on electronically inert d10 metal ions and conjugated organic ligands, have been shown to often display strong photoluminescence characteristics, and as such, these can be employed as responsive materials, for instance, for applications in sensing.15f,16a,31 With this in mind, and given the nature of the Tröger's base structure, which we have previously shown to be an excellent candidate for the luminescent sensing of for instance, polyanions,32 we investigated the use of TB-Zn-CP in the sensing of NACs. Initially, the absorption and the fluorescence emission spectra of the Tröger's base L was recorded in de-ionised water. The absorption spectra (Fig. S23, ESI†) exhibited a typical 1,8-naphthalimide Tröger's base spectra, which was blue shifted by ca., 60 nm compared to that seen for 2, as the 4-amino-moiety of the Tröger's base is less conjugated, giving rise to a smaller internal charge transfer (ICT) character than that seen for 2. This is in an agreement to that previously seen for other 1,8-naphthalimide Tröger's bases. Moreover, the fluorescence excitation and the emission spectra were recorded upon exciting at λ = 390 nm; the former structurally matching that seen in the absorption spectra. In the fluorescence emission spectra, a broad band centred at λ = 532 nm, Fig. 3, was observed, and this was assigned to the “push–pull” based ICT transition.26,32 Similarly, both the absorption and the emission spectra of the TB-Zn-CP colloidal particle were recorded dispersed in H2O. The results were similar to that seen above, except that the Zn(II) coordination gave rise to a high energy absorption band in the absorption spectra; the ICT band of the naphthalimide Tröger's base appeared slightly blue shifted (Fig. S24, ESI†). Upon excitation at 380 nm, a very intense emission band was observed, between 400–700 nm, with λmax = 520 nm, which was almost an order of magnitude more intense than seen for L, Fig. 3. This green emission was clear to the naked eye, even at low concentrations, as demonstrated in the inset in Fig. 3; the emission spectra of TB-Zn-CP was ca. 12 nm blue-shifted compared to L. The significant enhancement in the emission intensity (∼90%) of TB-Zn-CP can be ascribed to two main reasons. Firstly, long range electron communication between the adjacent fluorophores through Zn(II) ions10 and secondly, due to through-space depression of photoinduced electron transfer (PET) quenching from the aryl tetracarboxylic acid moiety which is close to being orthogonal to the naphthalimide moiety. Here, upon coordination to Zn(II), the oxidation potential of the receptor is enhanced, diminishing the thermodynamic pathway for PET to the naphthalimide centre, resulting in enhancement in naphthalimide emission. While such PET quenching is normally only seen for 4-amino-1,8-naphthalimide based structures, where the ion receptor is located, via a covalent spacer at the 4-amino moiety,26a–d,33 we have shown that upon incorporation of the diazocine moiety, the PET process for the resulting naphthalimide Tröger's base is modulated, allowing for such PET quenching to occur.33
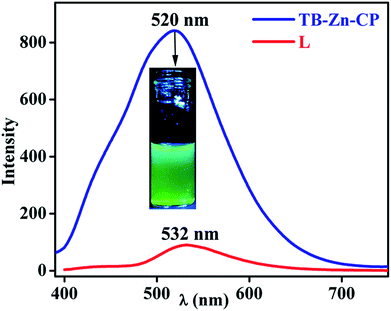 |
| | Fig. 3 Fluorescence emission spectra of TB-Zn-CP (λmax = 520 nm) and L (λmax = 532 nm) dispersed in water (inset: visual color of TB-Zn-CP taken under UV lamp). | |
Discriminative detection of nitroaromatics explosives
As discussed above, the strong luminescence characteristics of TB-Zn-CP in aqueous medium was the driving force for evaluating the polymer as a fluorescent sensor for NACs. The following NAC analytes were chosen for the fluorescence sensing studies: 4-nitrotoluene (4-NT), 2,6-dinitrotoluene (2,6-DNT), 3-nitrotoluene (3-NT), 2,4-dinitrotoluene (2,4-DNT), nitrobenzene (NB), 2-nitrotoluene (2-NT), 3-nitrophenol (3-NP), 2-nitrophenol (2-NP), 4-nitrophenol (4-NP), 2,4-dinitrophenol (2,4-DNP) 2,4,6-trinitrotoluene (TNT) and picric acid (PA). To ensure complete solubility, stock solutions of all the analytes were prepared in a water–ethanol (9:1) solvent mixture and all titrations were preformed in triplicate. From a practical point of view, the detection of nitroaromatics in an aqueous medium is highly desirable and hence all the fluorescence titration experiments were performed in aqueous medium.
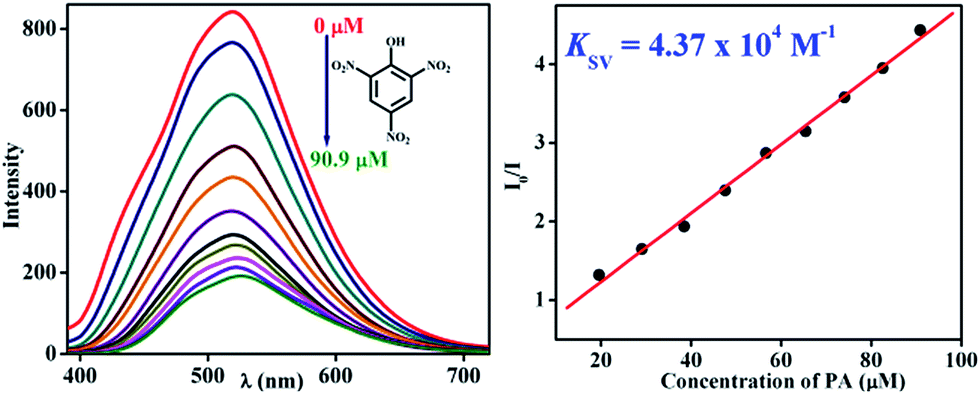
To demonstrate the ability of TB-Zn-CP for the detection of nitroaromatic explosives, we first performed fluorescence titration experiments of TB-Zn-CP with PA as representative example of NACs. Concomitantly, the absorption spectra were recorded, but no significant changes were seen in the ICT band. In contrast, the gradual addition (20–200 μL) of 1 mM PA solution to an aqueous suspension of TB-Zn-CP elicited significant quenching in the fluorescence emission, as demonstrated in Fig. 4; with no other significant spectral changes (such as shifts in λmax) being observed. The TB-Zn-CP emission responses were analysed by fitting the data to a Stern–Volmer equation:
where;
I0 is the initial fluorescence intensity before the addition of analyte,
I is the fluorescence intensity in the presence of analyte, [Q] is the molar concentration of analytes, and
KSV is the Stern–Volmer constant. The result for the PA titration is also shown in
Fig. 4, where a linear Stern–Volmer plot was obtained from the fluorescence quenching titration profile, and the calculated Stern–Volmer constant
KSV was determined as 4.37 × 10
4 M
−1. This demonstrates both high binding affinity and a significant improvement over the current state of the art, as previously reported binding values for other Zn(
II)-based luminescent coordination polymers are usually determined in less competitive media or in organic solvents, which makes them significantly less useful for practical applications.
15f,16a,31,34 The calculated higher rate of binding constant (
KSV) is most likely due to the long-range electronic communication between the adjacent fluorophores upon Zn(
II)-coordination.
14b,16c,35 The large spectral overlaps in the absorption spectrum of PA with the emission maximum of
TB-Zn-CP (see Fig. S25, ESI
†) confirms that an excited state energy transfer mechanism operates between
TB-Zn-CP and PA.
15e,24b We propose that the reason for the fluorescence quenching is due to efficient charge-transfer (CT) complex formation between the electron-rich Tröger's base coordination polymer and electron-poor PA, preferably
via hydrogen bonding.
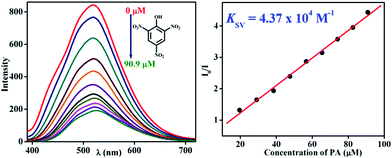 |
| | Fig. 4 Observed fluorescence quenching (left) of TB-Zn-CP upon addition of PA (μM) in water and its corresponding Stern–Volmer plot (right). | |
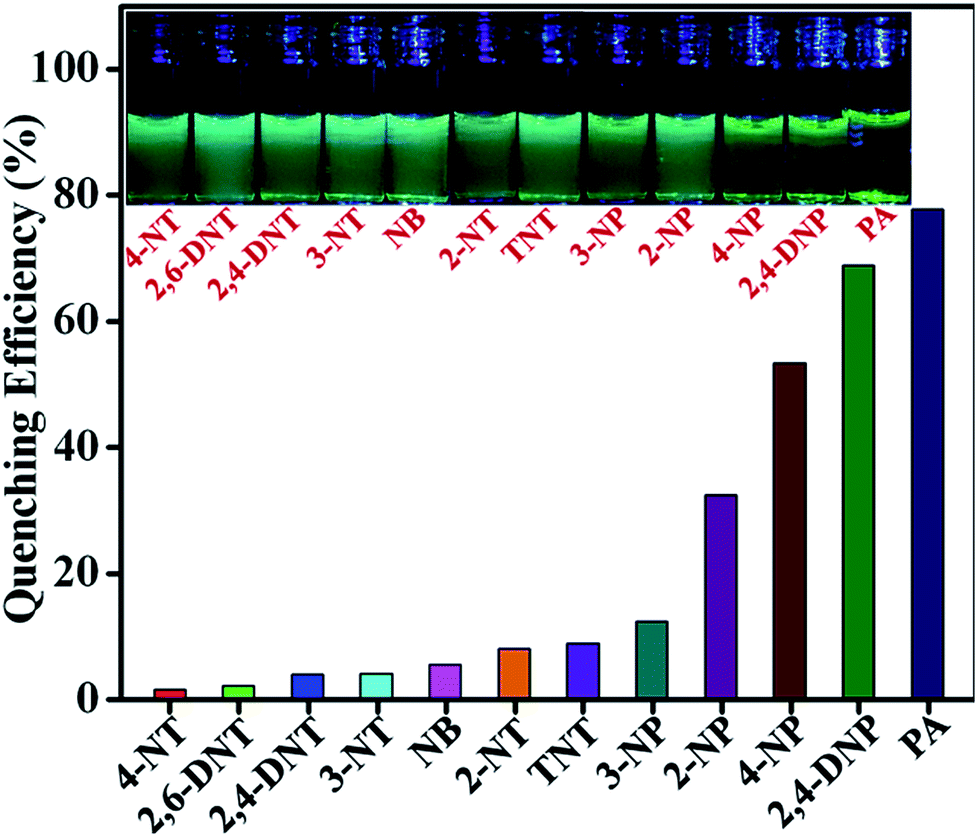
To explore the selectivity of TB-Zn-CP towards NACs, we performed similar fluorescence titrations, under identical spectroscopic conditions, with the other electron deficient NACs listed above. For these, TB-Zn-CP exhibited different fluorescence quenching responses, which allowed us to discriminate between structurally similar NACs. As shown in Fig. 5, the titrations with phenolic nitroaromatics such as PA, 2,4-DNP, 4-NP resulted in the highest quenching effect compared to other NACs; the changes also being clearly visible to the naked eye. Here, PA displayed the highest quenching response (78%) due to its more electron deficient nature because of the strong electron-withdrawing tendency of the substituted –NO2 groups. Notably, the non-phenolic NACs such as NB, 2-NT, 4-NT, 4-NT, 2,6-DNT, 2,4-DNT and TNT exhibited moderate to poor quenching effects (Fig S26–S35, ESI†). The descending order of quenching efficiencies are as follows: PA > 2,4-DNP > 4-NP > 2-NP > 3-NP > TNT > 2-NT > NB > 3-NT > 2,4-DNT > 2,6-DNT > 4-NT. These results clearly indicate the high selectivity of TB-Zn-CP towards phenolic nitroaromatics over other potentially interfering non-phenolic nitroaromatics. We believe that the highly selective binding of phenolic nitroaromatics is ascribed to the strong intermolecular interactions, particular due to the interactions between the –OH group and Lewis basic –N of the Tröger's base structural motif.5b The observed differential fluorescence quenching is also indicated by sharp visual colour changes as is evident from the inset in Fig. 5.
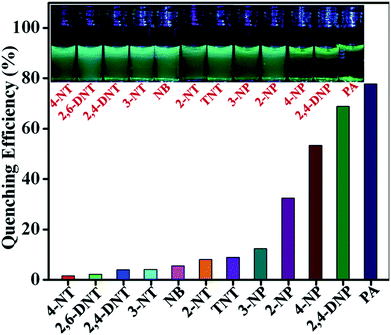 |
| | Fig. 5 Extent of fluorescence quenching of TB-Zn-CP observed upon the addition of various analytes. Inset: the colour changes observed. | |
The high selectivity of TB-Zn-CP towards these aromatics was further validated by carrying out a competitive fluorescence titration study using PA in the presence of other potentially competing nitroaromatic analytes. The results from this are shown in Fig. 6, where the initial emission (red bar) intensity of TB-Zn-CP did not change significantly, except for 4-NP and 2,4-DNP, upon mixing with other competing NACs (green bar). However, the subsequent addition of PA elicited a discernable decrease in the emission intensity (blue bar), which confirmed the discriminative sensing ability of TB-Zn-CP towards phenolic nitroaromatics in competing aqueous media. In general, the fluorescence quenching based detection of NACs follow two different mechanisms, e.g. static and dynamic or collisional quenching.9a,36 The static and dynamic quenching are simple PET or energy transfer from the excited state fluorophores to the analytes; this being characterised by a linear Stern–Volmer plot.36a In static quenching, the fluorophore binds with the analyte in the ground state through non-fluorescent charge-transfer complex formation and it solely depends on the extent of binding between the sensor and analyte. Dynamic quenching is an excited state process, in which the analyte binds with the fluorophore in the excited state and it depends on both the rate of collision of and lifetime of the fluorophore. Therefore, the static and dynamic quenching mechanisms can be distinguished by analysing the time-resolved fluorescence decay of the fluorophore at different analyte concentrations and the effect of temperature on the extent of fluorescence quenching.1b,3b,9a,37
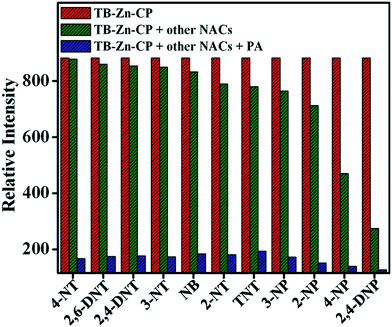 |
| | Fig. 6 Competitive selective binding affinity of TB-Zn-CP towards different NACs in the presence of PA in aqueous medium. | |
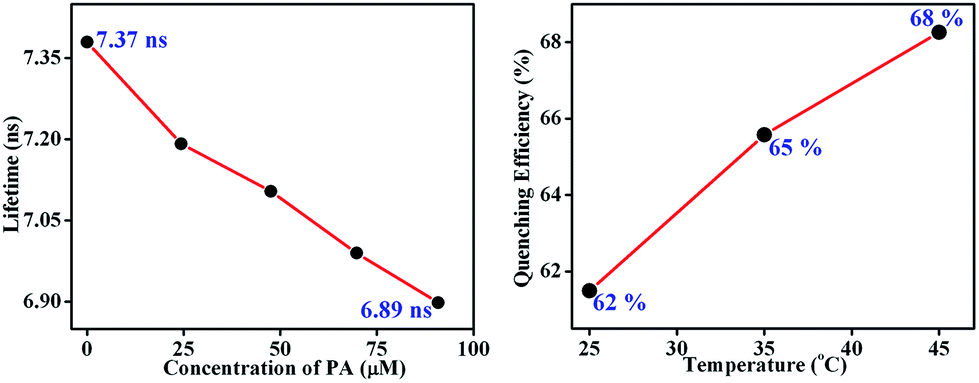
Here, the time-dependent fluorescent decay and temperature dependent fluorescence studies indicated that the observed fluorescence quenching of TB-Zn-CP, upon the addition of PA, was due to the dynamic quenching mechanism. As shown in Fig. 7 (left), the decrease in the lifetime (τ = 7.37 ns) of TB-Zn-CP upon increasing the concentration of PA (0 → 90.9 μM) revealed that PA binds with TB-Zn-CP in the excited state. Furthermore, the temperature dependent fluorescence titration study showed that the quenching enhanced as a function of increasing temperature, Fig. 7 (right). The estimated quenching efficiency of TB-Zn-CP at 25 °C was ca. 62%, being increased to 68% upon elevating the temperature to 45 °C. The increase in quenching efficiency at high temperature is also due to the increased molecular collision between the two species. These two results confirmed that the observed fluorescence quenching occurs through the dynamic quenching mechanistic pathway. The calculated quenching constant varies from 0.40 × 1011 M−1 S−1 to 59.29 × 1011 M−1 S−1 and the results are summarized in Table 1. The higher values of the quenching constant further corroborate the dynamic quenching pathway.18a,38
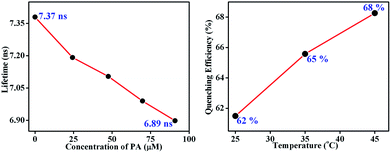 |
| | Fig. 7 Fluorescence decay profile (left) of the aqueous suspension of TB-Zn-CP upon the addition of PA in different concentrations (0–90.9 μM) (left) and extent of fluorescence quenching (right) observed after the addition of PA (47.6 μM) at different temperatures (25–45 °C). | |
Table 1 The quenching constants Kq and KSV of various nitroaromatics for the fluorescence quenching of TB-Zn-CP
| Analytes |
K
SV (M−1) × 103 |
K
q (M−1 S−1) × 1011 |
| 4-NT |
0.30 |
0.40 |
| 2,6-DNT |
0.33 |
0.44 |
| 2,4-DNT |
0.59 |
0.80 |
| 3-NT |
0.47 |
0.63 |
| NB |
1.17 |
1.58 |
| TNT |
1.09 |
1.47 |
| 2-NT |
1.53 |
2.07 |
| 3-NP |
1.71 |
2.32 |
| 2-NP |
5.29 |
7.17 |
| 4-NP |
12.6 |
17.09 |
| 2,4-DNP |
24.6 |
33.37 |
| PA |
43.7 |
59.29 |
Fluorescence quenching response time
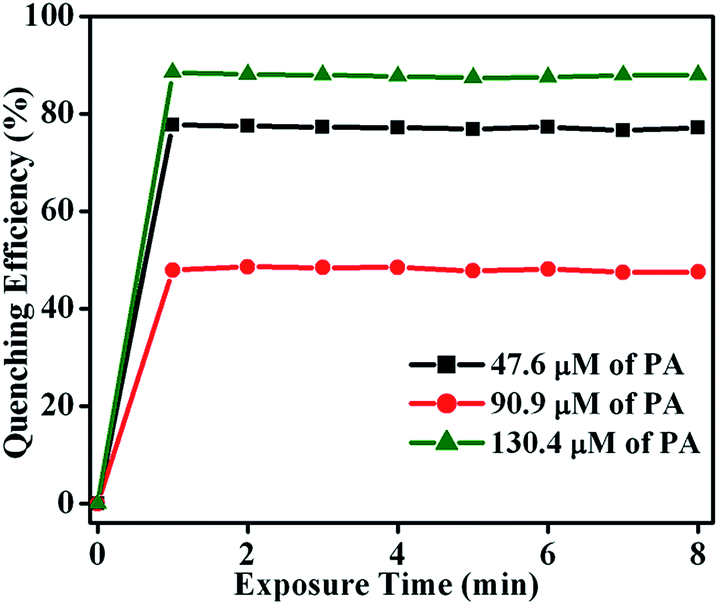
To determine the fluorescence quenching response time for the detection of NACs by TB-Zn-CP, time-dependent fluorescence titration experiments were carried using varyious concentrations of PA (47.6 → 130.4 μM). The plot of quenching efficiency vs. exposure time is shown in Fig. 8, demonstrating a fast response time for the sensing of PA. For instance, the addition of PA (130.4 μM) caused ∼90% fluorescence quenching of TB-Zn-CP within 60 s of contact time and thus TB-Zn-CP can be used as a potential sensor for the quick detection of NACs.
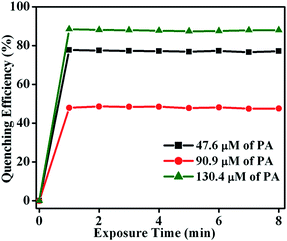 |
| | Fig. 8 Plot of quenching efficiency as a function of exposure time (0 → 8 min) monitored at different concentrations of PA (47.6 → 130.4 μM). | |
Sensitivity
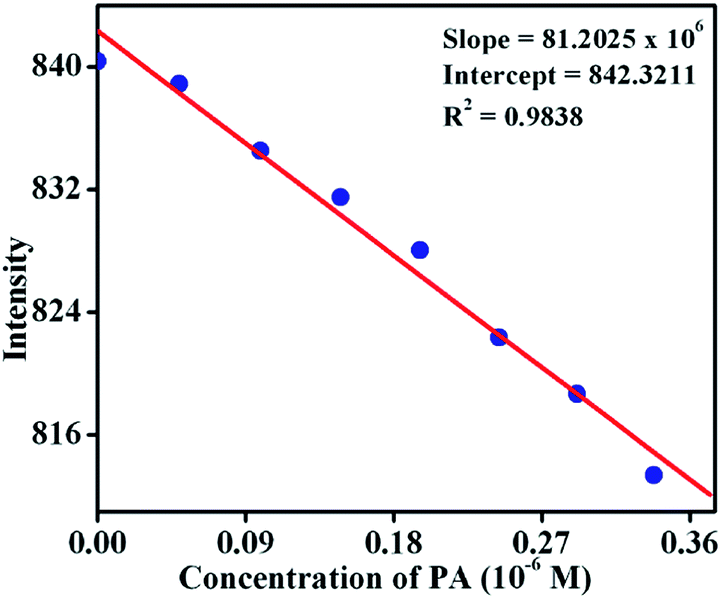
To establish the sensitivity of TB-Zn-CP towards phenolic-nitroaromatics, the fluorescence quenching titration studies were also performed at a very low concentration of PA; the detection limit being estimated by using:
where σ is the standard deviation of the initial emission intensity in the absence of the analyte and K is the slope of the linear calibration curve. As shown in Fig. 9, the plot of the change in fluorescence intensity versus PA concentration, resulted in a linear curve with slope K = 81.2 × 106 and a correlation coefficient of R2 = 0.9838. The quantitative analysis of the fluorescence titration profile revealed that TB-Zn-CP can respond to the presence of PA as low as the 26.3 ppb level of concentration which is comparable with other reported coordination polymer based fluorescence sensors when measured in organic solutions and this level of sensitivity comes under the allowed limit of NACs in drinking water as established by the US EPA.8,10a Hence, the results for TB-Zn-CP demonstrate that the material can function both in competitive media and within the recommended limit of detection which is highly significant for practical applications in the field.
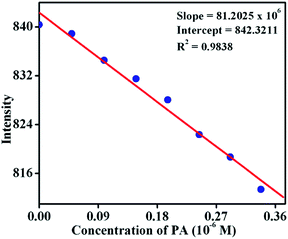 |
| | Fig. 9 Change in fluorescence intensity of TB-Zn-CP at different concentrations of PA. | |
Reversibility
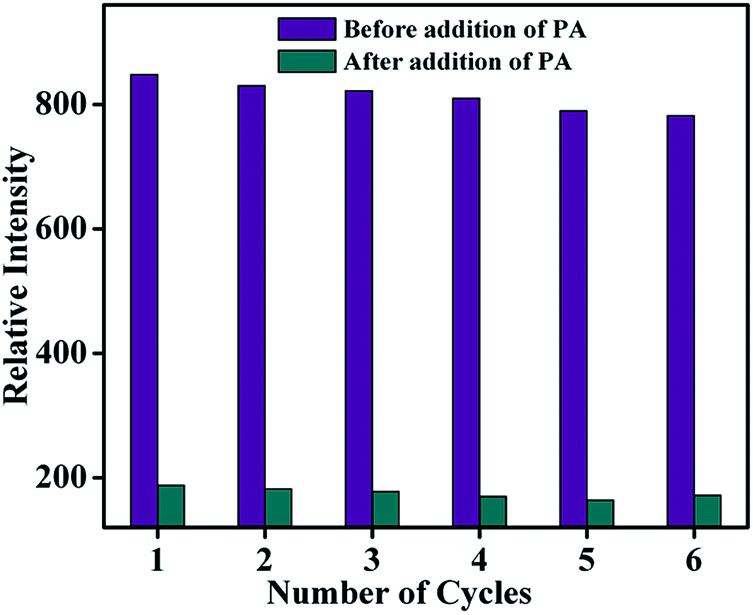
As far as a real-time application is concerned, the sensing ability of the sensor material must be reversible. The reproducibility of the sensing process of TB-Zn-CP towards PA was thus investigated. The emission intensity of TB-Zn-CP was first recorded in the presence of PA at 90.9 μM concentration and after each measurement, the material was collected by centrifugation and washed several times with water, or until the centrifugate became colourless. After six cycles of repetition, almost 92% of the initial emission intensity was retained and thus TB-Zn-CP showed excellent recyclability as demonstrated in Fig. 10. This study also revealed the excellent photostability of TB-Zn-CP demonstrating that the material can be used as a luminescent sensor for a significant number of sensing cycles.3a,6a,14c,18c
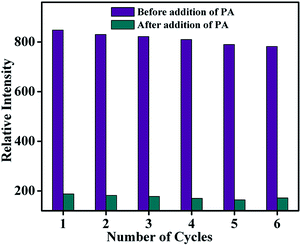 |
| | Fig. 10 Reproducibility of the quenching ability of TB-Zn-CP for PA dispersed in water. The material was recovered by centrifugation after each experiment and washed several times with water. | |
Conclusions
Herein we present our results on the design, synthesis and physical evaluation of a nitrogen-rich supramolecular Zn(II) coordination polymer TB-Zn-CP incorporating a 4-amino-1,8-naphthalimide derived Tröger's base structural motif as a sensory material for the detection of nitro-aromatic based explosives. The phase-purity including the morphological features were analysed by using various spectroscopy and microscopy techniques, as well as gas uptake studies. We show that the aqueous suspension of activated TB-Zn-CP showed strong fluorescence characteristics, this being assigned to the ICT character of the naphthalimide structure, and this property was used as a selective and discriminative fluorescence sensor for NACs. The gradual addition of NACs caused poor to significant quenching in the fluorescence emission of TB-Zn-CP; the observed quenching being ascribed to the excited state energy transfer from TB-Zn-CP to the nitroaromatics. The differential fluorescence quenching responses of TB-Zn-CP for closely related nitroaromatics, demonstrated significant selectivity of TB-Zn-CP for phenolic nitroaromatics.23b Notably, the ability of TB-Zn-CP to discriminate PA over TNT (both are marginally different in their chemical structure) was an interesting observation that has seldom been seen with other Zn2+ based coordination polymers. The time-resolved and temperature dependent fluorescence titration studies confirmed that the fluorescence quenching occurred through dynamic quenching; the detection limit for the sensing of PA being determined to be 26.3 ppb, which is comparable with other d10 (Zn2+ and Cd2+) metal based coordination polymers designed for such applications.7a,16a Recyclability experiments revealed that TB-Zn-CP was highly photo-stable and that the sensing was reversible; demonstrating that TB-Zn-CP can be used for long-term infield sensing of NACs. In summary, the high selectivity and discriminative sensing ability including superior sensitivity and reversibility make TB-Zn-CP a promising sensor material for the rapid detection of phenolic-nitroaromatics.
Experimental
Materials
Starting materials were obtained from Sigma Aldrich, Strem Chemicals and Fluka. Solvents used were HPLC grade unless otherwise stated. 1H NMR spectra were recorded at 400 MHz using a Bruker Spectrospin DPX-400, with chemical shifts expressed in parts per million (ppm or d) downfield from the standard. 13C NMR spectra were recorded at 100 MHz using a Bruker Spectrospin DPX-400 instrument. Infrared spectra were recorded on a Mattson Genesis II FTIR spectrophotometer equipped with a Gateway 2000 4DX2-66 workstation. Mass spectroscopy was carried out using HPLC grade solvents. Mass spectra were determined by detection using Electrospray on a Micromass LCT spectrometer, using a Water's 9360 to pump solvent. The system was controlled by Mass Lynx 3.5 on a Compaq Deskpro workstation.
Synthesis
N-(5-Dimethyl-isophthalate)-4-nitro-1,8-naphthalimide (1).
4-Nitro-1,8-naphthalic anhydride (3.50 g, 14.4 mmol, 1.0 eq.), dimethyl-5-aminoisophthalate (6.02 g, 28.8 mmol, 2.0 eq.) and imidazole (21.57 g, 316.8 mmol, 22.0 eq.) were dissolved in chloroform (150 mL) and stirred at reflux for 2 h at 80 °C. The solvents were removed under reduced pressure and the residue was suspended into ice-cold ethanol and sonicated for 15 min. The solution was filtered and the product was washed with ice cold ethanol (3 × 150 mL), dried via vacuum filtration and isolated as an off-white powder (4.9 g, 11.3 mmol, 78%) after triturating with CHCl3. Melting point 326–327 °C (decomp.). HRMS (m/z): calcd for C22H14N2O8 434.0828; found 434.2694 [M − H]−. 1H NMR (600 MHz, (CD3)2SO) δ 8.80 (1H, d, J = 8.3 Hz, Ar-H), 8.67 (1H, d, J = 7.6 Hz, Ar-H), 8.65 (1H, d, J = 7.8 Hz, Ar-H), 8.62 (1H, d, J = 7.8 Hz, Ar-H), 8.60 (1H, t, J = 6.6 Hz, Ar-H), 8.38 (2H, d, J = 1.54 Hz, Ar-H), 8.12 (1H, dd, J = 10.8 Hz, 8.3 Hz, Ar-H), 3.92 (6H, s, CH3–H); 13C NMR (151 MHz, (CD3)2SO) δ 170.1, 164.1, 163.2, 162.4, 161.2, 149.3, 136.7, 134.5, 131.6, 131.1, 130.1, 129.4, 128.9, 128.8, 127.2, 124.2, 123.4, 52.6. IR νmax (neat sample, cm−1) 3106, 3081, 2957, 1729, 1709, 1669, 1623, 1582, 1526, 1432, 1353, 1334, 1242, 1196, 1156, 1132, 988, 908, 882, 843, 784, 751, 682. UV-vis (DMSO) λmax(ε) [nm (M−1 cm−1)]: 350 (3953). Non-emissive due to the lack of internal charge transfer (ICT) transition.
N-(5-Dimethyl-isophthalate)-4-amino-1,8-naphthalimide (2).
Compound 1 (200 mg, 0.460 mmol, 1 eq.) was reduced by catalytic hydrogenation using Pd/C (10%, 20 mg) at 3 atm of H2 in C2H5OH:DMF (9:1, 10 mL) for 12 h. The mixture was diluted with DCM:CH3OH (1:1, 400 mL) and stirred in the dark for 1 h. The reaction mixture was then filtered through celite and washed several times with DCM:CH3OH (1:1) until the washings ran clear. The solvents were removed under reduced pressure to isolate compound 2 (48 mg, 0.119 mmol, 25%) as an orange solid after trituration with cold-diethylether. Melting point 317–319 °C (decomp.). HRMS (m/z): calcd for C22H15N2O6 403.1008; found 403.0932 (M − H)−; 1H NMR (600 MHz, (CD3)2SO) δ 8.64 (1H, d, J = 8.11 Hz, Ar-H), 8.53 (1H, s, Ar-H), 8.44 (1H, d, J = 7.70 Hz, Ar-H), 8.21 (2H, d, J = 7.50 Hz, Ar-H), 8.19 (1H, s, Ar-H), 7.69 (1H, t, Ar-H), 7.53 (2H, s, NH2), 6.89 (1H, d, J = 8.20 Hz, Ar-H), 3.91 (6H, s, CH3–H); 13C NMR (151 MHz, (CD3)2SO) δ 165.9, 164.9, 164.0, 154.0, 138.7, 135.7, 135.5, 135.1, 132.1, 131.8, 131.3, 130.6, 129.9, 124.9, 123.2, 120.5, 109.1, 108.6, 53.6. IR νmax (neat sample, cm−1) 3427, 3368, 3266, 1732, 1688, 1639, 1580, 1532, 1428, 1373, 1329, 1248, 1145, 996, 894, 835, 755, 685. UV-vis (DMSO) λmax(ε) [nm (M−1 cm−1)]: 273 (11423), 438 (7538). Emission (DMSO) λmax (nm) = 533.
Bis-[N-(5-dimethyl-isophthalate)]-9,18-methano-1,8-naphthalimide-[b,f][1,5]diazocine (3).
Compound 2 (150 mg, 0.371 mmol, 1 eq.) and paraformaldehyde (17 mg, 0.556 mmol, 1.5 eq.) were flushed with argon. Trifluoroacetic acid (3 mL) was added at 0 °C and the solution was stirred at room temperature for overnight under an inert atmosphere. The reaction mixture was added dropwise to aqueous ammonium hydroxide (100 mL) at 0 °C and ammonia solution was added until a pH > 10 was achieved. DCM (200 mL) was added and the organic solution was extracted and washed with saturated NaHCO3, brine, and H2O. The solution was dried over MgSO4 and the solvents were removed under reduced pressure. The solid was triturated in Et2O and isolated via filtration as an orange solid (64 mg, 0.076 mmol, 20%). Melting point 293–295 °C (decomp.). HRMS (m/z): calcd for C47H33N4O12 845.2017; found 845.2095 (M + H)+; 1H NMR (600 MHz, (CD3)2SO) δ 8.77 (2H, d, J = 7.90 Hz, Ar-H), 8.49 (2H, t, J = 6.00 Hz, Ar-H), 8.46 (2H, d, J = 7.40 Hz, Ar-H), 8.19 (4H, s, Ar-H), 8.12 (2H, s, Ar-H), 7.97 (2H, t, J = 12.00 Hz, Ar-H), 5.24 (2H, d, J = 17.57 Hz, CH2N–H), 4.79 (2H, s, NCH2N–H), 4.74 (2H, d, J = 17.47 Hz, CH2N–H), 3.89 (12H, s, CH3–H); 13C NMR (151 MHz, (CD3)2SO) δ 164.8, 163.6, 163.0, 149.2, 137.1, 134.6, 130.9, 130.5, 130.2, 129.2, 128.1, 127.1, 126.7, 126.1, 123.1, 118.2, 52.6; IR νmax (neat sample, cm−1) 2953, 1707, 1667, 1596, 1347, 1239, 1180, 998, 783, 753. UV-vis (DMSO) λmax(ε) [nm (M−1 cm−1)]: 347 (13058), 390 (15503). Emission (DMSO) λmax (nm) = 545.
Bis-[N-(1,3-benzenedicarboxylic acid)]-9,18-methano-1,8-naphthalimide-[b,f][1,5]diazocine (L).
Compound 3 (50 mg, 0.059 mmol, 1 eq.), was dissolved in THF:MeOH (4:1, 20 mL) and stirred for 10 min. KOH (aq.) (33 mg, 0.592 mmol, 10 mL, 10 eq.) was added and the solution was refluxed at 60 °C for 12 h. Solvents removed under reduced pressure, leaving water. The solution was acidified using HCl (1 M) until pH = 1 as achieved. The acidic solution was left to stir for 12 h and was filtered, washed with water, dried with diethyl ether and isolated as a yellow powder (30 mg, 0.038 mmol, 65%). Melting point 262–263 °C (decomp.). HRMS (m/z): calcd for C43H23N4O12 787.1319; found 787.1308 (M − H)−; 1H NMR (600 MHz, (CD3)2SO) δ 13.41 (4H, broad, COOH), 8.81 (2H, d, J = 8.51 Hz, Ar-H), 8.51 (2H, m, Ar-H), 8.47 (2H, s, Ar-H), 8.15 (2H, s, J = 10.00 Hz, Ar-H), 8.11 (4H, s, Ar-H), 7.97 (2H, t, J = 12.00 Hz, Ar-H), 5.24 (2H, d, J = 17.58 Hz, NCH2–H), 4.78 (2H, s, NCH2N), 4.74 (2H, d, J = 17.58 Hz, NCH2–H); 13C NMR (151 MHz, (CD3)2SO) δ 166.9, 164.6, 164.0, 150.1, 137.6, 135.2, 133.0, 131.5, 131.1, 130.5, 130.2, 129.0, 128.0, 127.7, 127.0, 124.0, 119.2, 67.0, 57.8. IR νmax (neat sample, cm−1) 3350, 2971, 1705, 1659, 1596, 1459, 1373, 1241, 1191, 1126, 948, 814, 785, 678. UV-vis (DMSO) λmax(ε) [nm (M−1 cm−1)]: 347 (13939), 392 (16650). Emission (DMSO) λmax (nm) = 532.
Synthesis of TB-Zn-CP.
A DMF (2 mL) solution of Zn(OAc)2·2H2O (16.7 mg, 0.08 mmol, 1.0 eq.) was added drop-wise to a clear DMF (2 mL) solution of L (30.0 mg, 0.04 mmol, 0.5 eq.) with continues stirring. Upon addition of Zn(OAc)2·2H2O, the clear orange solution of L gradually turned turbid, indicating the formation of coordination polymers, and the turbid solution was stirred gently for further 6 hours at room temperature to complete the reaction. The orange colloids formed were centrifuged, thoroughly washed with DMF (3 × 5 mL) to remove the unreacted starting materials, and then dried under vacuum for 6 h. The isolated orange colloidal particles were characterized by different techniques. Isolated yield = 28 mg (73%). Elemental analysis calculated for C43H20N4O12Zn·2H2O·DMF: C, 57.60; H, 3.26; N, 7.30; found: C, 57.54; H, 3.73; N, 7.33. FTIR νmax (neat sample, cm−1) 3434, 2981, 2157, 1657, 1573, 1458, 1402, 1371, 1249, 923, 785, 678. UV-vis (H2O) λmax(ε) [nm (M−1 cm−1)]: 351 (7485), 380 (8374). Emission (H2O) λmax (nm) = 520.
Acknowledgements
We would like to gratefully acknowledge the Irish Research Council for a postdoctoral fellowship (GOIPD/2013/442 to S. S.) and Science Foundation Ireland (SFI PI Awards 10/IN.1/B2999 and 13/IA/1865), and School of Chemistry, TCD, for the financial support. Authors are thankful to Advanced Microscopy Laboratory, CRANN for the SEM and EDX analysis. We thank in particular Dr Emma B. Veale (University of Dublin, Trinity College Dublin) for her help, stimulating discussions and support during this project, and for bringing the use of the 4-amino-1,8-naphthalimide Tröger's base idea to our laboratory. We also thank Dr J. E. O'Brien, Dr M. Ruether, Dr M. Feeney and Dr G. Hessman for carrying out NMR and MS characterisation of the intermediates and final products developed herein.
Notes and references
-
(a) D. Banerjee, Z. Hu and J. Li, Dalton Trans., 2014, 43, 10668–10685 RSC
 ;
(b) S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC ;
(c) D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC ;
(d) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed ;
(e) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed ;
(f) K. L. Diehl and E. V. Anslyn, Chem. Soc. Rev., 2013, 42, 8596–8611 RSC ;
(g) G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437 CrossRef CAS PubMed .
;
(b) S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC ;
(c) D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC ;
(d) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed ;
(e) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed ;
(f) K. L. Diehl and E. V. Anslyn, Chem. Soc. Rev., 2013, 42, 8596–8611 RSC ;
(g) G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437 CrossRef CAS PubMed .
-
(a) X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC ;
(b) Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC ;
(c) G. V. Zyryanov, D. S. Kopchuk, I. S. Kovalev, E. V. Nosova, V. L. Rusinov and O. N. Chupakhin, Russ. Chem. Rev., 2014, 83, 783 CrossRef .
-
(a) S. Shanmugaraju, H. Jadhav, R. Karthik and P. S. Mukherjee, RSC Adv., 2013, 3, 4940–4950 RSC ;
(b) S. Shanmugaraju and P. S. Mukherjee, Chem.–Eur. J., 2015, 21, 6656–6666 CrossRef CAS PubMed ;
(c) S. Ghosh and P. S. Mukherjee, Organometallics, 2008, 27, 316–319 CrossRef CAS ;
(d) S. Ghosh, B. Gole, A. K. Bar and P. S. Mukherjee, Organometallics, 2009, 28, 4288–4296 CrossRef CAS .
-
(a) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC ;
(b) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, CrystEngComm, 2016, 18, 2994–3007 RSC ;
(c) N. Venkatramaiah, S. Kumar and S. Patil, Chem. Commun., 2012, 48, 5007–5009 RSC ;
(d) M. K. Chahal and M. Sankar, Anal. Methods, 2015, 7, 10272–10279 RSC .
-
(a)
P. S. Mukherjee, P. Sunipa and S. Shanmugaraju, in Molecular Self-Assembly, Pan Stanford Publishing, 2012, pp. 259–299 Search PubMed ;
(b) M. A. Ivy, L. T. Gallagher, A. D. Ellington and E. V. Anslyn, Chem. Sci., 2012, 3, 1773–1779 RSC ;
(c) S. Pramanik, Z. Hu, X. Zhang, C. Zheng, S. Kelly and J. Li, Chem.–Eur. J., 2013, 19, 15964–15971 CrossRef CAS PubMed .
-
(a) S. Shanmugaraju, S. A. Joshi and P. S. Mukherjee, J. Mater. Chem., 2011, 21, 9130–9138 RSC ;
(b) S. Singh, J. Hazard. Mater., 2007, 144, 15–28 CrossRef CAS PubMed ;
(c)
S. Talmage, D. Opresko, C. Maxwell, C. E. Welsh, F. M. Cretella, P. Reno and F. B. Daniel, in Rev. Environ. Contam T., ed. G. Ware, Springer, New York, 1999, vol. 161, pp. 1–156 Search PubMed ;
(d) S. Barman, J. Anand Garg, O. Blacque, K. Venkatesan and H. Berke, Chem. Commun., 2012, 48, 11127–11129 RSC .
-
(a) L. Zhang, Z. Kang, X. Xin and D. Sun, CrystEngComm, 2016, 18, 193–206 RSC ;
(b) Q.-H. Tan, Y.-Q. Wang, X.-Y. Guo, H.-T. Liu and Z.-L. Liu, RSC Adv., 2016, 6, 61725–61731 RSC ;
(c) R.-B. Fu, S.-M. Hu and X.-T. Wu, Cryst. Growth Des., 2016, 16, 5074–5083 CrossRef CAS .
-
(a)
Environmental Protection Agency, innovative treatment technologies: Annual status report, 8th edn, 1996 Search PubMed ;
(b) Y. H. Lee, H. Liu, J. Y. Lee, S. H. Kim, S. K. Kim, J. L. Sessler, Y. Kim and J. S. Kim, Chem.–Eur. J., 2010, 16, 5895–5901 CrossRef CAS PubMed .
-
(a) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC ;
(b) K. K. Kartha, A. Sandeep, V. K. Praveen and A. Ajayaghosh, Chem. Rec., 2015, 15, 252–265 CrossRef CAS PubMed ;
(c) S. Shanmugaraju, H. Jadhav and P. Mukherjee, Proc. Natl. Acad. Sci., India, Sect. A, 2014, 84, 197–203 CrossRef CAS .
-
(a) A. Buragohain, M. Yousufuddin, M. Sarma and S. Biswas, Cryst. Growth Des., 2016, 16, 842–851 CrossRef CAS ;
(b) M. Sk and S. Biswas, CrystEngComm, 2016, 18, 3104–3113 RSC .
-
(a) D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499–2512 CrossRef CAS ;
(b) K. Håkansson, R. V. Coorey, R. A. Zubarev, V. L. Talrose and P. Håkansson, J. Mass Spectrom., 2000, 35, 337–346 CrossRef ;
(c) R. D. Luggar, M. J. Farquharson, J. A. Horrocks and R. J. Lacey, X-Ray Spectrom., 1998, 27, 87–94 CrossRef CAS ;
(d) J. M. Sylvia, J. A. Janni, J. D. Klein and K. M. Spencer, Anal. Chem., 2000, 72, 5834–5840 CrossRef CAS PubMed ;
(e) V. P. Anferov, G. V. Mozjoukhine and R. Fisher, Rev. Sci. Instrum., 2000, 71, 1656–1659 CrossRef CAS .
-
(a)
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed ;
(b) T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589–598 CAS ;
(c) E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed ;
(d) S. J. Bradberry, A. J. Savyasachi, M. Martinez-Calvo and T. Gunnlaugsson, Coord. Chem. Rev., 2014, 273–274, 226–241 CrossRef CAS ;
(e) G. Tobin, S. Comby, N. Zhu, R. Clerac, T. Gunnlaugsson and W. Schmitt, Chem. Commun., 2015, 51, 13313–13316 RSC .
-
(a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed ;
(b) W. Guan, W. Zhou, J. Lu and C. Lu, Chem. Soc. Rev., 2015, 44, 6981–7009 RSC .
-
(a) A. K. Bar, S. Shanmugaraju, K.-W. Chi and P. S. Mukherjee, Dalton Trans., 2011, 40, 2257–2267 RSC ;
(b) B. Gole, S. Shanmugaraju, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 10046–10048 RSC ;
(c) S. Shanmugaraju, H. Jadhav, Y. P. Patil and P. S. Mukherjee, Inorg. Chem., 2012, 51, 13072–13074 CrossRef CAS PubMed ;
(d) S. Shanmugaraju, S. A. Joshi and P. S. Mukherjee, Inorg. Chem., 2011, 50, 11736–11745 CrossRef CAS PubMed ;
(e) S. Shanmugaraju, D. Samanta, B. Gole and P. S. Mukherjee, Dalton Trans., 2011, 40, 12333–12341 RSC ;
(f) M. Wang, V. Vajpayee, S. Shanmugaraju, Y.-R. Zheng, Z. Zhao, H. Kim, P. S. Mukherjee, K.-W. Chi and P. J. Stang, Inorg. Chem., 2011, 50, 1506–1512 CrossRef CAS PubMed .
-
(a) S. Rochat and T. M. Swager, ACS Appl. Mater. Interfaces, 2013, 5, 4488–4502 CrossRef CAS PubMed ;
(b) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed ;
(c) S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC ;
(d) A. McCluskey, C. I. Holdsworth and M. C. Bowyer, Org. Biomol. Chem., 2007, 5, 3233–3244 RSC ;
(e) S. Mukherjee, A. V. Desai, B. Manna, A. I. Inamdar and S. K. Ghosh, Cryst. Growth Des., 2015, 15, 4627–4634 CrossRef CAS ;
(f) B. Gole, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137–12139 RSC ;
(g) G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC ;
(h) G. Tang, S. S. Y. Chen, P. E. Shaw, K. Hegedus, X. Wang, P. L. Burn and P. Meredith, Polym. Chem., 2011, 2, 2360–2368 RSC ;
(i) V. Balzani, P. Ceroni, A. Juris, M. Venturi, S. Campagna, F. Puntoriero and S. Serroni, Coord. Chem. Rev., 2001, 219–221, 545–572 CrossRef CAS .
-
(a) T. Cheng, J. Hu, C. Zhou, Y. Wang and M. Zhang, Sci. China: Chem., 2016, 59, 929–947 CrossRef CAS ;
(b) B. Gole, A. K. Bar and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 13321–13336 CrossRef CAS PubMed ;
(c) B. Gole, A. K. Bar and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 2276–2291 CrossRef CAS PubMed ;
(d) D. K. Singha, S. Bhattacharya, P. Majee, S. K. Mondal, M. Kumar and P. Mahata, J. Mater. Chem. A, 2014, 2, 20908–20915 RSC ;
(e) A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed ;
(f) J. A. Kitchen, N. Zhang, A. B. Carter, A. J. Fitzpatrick and G. G. Morgan, J. Coord. Chem., 2016, 69, 2024–2037 CrossRef CAS ;
(g) J. A. Kitchen, P. N. Martinho, G. G. Morgan and T. Gunnlaugsson, Dalton Trans., 2014, 43, 6468–6479 RSC ;
(h) E. B. Veale, J. A. Kitchen and T. Gunnlaugsson, Supramol. Chem., 2013, 25, 101–108 CrossRef CAS .
-
(a) C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed ;
(b) I. Imaz, M. Rubio-Martinez, J. An, I. Sole-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287–7302 RSC ;
(c) A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218–1227 RSC .
-
(a) Y. Li, K. Liu, W.-J. Li, A. Guo, F.-Y. Zhao, H. Liu and W.-J. Ruan, J. Phys. Chem. C, 2015, 119, 28544–28550 CrossRef CAS ;
(b) H. Xu, F. Liu, Y. Cui, B. Chen and G. Qian, Chem. Commun., 2011, 47, 3153–3155 RSC ;
(c) P. Kumar, A. K. Paul and A. Deep, Anal. Methods, 2014, 6, 4095–4101 RSC .
-
(a) V. M. Suresh, S. Chatterjee, R. Modak, V. Tiwari, A. B. Patel, T. K. Kundu and T. K. Maji, J. Phys. Chem. C, 2014, 118, 12241–12249 CrossRef CAS ;
(b) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed .
- C. Zhang, Y. Che, Z. Zhang, X. Yang and L. Zang, Chem. Commun., 2011, 47, 2336–2338 RSC .
- R. Li, Y.-P. Yuan, L.-G. Qiu, W. Zhang and J.-F. Zhu, Small, 2012, 8, 225–230 CrossRef CAS PubMed .
- N. Shi, Y. Zhang, D. Xu, C. Song, X. Jin, D. Liu, L. Xie and W. Huang, New J. Chem., 2015, 39, 9275–9280 RSC .
-
(a) M.-M. Chen, X. Zhou, H.-X. Li, X.-X. Yang and J.-P. Lang, Cryst. Growth Des., 2015, 15, 2753–2760 CrossRef CAS ;
(b) Y. Xu, B. Li, W. Li, J. Zhao, S. Sun and Y. Pang, Chem. Commun., 2013, 49, 4764–4766 RSC ;
(c) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881–2885 CrossRef CAS PubMed .
-
(a) B. Joarder, A. V. Desai, P. Samanta, S. Mukherjee and S. K. Ghosh, Chem.–Eur. J., 2015, 21, 965–969 CrossRef CAS PubMed ;
(b) S. S. Nagarkar, A. V. Desai, P. Samanta and S. K. Ghosh, Dalton Trans., 2015, 44, 15175–15180 RSC ;
(c) B.-Q. Song, C. Qin, Y.-T. Zhang, X.-S. Wu, L. Yang, K.-Z. Shao and Z.-M. Su, Dalton Trans., 2015, 44, 18386–18394 RSC ;
(d) S. Mukherjee, A. V. Desai, A. I. Inamdar, B. Manna and S. K. Ghosh, Cryst. Growth Des., 2015, 15, 3493–3497 CrossRef CAS ;
(e) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, Chem. Commun., 2014, 50, 8915–8918 RSC .
-
(a) M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed ;
(b) M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, M. Lee, I. Rose and N. B. McKeown, Polym. Chem., 2014, 5, 5267–5272 RSC ;
(c) M. Carta, M. Croad, K. Bugler, K. J. Msayib and N. B. McKeown, Polym. Chem., 2014, 5, 5262–5266 RSC ;
(d) M. Carta, M. Croad, J. C. Jansen, P. Bernardo, G. Clarizia and N. B. McKeown, Polym. Chem., 2014, 5, 5255–5261 RSC .
-
(a) E. B. Veale, D. O. Frimannsson, M. Lawler and T. Gunnlaugsson, Org. Lett., 2009, 11, 4040–4043 CrossRef CAS PubMed ;
(b) E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2010, 75, 5513–5525 CrossRef CAS PubMed ;
(c) S. Banerjee, S. A. Bright, J. A. Smith, J. Burgeat, M. Martinez-Calvo, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, J. Org. Chem., 2014, 79, 9272–9283 CrossRef CAS PubMed ;
(d) S. Murphy, S. A. Bright, F. E. Poynton, T. McCabe, J. A. Kitchen, E. B. Veale, D. C. Williams and T. Gunnlaugsson, Org. Biomol. Chem., 2014, 12, 6610–6623 RSC ;
(e) S. Banerjee, J. A. Kitchen, S. A. Bright, J. E. O'Brien, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Commun., 2013, 49, 8522–8524 RSC .
- C. S. Hawes, C. M. Fitchett, S. R. Batten and P. E. Kruger, Inorg. Chim. Acta, 2012, 389, 112–117 CrossRef CAS .
- Examples of naphthalimide structures developed for Zn(II) coordination and sensing:
(a) R. Parkesh, T. C. Lee and T. Gunnlaugsson, Org. Biomol. Chem., 2007, 5, 310 RSC ;
(b) T. Gunnlaugsson, T. C. Lee and R. Parkesh, Org. Biomol. Chem., 2003, 1, 3265 RSC ;
(c) E. Tamanini, A. Katewa, L. M. Sedger, M. H. Todd and M. Watkinson, Inorg. Chem., 2009, 48, 319 CrossRef CAS PubMed ;
(d) E. Tamanini, K. Flavin, M. Motevalli, S. Piperno, L. A. Gheber, M. H. Todd and M. Watkinson, Inorg. Chem., 2010, 49, 3789 CrossRef CAS PubMed ;
(e) K. Jobe, C. H. Brennan, M. Motevalli, S. M. Goldup and M. Watkinson, Chem. Commun., 2011, 47, 6036–6038 RSC ;
(f) J. Pancholi, D. J. Hodson, K. Jobe, G. A. Rutter, S. M. Goldup and M. Watkinson, Chem. Sci., 2014, 5, 3528–3535 RSC .
-
(a) V. M. Suresh, S. J. George and T. K. Maji, Adv. Funct. Mater., 2013, 23, 5585–5590 CrossRef CAS ;
(b) D. Banerjee, Z. Hu, S. Pramanik, X. Zhang, H. Wang and J. Li, CrystEngComm, 2013, 15, 9745–9750 RSC ;
(c) C. C. R. Sutton, G. D. Silva and G. V. Franks, Chem.–Eur. J., 2015, 21, 6801–6805 CrossRef CAS PubMed ;
(d) D. Martinez, M. Motevalli and M. Watkinson, Dalton Trans., 2010, 39, 446–455 RSC ;
(e) G. B. Deacon and R. J. Philips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS .
-
(a) Z.-Z. Yang, H. Zhang, B. Yu, Y. Zhao, G. Ji and Z. Liu, Chem. Commun., 2015, 51, 1271–1274 RSC ;
(b) X. Du, Y. Sun, B. Tan, Q. Teng, X. Yao, C. Su and W. Wang, Chem. Commun., 2010, 46, 970–972 RSC ;
(c) J. Byun, S.-H. Je, H. A. Patel, A. Coskun and C. T. Yavuz, J. Mater. Chem. A, 2014, 2, 12507–12512 RSC ;
(d) S. Ast, S. Kuke, P. J. Rutledge and M. H. Todd, Eur. J. Inorg. Chem., 2015, 58–66 CrossRef CAS .
- M. Guo and Z.-M. Sun, J. Mater. Chem., 2012, 22, 15939–15946 RSC .
-
(a) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC ;
(b) S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC .
-
(a) A. Prasanna de Silva and T. E. Rice, Chem. Commun., 1999, 163–164 RSC ;
(b) B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC .
-
(a) D. Ma, B. Li, X. Zhou, Q. Zhou, K. Liu, G. Zeng, G. Li, Z. Shi and S. Feng, Chem. Commun., 2013, 49, 8964–8966 RSC ;
(b) G.-l. Liu, Y.-j. Qin, L. Jing, G.-y. Wei and H. Li, Chem. Commun., 2013, 49, 1699–1701 RSC ;
(c) D. Singh and C. M. Nagaraja, Dalton Trans., 2014, 43, 17912–17915 RSC ;
(d) X. Jiang, Y. Liu, P. Wu, L. Wang, Q. Wang, G. Zhu, X.-l. Li and J. Wang, RSC Adv., 2014, 4, 47357–47360 RSC ;
(e) S. Sanda, S. Parshamoni, S. Biswas and S. Konar, Chem. Commun., 2015, 51, 6576–6579 RSC ;
(f) M. Jurcic, W. J. Peveler, C. N. Savory, D. O. Scanlon, A. J. Kenyon and I. P. Parkin, J. Mater. Chem. A, 2015, 3, 6351–6359 RSC .
-
(a) B. Gole, W. Song, M. Lackinger and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 13662–13680 CrossRef CAS PubMed ;
(b) Z.-Q. Shi, Z.-J. Guo and H.-G. Zheng, Chem. Commun., 2015, 51, 8300–8303 RSC .
-
(a) M. E. Germain and M. J. Knapp, J. Am. Chem. Soc., 2008, 130, 5422–5423 CrossRef CAS PubMed ;
(b) Z. Hu, S. Pramanik, K. Tan, C. Zheng, W. Liu, X. Zhang, Y. J. Chabal and J. Li, Cryst. Growth Des., 2013, 13, 4204–4207 CrossRef CAS ;
(c) H. He, Y. Song, F. Sun, N. Zhao and G. Zhu, Cryst. Growth Des., 2015, 15, 2033–2038 CrossRef CAS ;
(d) D. Tian, Y. Li, R.-Y. Chen, Z. Chang, G.-Y. Wang and X.-H. Bu, J. Mater. Chem. A, 2014, 2, 1465–1470 RSC .
-
(a) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC ;
(b) F.-Y. Yi, Y. Wang, J.-P. Li, D. Wu, Y.-Q. Lan and Z.-M. Sun, Mater. Horiz., 2015, 2, 245–251 RSC ;
(c) S.-R. Zhang, D.-Y. Du, J.-S. Qin, S.-J. Bao, S.-L. Li, W.-W. He, Y.-Q. Lan, P. Shen and Z.-M. Su, Chem.–Eur. J., 2014, 20, 3589–3594 CrossRef CAS PubMed .
-
(a) K. S. Asha, G. S. Vaisakhan and S. Mandal, Nanoscale, 2016, 8, 11782–11786 RSC ;
(b) K. S. Asha, K. Bhattacharyya and S. Mandal, J. Mater. Chem. C, 2014, 2, 10073–10081 RSC .
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic (multinuclear NMR, IR HRMS) characterizations, optimized structure, TGA, powder diffraction and fluorescence titration profiles. See DOI: 10.1039/c6sc04367d |
| ‡ M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Wallingford CT, 2009. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a