
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Heterocyclic carbene-catalyzed oxidative [3 + 2] annulation of dioxindoles and enals: cross coupling of homoenolate and enolate†
Xiang-Yu
Chen‡
ac,
Kun-Quan
Chen‡
ab,
De-Qun
Sun
*b and
Song
Ye
*ac
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: songye@iccas.ac.cn; Fax: +86-10-62554449
bMarine College, Shandong University at Weihai, Wenhua West Road, No. 180, Weihai 264209, China. E-mail: dequn.sun@sdu.edu.cn; Fax: +86-631-5688303
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 9th November 2016
Abstract
The N-heterocyclic carbene-catalyzed oxidative [3 + 2] annulation of dioxindole and enals was developed, giving the corresponding spirocyclic oxindole-γ-lactones in good yields with high to excellent diastereo- and enantioselectivities. The challenging aliphatic enals worked effectively using this strategy. The oxidative cross coupling of homoenolate and enolate via single electron transfer was proposed as the key step for the reaction.
Introduction
Being a step- and atom-economical process via C–H functionalization, the oxidative cross coupling reaction is of great value in modern organic synthesis.1 Among the different types, the oxidative coupling of two enolates is a powerful route to 1,4-dicarbonyl compounds, which has been well established.2 However, to the best of our knowledge, the oxidative cross coupling of homoenolate and enolate remains unexplored but is very useful for 1,5-dicarbonyl and related compounds.Initiated half century ago,3 N-heterocyclic carbene (NHC) catalysis has witnessed great success in recent years.4 In 2004, Bode et al. and Glorius et al. reported the elegant NHC-catalyzed reaction of enals involving homoenolate as the key intermediate (Scheme 1, reaction a).5 The homoenolate intermediate could be oxidized to give α,β-unsaturated acyl azolium, which worked as a versatile 1,3-biselectrophile (Scheme 1, reaction b).6 Single electron oxidation could open new ways for organic reactions. In 2008, Studer et al.7 reported the NHC-catalyzed conversion of enals to esters using TEMPO as a single-electron oxidant (Scheme 1, reaction c). During our investigation of this work, the single electron oxidation of the homoenolate was pioneered by Rovis et al.8 and Chi et al.9 (Scheme 1, reaction d). The oxidative homo and cross-coupling of the two homoenolates was established by Rovis et al. (Scheme 1, reaction e).10 In this paper, we demonstrated that the challenge of the cross-coupling of homoenolate and enolate was solved well when a proper oxidant and proper enolates were employed (Scheme 1, reaction f).
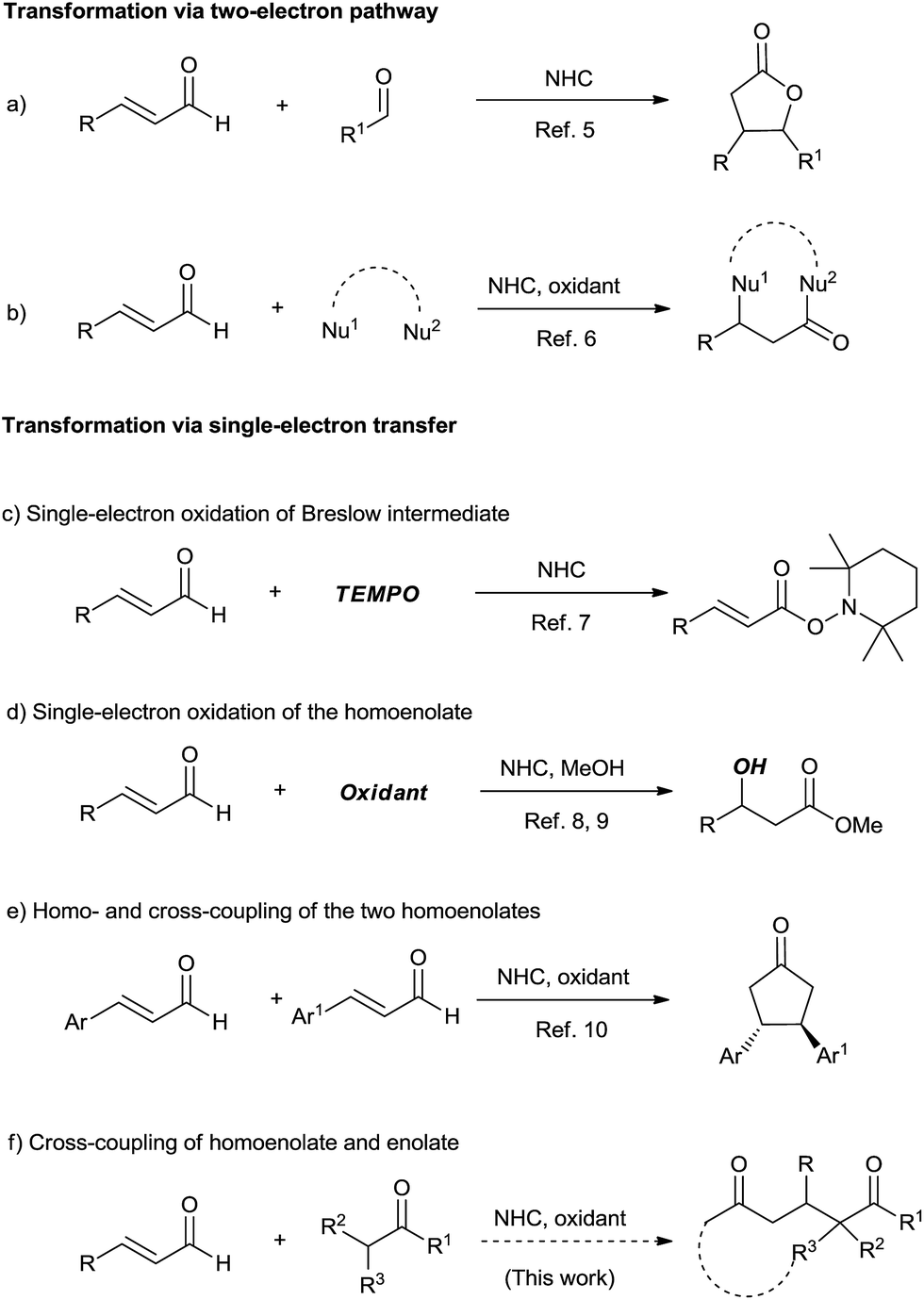 | ||
| Scheme 1 Oxidative NHC-catalyzed transformations of enals. | ||
Spirooxindole plays a valuable role in the synthesis of biologically active natural products and pharmaceuticals.11 Therefore, its direct and catalytic asymmetric construction is especially attractive. Recently, several catalytic asymmetric approaches have been demonstrated as an efficient method for the synthesis of spirooxindole bearing β-aryl-substituted lactones.12–14 However, the corresponding spirooxindole with β-alkyl-substituted lactones failed due to substrate-scope limitations, and/or low yield, diastereo- and enantioselectivities.
The enolate of dioxindole and its radical intermediate13,15 are readily available, and have been widely applied for the synthesis of pharmaceuticals and bioactive natural products with an indole motif.16 We envisioned that the oxidative cross-coupling of the enolate of dioxindole and the homoenolate from enal would be a solution for the [3 + 2] annulation of dioxindole and alkylenals.
Results and discussion
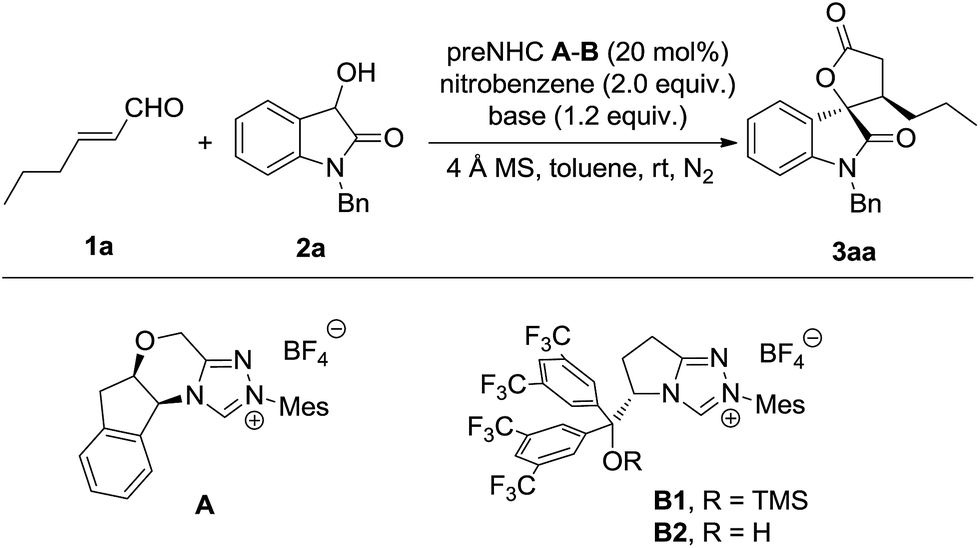
The model reaction of dioxindole and alkylenal was carried out under NHC catalysis in the presence of nitrobenzene as a single electron oxidant (Table 1).17 It was found that the reaction catalysed by triazolium preNHC A gave the desired product 3aa in good yield with exclusive diastereoselectivity but very low enantioselectivity (entry 1). The mixed base of DBU and DABCO resulted in high yield but no improvement of the enantioselectivity (entry 2). The preNHCs B1–B2 derived from L-pyroglutamic acid were then tested.18 We were encouraged to find that preNHC B1 with trimethylsilyl ether resulted in better enantioselectivity albeit with decreased yield (entry 3). The yield and enantioselectivity were improved when NHC B2 with a free hydroxyl group was used (entry 4), possibly owing to the H-bonding between the hydroxyl and the enal. Interestingly, excellent enantioselectivity was reached without compensation of the yield and diastereoselectivity when DABCO was used as the base instead of the mixed base (entry 5). Some increase of the yield was observed when the oxidant was slowly added (entry 6). It should be noted that no reaction was observed for the reaction in the absence of nitrobenzene (entry 7).|
|
|||||
|---|---|---|---|---|---|
| Entry | NHC | Base | Yielda (%) | Drb | Eec (%) |
| a Isolated yield of the mixture of two diastereoisomers. b Determined by 1H NMR (400 MHz) spectroscopy of the raw product. c Determined by HPLC using a chiral stationary phase. d DBU (0.2 equiv.) and DABCO (1.0 equiv.). e The oxidant was slowly added over 30 minutes. f No nitrobenzene was added. | |||||
| 1 | A | DABCO | 58 | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
5 |
| 2 | A | DBU/DABCOd | 97 | >20:1 |
5 |
| 3 | B1 | DBU/DABCOd | 45 | >20:1 |
35 |
| 4 | B2 | DBU/DABCOd | 71 | >20:1 |
73 |
| 5 | B2 | DABCO | 72 | >20:1 |
95 |
| 6e | B2 | DABCO | 77 | >20:1 |
95 |
| 7f | B2 | DABCO | NR | — | — |
The scope of oxidative [3 + 2] annulation with alkylenals was then briefly investigated under the optimized conditions (Table 2). Both enals with β-n-ethyl and n-propyl reacted well with dioxindole to give the desired annulation products in good yields with high enantioselectivities (3aa19 and 3ba). The reactions of enals with a longer alkyl chain were also successful (3ca and 3da). Dioxindoles with different substituents (4-Br, 5-MeO, 6-Br) all reacted well with alkylenals to give the annulation products (3ab, 3cb, 3ac and 3ad) in high yields with good enantioselectivities. It is noteworthy that the β-alkenylenal 1e also worked well with both N-benzyl and N-hydrogen dioxindoles (2a and 2a′), giving the annulation products 3ea and 3ea′ in good yield with high enantio- and diastereoselectivities.
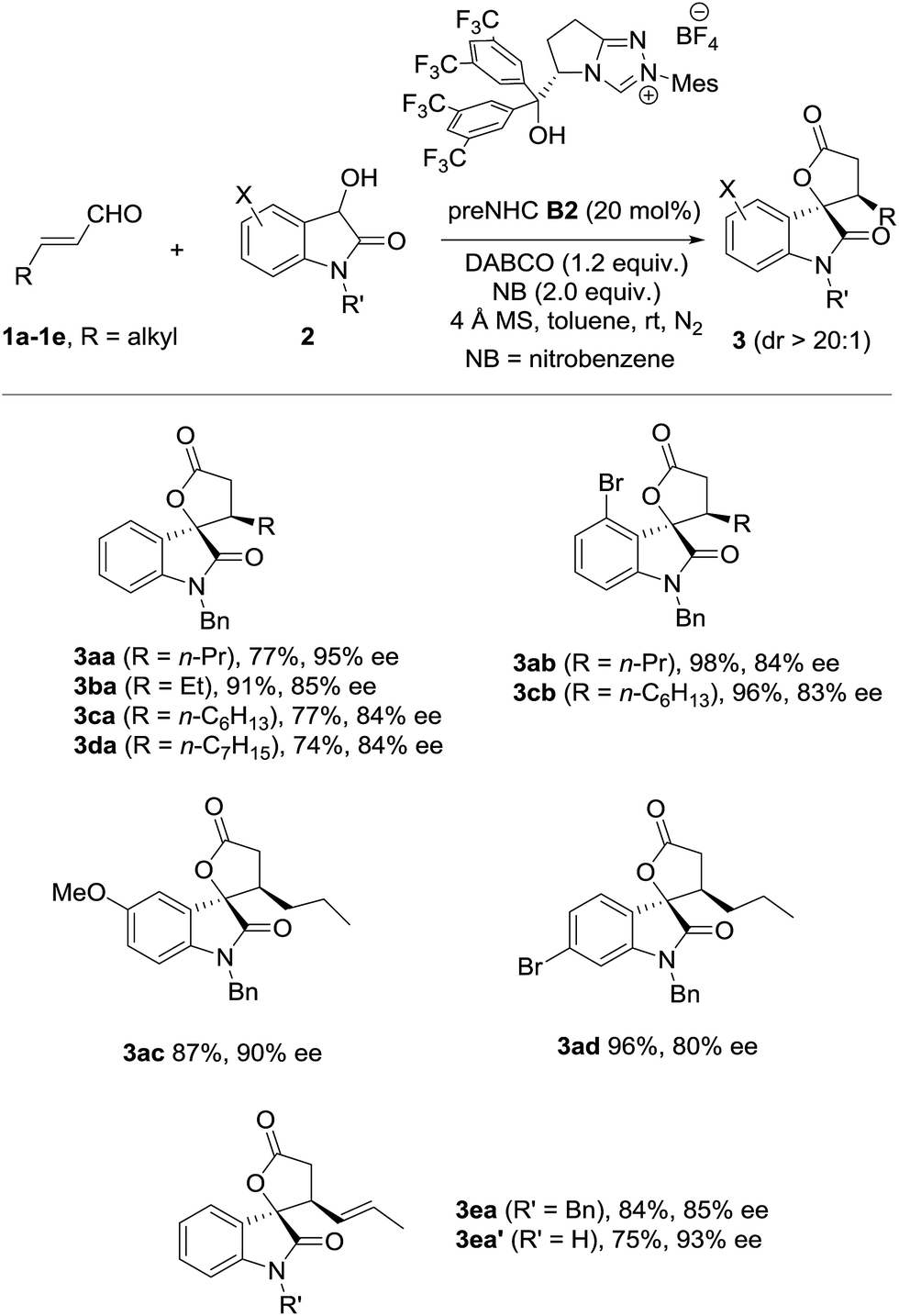
|
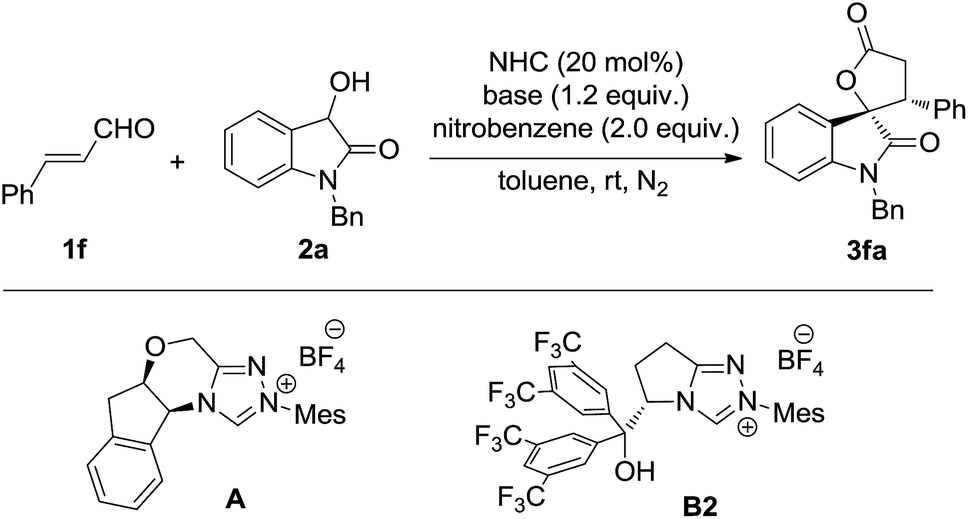
The oxidative [3 + 2] annulation of dioxindole and arylenal was then explored under different conditions (Table 3). Interestingly, the reaction catalyzed by the tetracyclic NHC precursor A gave the desired product 3fa in good yield with moderate diastereo- and enantioselectivity (entry 1), while only a trace amount of 3fa was observed when the NHC precursor B2 was used (entry 2). The different reactivity may be caused by the increased stability of the arylenals and their homoenol radical compared to the alkyl ones. Thus the more nucleophilic and less sterically hindered NHC presusor A performed much better than precursor B for the less reactive arylenals. The screening of bases revealed that DBU offered the best yield, while DABCO resulted in better diastereo-and enantioselectivity (entries 2–5). Thus, the mixed base of DBU and DABCO was then used, which gave the product in 70% yield with 8:1 dr and 91% ee (entry 6). Several single electron oxidants other than nitrobenzene were also investigated, but showed no better results. Further improvement was realized when 4 Å molecular sieves were added as the additive (entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | NHC | Base | Yielda (%) | drb | eec (%) |
| a Isolated yield of the mixture of two diastereoisomers. b Determined by 1H NMR (400 MHz) spectroscopy of the raw product. c Determined by HPLC using a chiral stationary phase. d DBU (0.2 equiv.) and DABCO (1.0 equiv.). e 4 Å molecular sieves were added. | |||||
| 1 | A | DBU | 68 | 3:1 |
76 |
| 2 | B2 | DBU | Trace | — | — |
| 3 | A | DABCO | 32 | 10:1 |
81 |
| 4 | A | Cs2CO3 | 56 | 7:1 |
75 |
| 5 | A | DIPEA | 31 | 5:1 |
59 |
| 6 | A | DBU/DABCOd | 70 | 8:1 |
91 |
| 7e | A | DBU/DABCOd | 78 | 8:1 |
95 |
With the optimized conditions in hand, the scope of dioxindoles with various groups was briefly investigated (Table 4). The reaction of dioxindole with substituents at different positions (Ar = 4-BrC6H3, 5-MeOC6H3 and 6-BrC6H3) was well tolerated and gave the corresponding spirooxindole-γ-lactones in good yields with good diastereoselectivities and high enantioselectivities (3fb, 3fc and 3fd). Both enals with electron-donating (Ar = 4-MeOC6H4 and 4-MeC6H4) and electron-withdrawing groups (Ar = 4-FC6H4, 4-ClC6H4, 4-BrC6H4 and 4-NO2C6H4) worked well to afford the desired product in high yields with good to excellent enantioselectivities (3ga–3la). Both cinnamaldehydes with a meta-substituent (Ar = 3-ClC6H4) and ortho-substituent (Ar = 2-ClC6H4) worked well (3ma and 3na). The reaction of an enal with a 2-furyl group was also successful and provided the product in 89% yield with 20:1 dr and 90% ee (3oa).
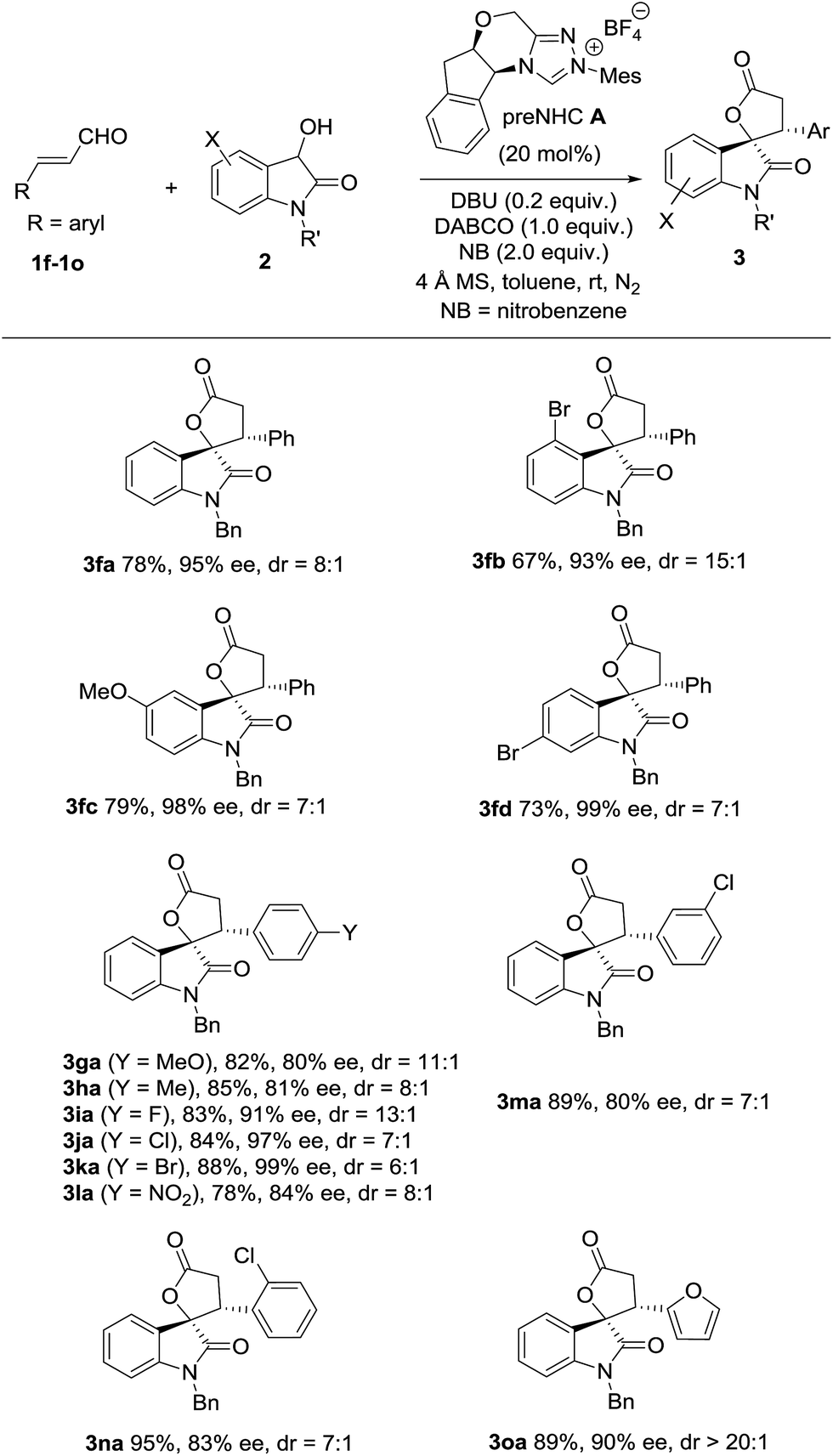
|
The (2S,3R)-configuration of annulation product 3fb20 was determined by the X-ray analysis of its crystal.
A series of control experiments were carried out to clarify the possible reaction pathways (Schemes 2–4). Pathway A, involving the oxidation of dioxindole to isatin followed by annulation with enals was ruled out by the control experiment in which the oxidation of dioxindole 2a′ under the reaction conditions gave only a trace amount of isatin but a majority amount of isatide via homocoupling of the radical of dioxindole (Scheme 2, reaction a).14,15 The formation of isatide was found to be not reversible (Scheme 2, reaction b).
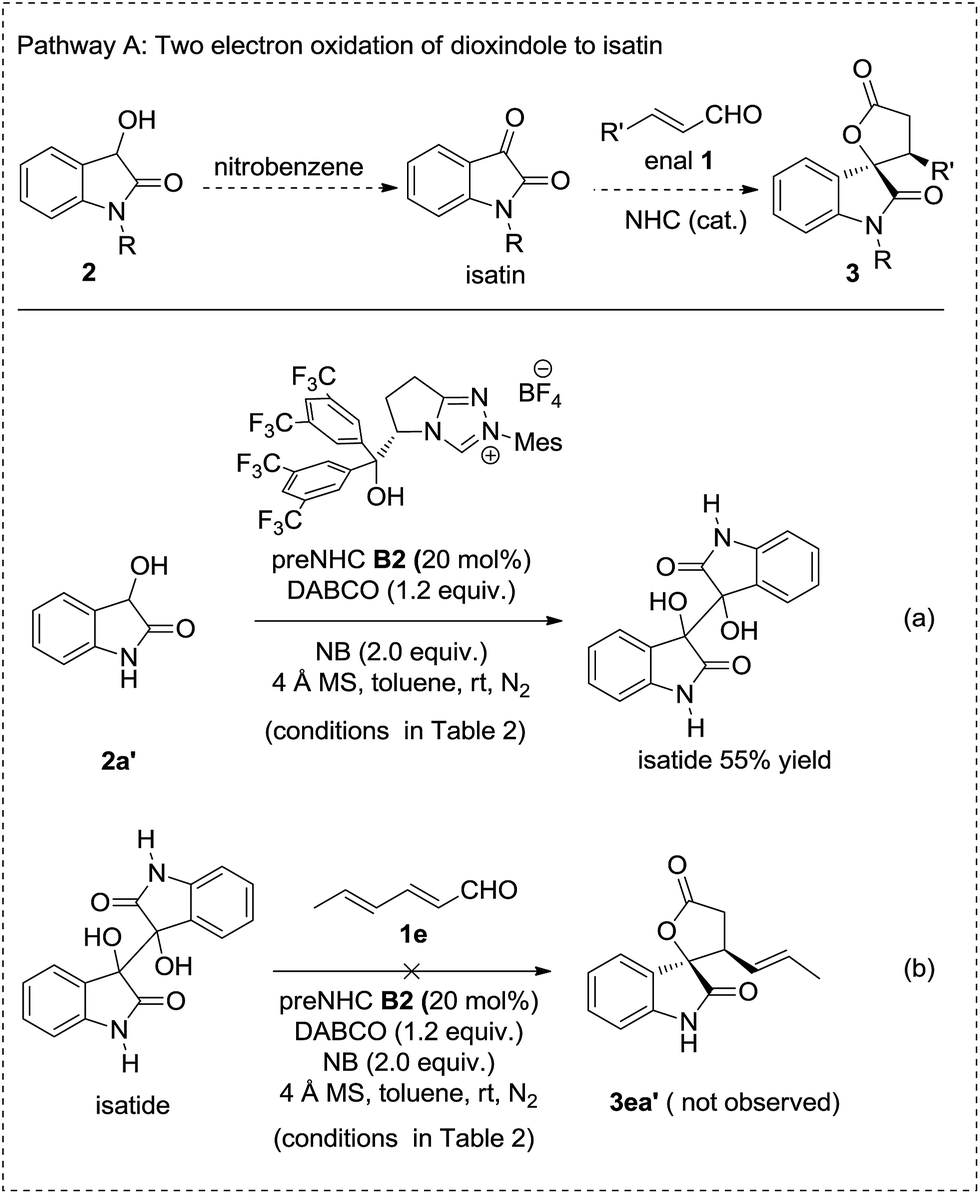 | ||
| Scheme 2 Ruled out pathway A by control experiments. | ||
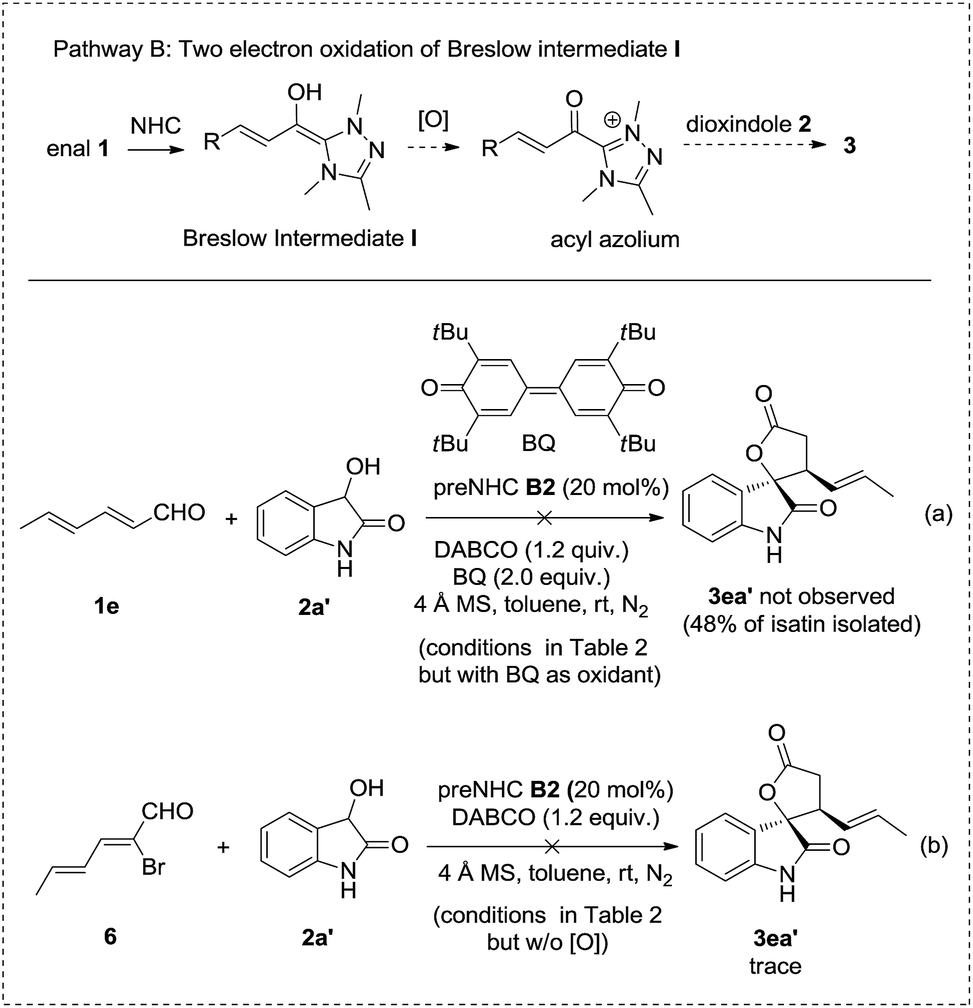 | ||
| Scheme 3 Ruled out pathway B by control experiments. | ||
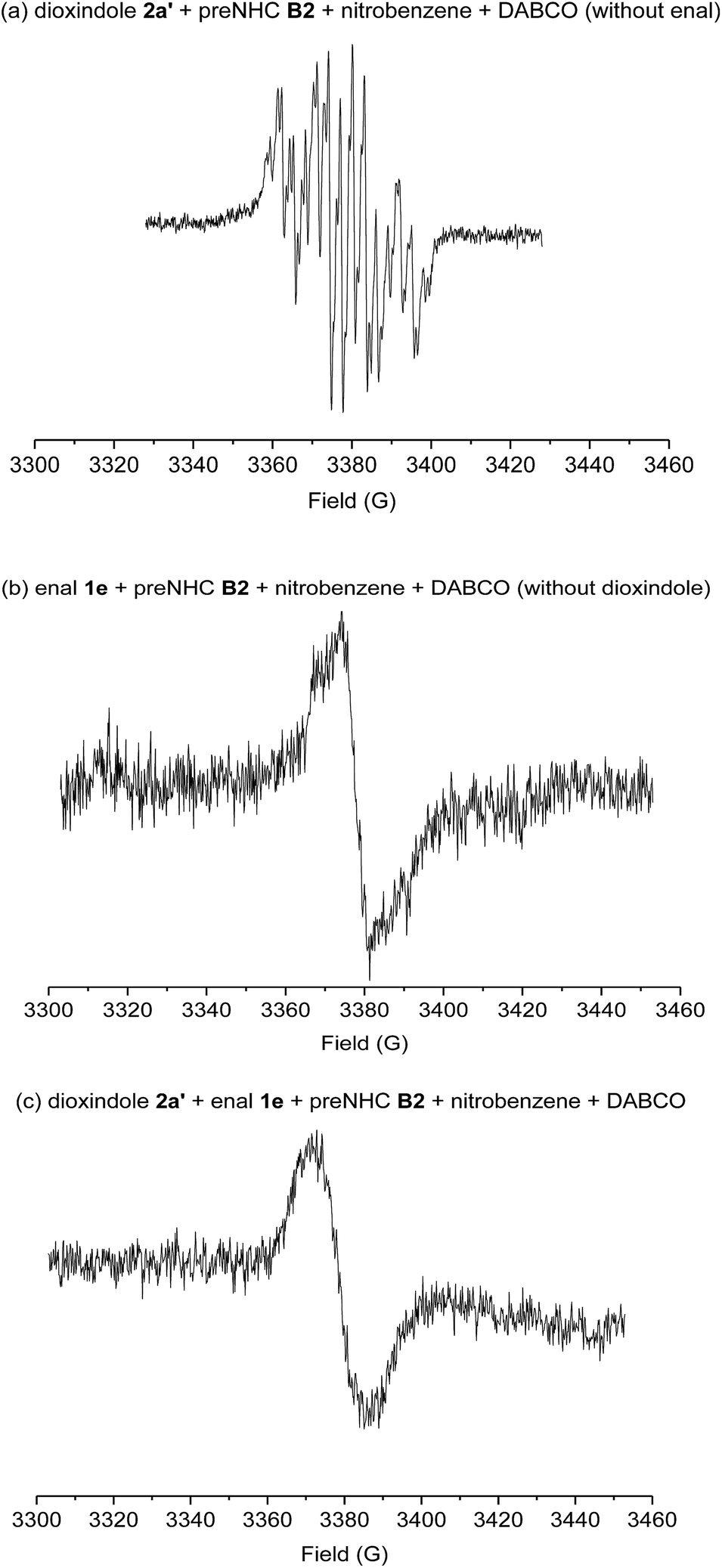 | ||
| Scheme 4 The electron paramagnetic resonance (EPR) spectra. | ||
Pathway B involves the two electron oxidation of the Breslow intermediate to acyl azolium followed by annulation with dioxindole. However, the reaction using bisquinone, which is a well established two electron oxidant to transform the Breslow intermediate to acyl azolium, afforded no desired cycloadduct 3ea′ but isatin in 48% yield with 70% enal 1e recovered (Scheme 3, reaction a). In addition, the reaction of bromoenal 6, which is a reported precursor for acyl azolium, gave only a trace amount of cycloadduct 3ea′ (Scheme 3, reaction b). In addition, no oxidative cyclization product was observed when the reaction was carried out under N2 or an aerobic atmosphere in the absence of nitrobenzene as the oxidant.
When TEMPO, a radical-trapping reagent, was added to the reaction, only a trace amount of the corresponding product was observed. This result indicated that the radical intermediates were involved in the reaction. Several electron paramagnetic resonance (EPR) spectra were measured to identify the possible radical intermediates (Scheme 4). As expected, the reported radical intermediate from dioxindole was observed under our standard conditions but without the addition of enal (Scheme 4, spectrum a). Interestingly, a radical species was also observed when the enal was subjected to the standard conditions but without the addition of dioxindole (Scheme 4, spectrum b), which confirms the generation of the homoenolate radical from enal in the presence of NHC and nitrobenzene.8,17 In addition, an essentially identical EPR signal was observed for the real reaction mixture (Scheme 4, spectrum c).
Based on the control experiments and EPR signals observed, we propose a radical/radical cross-coupling pathway for this NHC-catalyzed oxidative [3 + 2] annulation reaction of dioxindoles and enals (Fig. 1). Firstly, the addition of NHC to enals gives the corresponding Breslow intermediate I, which is partially oxidized to the radical cation intermediate II in the presence of nitrobenzene as the single electron oxidant.8,17 In the meantime, the generated radical anion III could abstract a hydrogen from dioxindole 2 to give its enolate radical IV. The cross-coupling of the enolate radical II and the homoenolate radical IV affords adduct V, which is tautomerized to γ-hydroxy acylazolium VI. The lactonization of acyl azolium VI under basic conditions gives the final cycloadduct 3 and regenerates the NHC catalyst.
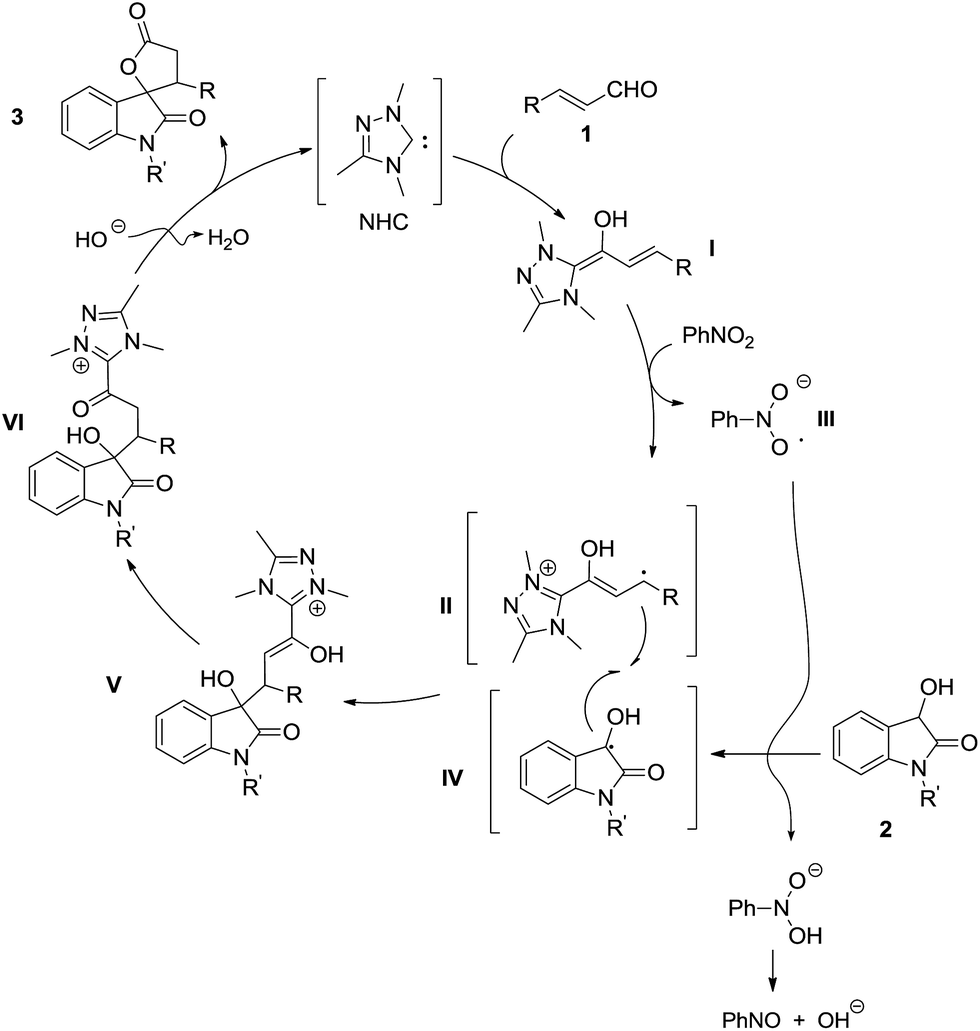 | ||
| Fig. 1 Plausible catalytic cycle. | ||
Conclusions
In summary, the NHC-catalyzed oxidative [3 + 2] annulation reaction of dioxindoles and enals was developed. Both challenging alkyl enals and aryl enals worked well to give the corresponding spirooxindole-γ-lactones in good yields with high to excellent diastereo- and enantioselectivities. Both radicals from enolate and homoenolate were observed by EPR spectra. The reaction represents an unprecedented catalytic oxidative cross coupling of homoenolate and enolate, which is highly interesting mechanistically and synthetically.Acknowledgements
Financial support from National Natural Science Foundation of China (21425207, 21521002, 21672216) and the Chinese Academy of Sciences are greatly acknowledged.Notes and references
- (a) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 Search PubMed; (b) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 Search PubMed; (c) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 Search PubMed; (d) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761 Search PubMed; (e) R. Narayan, K. Matcha and A. P. Antonchick, Chem.–Eur. J., 2015, 21, 14678 Search PubMed.
- (a) F. Guo, M. D. Clift and R. J. Thomson, Eur. J. Org. Chem., 2012, 2012, 4881 Search PubMed and references therein; (b) P. S. Baran and M. P. DeMartino, Angew. Chem., Int. Ed., 2006, 45, 7083 Search PubMed; (c) M. P. DeMartino, K. Chen and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 11546 Search PubMed; (d) T. Amaya, Y. Maegawa, T. Masuda, Y. Osafune and T. Hirao, J. Am. Chem. Soc., 2015, 137, 10072 Search PubMed.
- (a) T. Ugai, S. Tanaka and S. Dokawa, J. Pharm. Soc. Jpn., 1943, 63, 296 Search PubMed; (b) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 Search PubMed.
- For selected reviews, see: (a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 Search PubMed; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 Search PubMed; (c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 Search PubMed; (d) D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53 RSC; (e) J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 44, 2295 Search PubMed; (f) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS PubMed; (g) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686 Search PubMed; (h) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617 Search PubMed; (i) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696 CrossRef CAS PubMed; (j) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 Search PubMed; (k) M. D. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 Search PubMed; (l) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040 Search PubMed.
- (a) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 Search PubMed; (b) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 Search PubMed . For other selected papers, see: ; (c) A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 5334 Search PubMed; (d) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 Search PubMed; (e) H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 Search PubMed; (f) C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232 Search PubMed; (g) S. R. Yetra, S. Mondal, S. Mukherjee, R. G. Gonnade and A. T. Biju, Angew. Chem., Int. Ed., 2016, 55, 268 Search PubMed.
- (a) S. De Sarkar and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 9266 Search PubMed; (b) J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810 Search PubMed; (c) Z.-Q. Rong, M.-Q. Jia and S.-L. You, Org. Lett., 2011, 13, 4080 Search PubMed; (d) F.-G. Sun, L.-H. Sun and S. Ye, Adv. Synth. Catal., 2011, 353, 3134 Search PubMed; (e) Z.-Q. Zhu, X.-L. Zheng, N.-F. Jiang, X. Wan and J.-C. Xiao, Chem. Commun., 2011, 47, 8670 Search PubMed; (f) C. Yao, D. Wang, J. Lu, T. Li, W. Jiao and C. Yu, Chem.–Eur. J., 2012, 18, 1914 Search PubMed; (g) D. Du, Z. Hu, J. Jin, Y. Lu, W. Tang, B. Wang and T. Lu, Org. Lett., 2012, 14, 1274 Search PubMed; (h) Y. Lu, W. Tang, Y. Zhang, D. Du and T. Lu, Adv. Synth. Catal., 2013, 355, 321 CrossRef CAS; (i) Q. Ni, X. Song, G. Raabe and D. Enders, Chem.–Asian J., 2014, 9, 1535 CrossRef CAS PubMed.
- J. Guin, S. De Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727 Search PubMed.
- N. A. White and T. Rovis, J. Am. Chem. Soc., 2014, 136, 14674 CrossRef CAS PubMed.
- Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, J. Am. Chem. Soc., 2015, 137, 2416 CrossRef CAS PubMed.
- N. A. White and T. Rovis, J. Am. Chem. Soc., 2015, 137, 10112 CrossRef CAS PubMed.
- (a) G. Buchi, P. R. DeShong, S. Katsumura and Y. Sugimura, J. Am. Chem. Soc., 1979, 101, 5084 Search PubMed; (b) M. Nakagawa, M. Taniguchi, M. Sodeoka, M. Ito, K. Yamaguchi and T. Hino, J. Am. Chem. Soc., 1983, 105, 3709 CrossRef CAS; (c) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 Search PubMed; (d) T. Ueda, M. Inada, I. Okamoto, N. Morita and O. Tamura, Org. Lett., 2008, 10, 2043 CrossRef CAS PubMed; (e) B. M. Trost and M. K. Brennan, Synthesis, 2009, 2009, 3003 Search PubMed; (f) J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2010, 13, 758 Search PubMed; (g) R. Rios, Chem. Soc. Rev., 2012, 41, 1060 Search PubMed.
- For NHC catalysis, see: (a) V. Nair, S. Vellalath, M. Poonoth, R. Mohan and E. Suresh, Org. Lett., 2006, 8, 507 Search PubMed; (b) L.-H. Sun, L.-T. Shen and S. Ye, Chem. Commun., 2011, 47, 10136 Search PubMed; (c) J. Dugal-Tessier, E. A. O'Bryan, T. B. H. Schroeder, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 4963 Search PubMed; (d) J.-L. Li, B. Sahoo, C.-G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10515 Search PubMed; (e) Z. Jin, K. Jiang, Z. Fu, J. Torres, P. Zheng, S. Yang, B.-A. Song and Y. R. Chi, Chem.–Eur. J., 2015, 21, 9360 Search PubMed; (f) Y. Xie, C. Yu, T. Li, S. Tu and C. Yao, Chem.–Eur. J., 2015, 21, 5355 Search PubMed.
- For metal catalysis, see: B. M. Trost and K. Hirano, Org. Lett., 2012, 14, 2446 Search PubMed.
- For enamine strategy, see: G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 971 Search PubMed.
- (a) H. Hellmann, G. Hallmann and F. Lingens, Chem. Ber., 1953, 86, 1346 Search PubMed; (b) P. L. Julian, H. C. Printy and E. E. Dailey, J. Am. Chem. Soc., 1956, 78, 3501 Search PubMed; (c) G. Hallmann, Chem. Ber., 1962, 95, 1138 Search PubMed; (d) F. Ziegler, T. Kappe and R. Salvador, Monatsh. Chem., 1963, 94, 453 Search PubMed; (e) G. A. Russell, C. L. Myers, P. Bruni, F. A. Neugebauer and R. Blankespoor, J. Am. Chem. Soc., 1970, 92, 2762 Search PubMed.
- (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 Search PubMed; (b) P. Satyamaheshwar, Curr. Bioact. Compd., 2009, 5, 20 Search PubMed; (c) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 Search PubMed; (d) S.-H. Hao, X.-Y. Zhang, D.-Q. Dong and Z.-L. Wang, Chin. Chem. Lett., 2015, 26, 599 Search PubMed.
- A radical species generated by the oxidation of nitrobenzene for thiazolium NHC-catalyzed esterification of aldehyde was reported: J. Castells, F. Pujol, H. Llitjós and M. Moreno-Mañas, Tetrahedron, 1982, 38, 337 Search PubMed.
- (a) D. Enders, O. Niemeier and T. Balensiefer, Angew. Chem., Int. Ed., 2006, 45, 1463 CrossRef CAS PubMed; (b) Y.-R. Zhang, L. He, X. Wu, P.-L. Shao and S. Ye, Org. Lett., 2008, 10, 277 Search PubMed; (c) L. He, Y.-R. Zhang, X.-L. Huang and S. Ye, Synthesis, 2008, 17, 2825 Search PubMed; (d) H.-L. Sun, Z.-Q. Liang and S. Ye, Acta Chim. Sin., 2014, 72, 841 Search PubMed.
- The absolute structure of product 3aa was established by comparison of its optical oration and HPLC spectrum with our previous report (ref. 12b).
- See ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterizations (PDF). CCDC 1041425 (3fb). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04135c |
| ‡ X.-Y. Chen and K.-Q. Chen contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |