
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoselective oxidation of aryl organoboron systems enabled by boronic acid-selective phase transfer†
John J.
Molloy
a,
Thomas A.
Clohessy
ab,
Craig
Irving
a,
Niall A.
Anderson
b,
Guy C.
Lloyd-Jones
c and
Allan J. B.
Watson
 *a
*a
aDepartment of Pure and Applied Chemistry, WestCHEM, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: allan.watson.100@strath.ac.uk
bGlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK
cSchool of Chemistry, University of Edinburgh West Mains Road, Edinburgh, EH9 3JJ, UK
First published on 27th October 2016
Abstract
We report the direct chemoselective Brown-type oxidation of aryl organoboron systems containing two oxidizable boron groups. Basic biphasic reaction conditions enable selective formation and phase transfer of a boronic acid trihydroxyboronate in the presence of boronic acid pinacol (BPin) esters, while avoiding speciation equilibria. Spectroscopic investigations validate a base-promoted phase-selective discrimination of organoboron species. This phenomenon is general across a broad range of organoboron compounds and can also be used to invert conventional protecting group strategies, enabling chemoselective oxidation of BMIDA species over normally more reactive BPin substrates. We also demonstrate the selective oxidation of diboronic acid systems with chemoselectivity predictable a priori. The utility of this method is exemplified through the development of a chemoselective oxidative nucleophile coupling.
Introduction
The use of di- and multi-boron containing systems has become a powerful approach for the rapid synthesis of highly functionalized molecules from readily accessible starting materials.1,2 Chemoselectivity in these systems is currently achieved using only two approaches. Firstly, B-protecting groups render a specific boron unit unreactive under the prevailing reaction conditions. Widely used B-protecting groups include N-methyliminodiacetic acid (MIDA) esters,3 diaminonaphthalene (DAN)-based aminoboranes,4 and potassium organotrifluoroborates (RBF3K) (Scheme 1a).5–7 Secondly, self-activation/protection mechanisms allow discrimination of geminal and vicinal diboron compounds, enabling chemoselectivity within superficially equivalent systems (Scheme 1b).8–10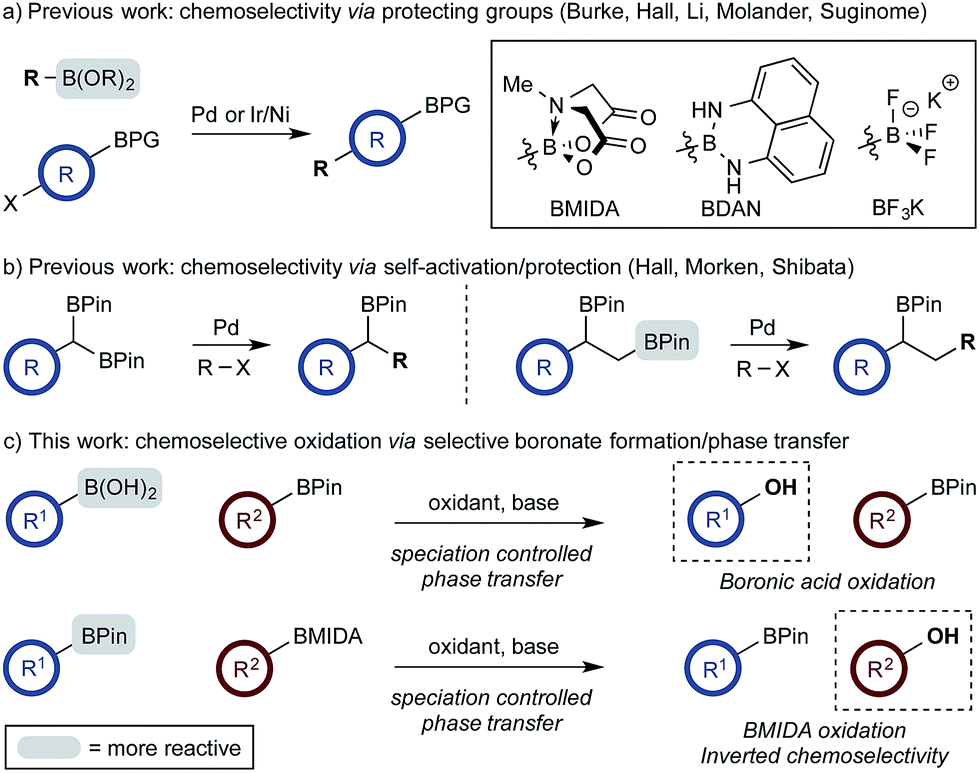 | ||
| Scheme 1 Chemoselective reactions of diboron systems. | ||
B-protecting groups are the most widely adopted strategy and are compatible with sp, sp2, and sp3 organoborons,1–7 while self-activation/protection is only applicable with sp3 organoborons.1,8,9 Accordingly, chemoselectivity within systems containing more than one aryl organoboron compound is currently only achievable by employing a suitable protecting group strategy. Based on the broadly similar reactivity profiles of boronic acids and esters,11 and the added complication of speciation equilibria,11–13 establishing chemoselective control within mixed organoboron systems represents a significant challenge. However, the identification of new chemoselective control mechanisms would be a fundamental advance, enabling the design and development of new synthetic methods for systems containing more than one reactive organoboron compound.
Here, we establish a new method for achieving chemoselectivity within systems containing two unprotected aryl organoboron compounds. Specifically, we show that chemoselective oxidation of aryl boronic acid/BPin systems can be achieved by selective boronate phase transfer while controlling potential solution speciation processes (Scheme 1c). In addition, we show that this approach can formally invert conventional chemoselectivity profiles using established MIDA protecting group chemistry. Spectroscopic investigations of the biphasic reactions provide insight into the mechanism by which chemoselectivity is achieved and how this may be predicted a priori.
Results and discussion
To probe chemoselectivity in systems containing two non-protected aryl organoboron compounds we selected a workhorse reaction. The Brown oxidation of an organoboron compound to the corresponding alcohol or phenol is a fundamental method within the synthetic organic chemistry toolbox.11,14 Boronic acids and esters are typically rapidly and indiscriminately oxidized and, consequently, chemoselective oxidation of a system containing two reactive organoboron compounds is unknown.In the general sense, small differences in reactivity of boronic acids and esters have been observed, albeit in non-competitive systems.15 Accordingly, to initiate this study, we examined the oxidation of naphthyl boronic acid 1a and BPin ester 1b under a variety of reaction conditions.‡ Common oxidants such as H2O2, NaBO3, m-CPBA provided an uncontrollable oxidation from which no useful rate discrimination was observed (a range of oxidants and reaction conditions were surveyed, see ESI†). However, milder oxidants were useful and, in particular, a small rate difference favoring a more rapid oxidation of 1a was found using Oxone® under biphasic reaction conditions (Scheme 2 and Chart 1).§ Specifically, the oxidation of 1a appeared to show a significant conversion in the burst phase while the oxidation of 1b exhibited a more linear profile.
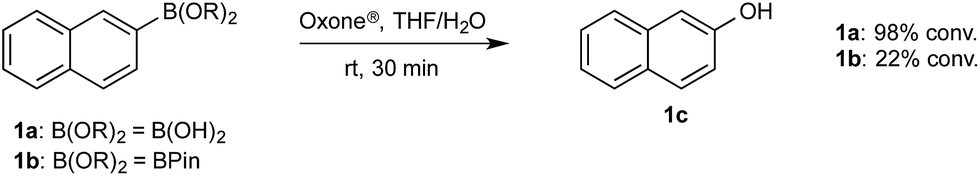 | ||
| Scheme 2 Oxidation of 1a and 1b using Oxone®. | ||
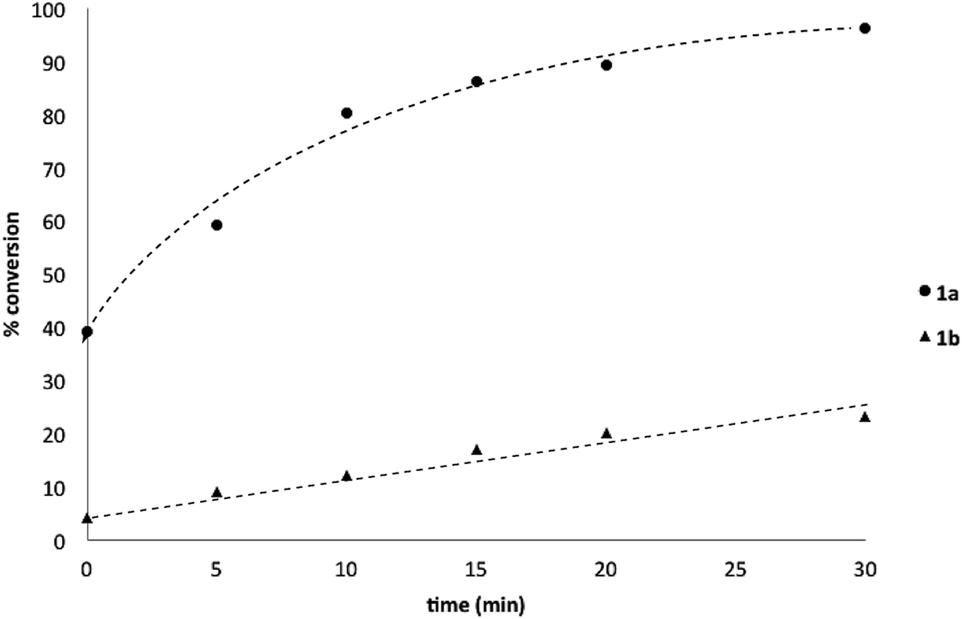 | ||
| Chart 1 Oxidation of 1a and 1b using Oxone® under biphasic reaction conditions (THF/H2O) over 30 min at 350 rpm (vide infra). Determined by HPLC using an internal standard, see ESI.†§ | ||
Based on the observed reactivity profiles in the non-optimized, non-competitive system, we considered it might be possible to leverage chemoselectivity in the corresponding mixed system (i.e., containing both a boronic acid and BPin ester). However, translating these reaction conditions to a model system consisting of 1a and BPin 2b provided high conversion but with only trace levels of chemoselectivity (Table 1, entry 1).
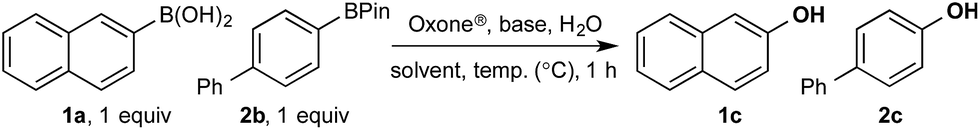
In a purely organic medium (THF), no conversion was observed either in the absence or presence of base (entries 2 and 3 – a range of bases was evaluated, see ESI†), likely due to the poor solubility of Oxone®.16 However, upon addition of K3PO4 to the original biphasic system (i.e., entry 1), we immediately noted moderate conversion, now with significant levels of chemoselectivity for the desired oxidation of 1a (entry 4). A systematic evaluation of the reaction medium composition and temperature (see ESI†) revealed that conversion and chemoselectivity could both be improved using additional H2O at 60 °C (entry 5). A solvent survey revealed CPME as the optimum organic phase that allowed quantitative oxidation of the boronic acid and with very high levels of chemoselectivity at 70 °C under basic biphasic reaction conditions (entry 6 – a range of solvents was evaluated, see ESI†).16
Determination of the origin of chemoselectivity
Basic biphasic reaction conditions allow chemoselective oxidation of boronic acid 1a over BPin 2b. While a small difference in rate using 1a and 1b was observed in the non-competitive system (Chart 1), oxidation was non-selective in the equivalent mixed organoboron system (Table 1, entry 1), suggesting that kinetic discrimination based on reactivities of the organoboron reagents with the oxidant was not the origin of the observed selectivity.Several other data were notable: (i) Oxone® is poorly soluble in organic solvents17 and in the absence of H2O, no reaction was observed (Table 1, entries 2 and 3) suggesting a phase transfer process; (ii) speciation behavior of 1a and 2b was observed in similar basic biphasic media resulting in pinacol transfer to produce a mixture of 1a, 1b, 2a, and 2b in approx. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 ratio (Scheme 3), accordingly, pinacol exchange is avoided under the optimized reaction conditions;11–13,18 (iii) chemoselectivity counter-intuitively increased with increasing temperature (Table 1); and (iv) shearing profoundly impacted the chemoselectivity of oxidation with high stirring rates resulting in lower chemoselectivity and vice versa. The impact of stirring rate was clearly seen in the change of reaction profile of BPin oxidation where increasing the stirring rate changed the reaction profile from linear at 350 rpm to exhibiting a burst phase at 900 rpm similar to the oxidation of 1a (Chart 2).19
:1:1:1 ratio (Scheme 3), accordingly, pinacol exchange is avoided under the optimized reaction conditions;11–13,18 (iii) chemoselectivity counter-intuitively increased with increasing temperature (Table 1); and (iv) shearing profoundly impacted the chemoselectivity of oxidation with high stirring rates resulting in lower chemoselectivity and vice versa. The impact of stirring rate was clearly seen in the change of reaction profile of BPin oxidation where increasing the stirring rate changed the reaction profile from linear at 350 rpm to exhibiting a burst phase at 900 rpm similar to the oxidation of 1a (Chart 2).19
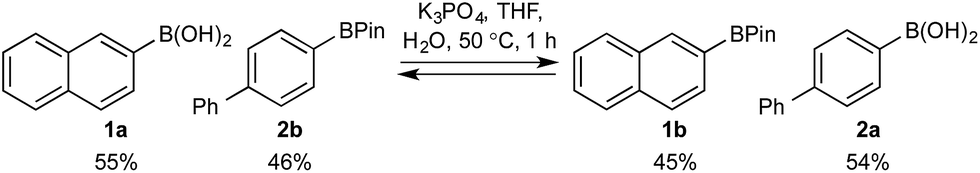 | ||
| Scheme 3 Speciation equilibria of 1a and 2b in a basic biphasic medium. | ||
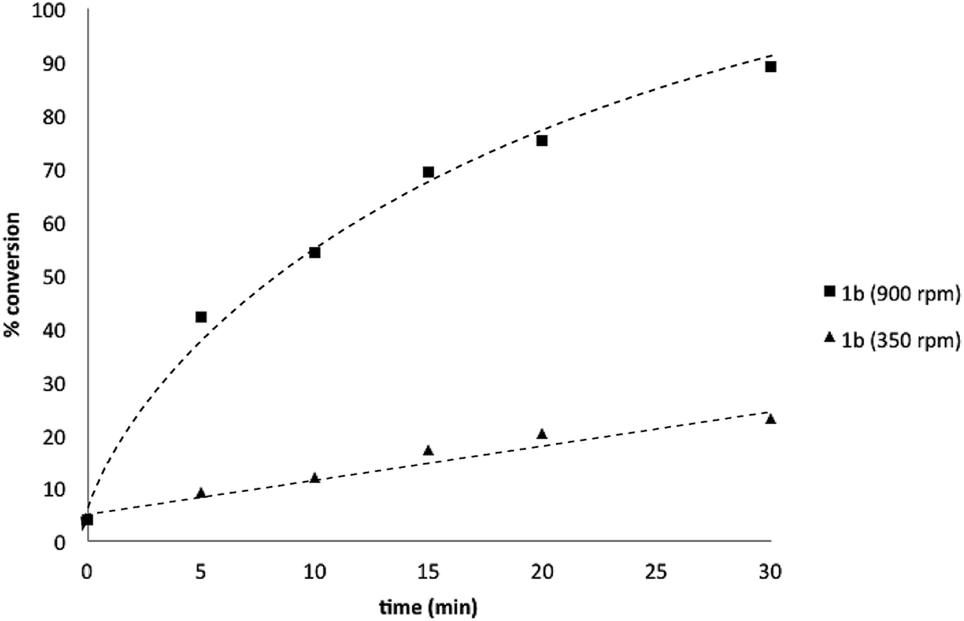 | ||
| Chart 2 Oxidation of 1b to 1c using Oxone® under biphasic reaction conditions (THF/H2O) over 30 min at 350 and 900 rpm. Determined by HPLC using an internal standard, see ESI.†§ | ||
In relation to speciation (Scheme 3), full equilibration was observed to occur in ca. 1 h. Since the oxidation reaction also proceeds to completion in 1 h, we surmise that chemoselectivity is aided by Le Chateliers's principle, i.e., consumption of 1avia oxidation inhibits diol exchange and enforces high levels of chemoselectivity.
Based on all of the above, we suspected that oxidation was taking place via a phase transfer process where the boronic acid was selectively transported to and oxidized in the aqueous phase with the equivalent process for the BPin ester much slower in comparison.
Hall has shown that various polyols can be used to stoichiometrically transfer boronic acids to an aqueous phase as their boronate derivatives to allow purification by phase separation20 as well as providing a method for bioconjugation.21 No such phase-transfer catalyst was employed in the present oxidation; however, boronates are considerably more soluble in aqueous media than organic,10 suggesting chemoselective boronate formation could be taking place (boronic acid over BPin)22 while avoiding speciation processes in the basic biphasic medium. Boronic esters are more Lewis acidic than boronic acids;23 therefore, selective boronic acid trihydroxyboronate formation must be under kinetic control – this is typically a very rapid (practically barrier-less) process.24 To confirm this hypothesis, we undertook detailed analysis of the basic biphasic reaction mixture.
|
|
||||
|---|---|---|---|---|
| Entry | Inorganics | Temp. (°C) | Organic:aqueousa (%) |
|
| 1a | 2b | |||
| a Determined by HPLC analysis using an internal standard. See ESI.† | ||||
| 1 | — | 20, 50, 70 | >99:1 |
>99:1 |
| 2 | K3PO4 | 20 | 54:46 |
>99:1 |
| 3 | K3PO4 | 50 | 46:54 |
96:4 |
| 4 | K3PO4 | 70 | 29:71 |
98:2 |
| 5 | KHSO4, K2SO4 | 20 | >99:1 |
>99:1 |
| 6 | KHSO4, K2SO4 | 50 | >99:1 |
>99:1 |
| 7 | KHSO4, K2SO4 | 70 | 98:2 |
>99:1 |
| 8 | K3PO4, KHSO4, K2SO4 | 20 | 67:33 |
>99:1 |
| 9 | K3PO4, KHSO4, K2SO4 | 50 | 59:41 |
>99:1 |
| 10 | K3PO4, KHSO4, K2SO4 | 70 | 54:46 |
>99:1 |
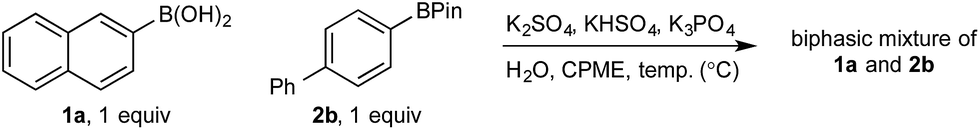
In the absence of any inorganics, both 1a and 2b were confined to the organic phase (entry 1). However, addition of K3PO4 immediately distorted this distribution, with ca. 1:1 distribution of 1a in each phase but with no effect on the distribution of 2b (entry 2). The concentration of 1a in the aqueous phase increased with temperature, reaching ca. 70% at the optimum reaction temperature of 70 °C, with the distribution of 2b again remaining unchanged throughout (entries 2–4). Addition of Oxone®-relevant inorganics (without the active oxidant, KHSO5)25 had no effect on the distribution of either 1a or 2b at any temperature, with similar results to that observed in the absence of any inorganics (entries 5–7 vs. entry 1). In the presence of K3PO4, KHSO4, and K2SO4, 1a was once again observed to distribute in both phases, up to ca. 1:1 at 70 °C, while 2b remained confined to the organic phase, even at elevated temperatures (entries 8–10). The lower concentration of 1a in the aqueous phase in the presence of all inorganics may be attributable to buffering. Lastly, no speciation behavior (i.e., diol transfer, see Scheme 3) was observed throughout. However, it should be noted that speciation (diol transfer) was observed when mixtures of 1a and 2a were left for extended time periods at elevated temperatures.
Initial control experiments were informative and provided further empirical evidence to support selective hydroxyboronate formation. Specifically, while trihydroxyboronate 1d could be formed using aq. K3PO4, BPin hydroxyboronate 2e was not observed under similar conditions. Indeed, 2e was only observed upon treatment with aq. KOH, which also led to extensive hydrolysis,26 generating the corresponding boronic acid 2a and, consequently, its trihydroxyboronate derivative. This supported the hypothesis that under the reaction conditions, boronic acid trihydroxyboronates may be formed selectively, thereby allowing selective phase transport and subsequent chemoselective oxidation.
11B NMR analysis of the aqueous phase of the biphasic monoboron system containing 1a and relevant inorganics (again without KHSO5) showed the presence of a single boron species with a resonance at 3.7 ppm, consistent with trihydroxyboronate 1d (Scheme 4a) while no signal was detected in the aqueous phase for the equivalent experiment using only 2b (Scheme 4b). Analysis of the corresponding model system containing both 1a and 2b revealed a single signal in the aqueous phase at 3.7 ppm, consistent with 1d (Scheme 4c). This analysis agreed with the HPLC data (Table 2) and also supported selective phase transport of 1a to the aqueous phase as its trihydroxyboronate derivative, 1d.27 No charged species (1d or 2e) or anhydride formation were observed in a complementary analysis of the organic phase – only the neutral species (1a and 2b) were observed.
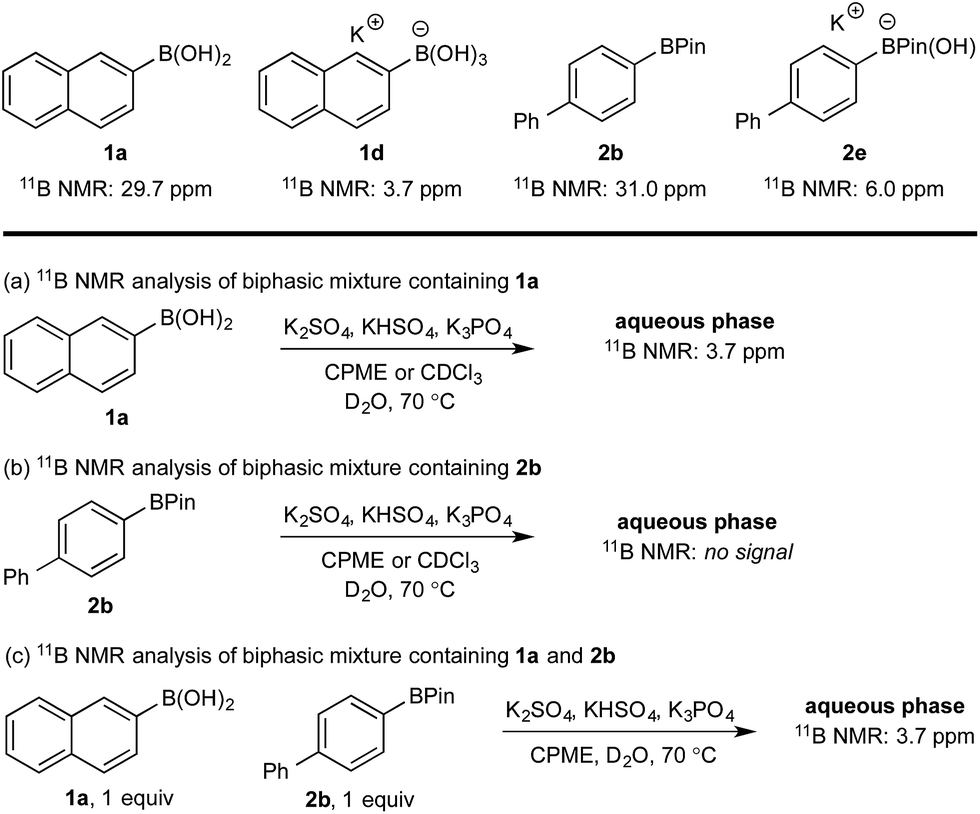 | ||
| Scheme 4 11B NMR analysis of mono- and diboron systems of 1a and 2b under representative biphasic conditions. | ||
Hydroxyboronates 1d and 2e are distinguishable by 11B NMR (see ESI†) and the assignment of the observed 11B NMR signal at 3.7 ppm was attributed to 1d. However, it is conceivable that in situ hydrolysis of 2b could occur to deliver 2a and ultimately its trihydroxyboronate derivative (2d), which has a similar 11B NMR resonance to 1d (3.6 ppm, see ESI†). The 2d signal may be obscured by 1d, preventing detection at low concentration. To ensure a robust assignment, we analyzed two mono-fluorinated diboron systems by 11B and 19F NMR analysis (Scheme 5).
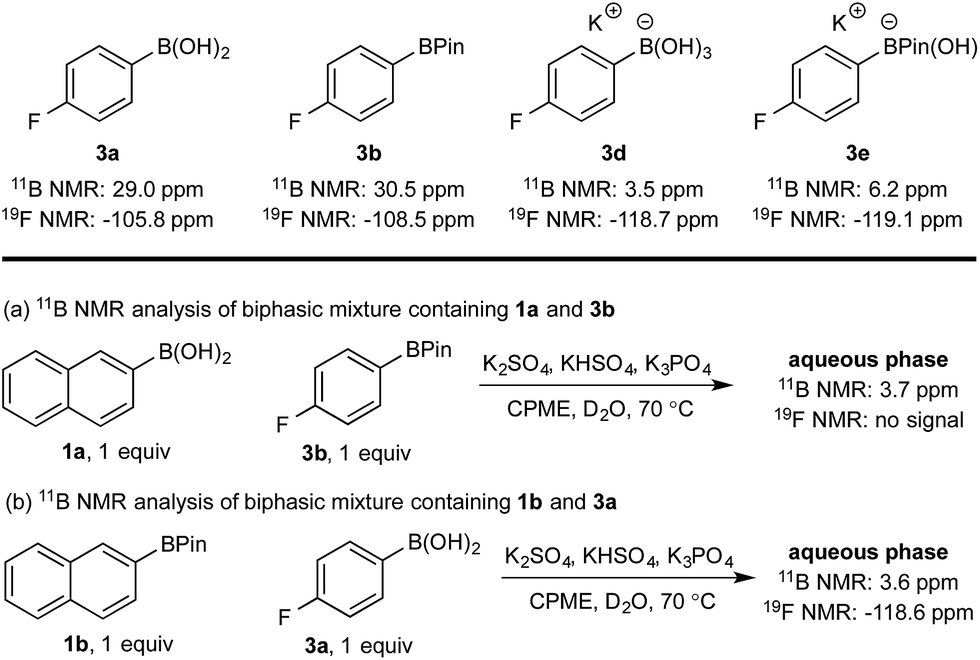 | ||
| Scheme 5 11B and 19F NMR analysis of mono-fluorinated diboron systems under representative biphasic conditions. | ||
11B and 19F NMR analysis of the aqueous phase of the system containing 1a and 3b revealed a single 11B NMR signal at 3.7 ppm, consistent with 1d; no 19F NMR signals were detected (Scheme 5a). Conversely, analysis of the mixture of 1b and 3a showed one 11B NMR signal at 3.6 ppm and one 19F NMR signal at −118.6 ppm, both of which were consistent with trihydroxyboronate 3d (Scheme 5b). Thus, these experiments support the hypothesis of a selective boronic acid trihydroxyboronate formation and that diol transfer is inhibited.
Temporal profiling of the aqueous phase via variable temperature NMR provided further data to assist in explaining the observed trends (Fig. 1 – for temperature/temporal profiling of all systems, see ESI†). Consistent with the HPLC analysis (Table 2), variable temperature 11B NMR revealed that [1d] increased with temperature, with no detectable increase in [1a], [2b] or [2e].28 [1d] also increased over time (see ESI†). This combined HPLC and NMR data set assists with the interpretation of the non-intuitive temperature-proportional increase in chemoselectivity.29
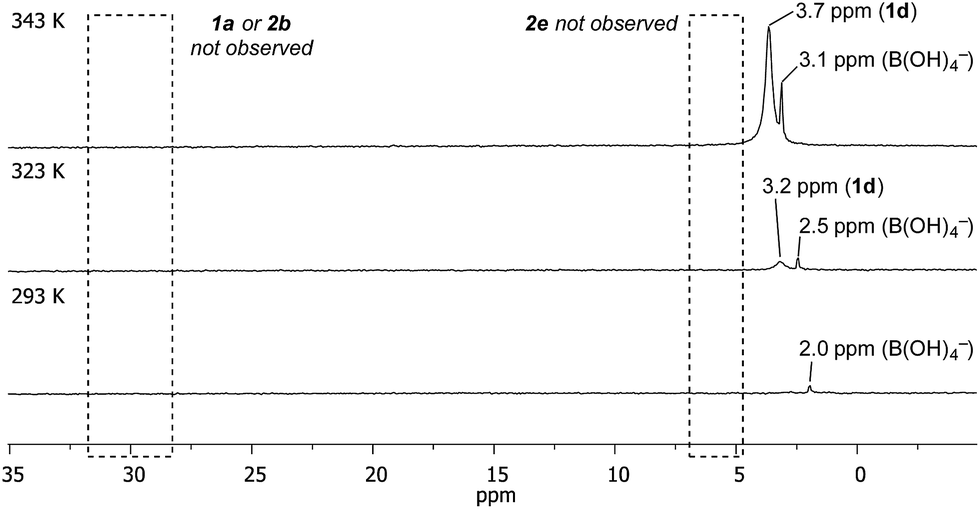 | ||
| Fig. 1 Temperature- and time-proportional concentration of 1d in the aqueous phase by 11B NMR analysis. | ||
The rate of oxidation was found to be rapid in the burst phase (see Chart 1 and ESI†). Therefore, the rate determining process for oxidation under the developed biphasic reaction conditions appears to be phase transfer of the organoboron species to the aqueous phase, which is assisted by trihydroxyboronate formation. Since oxidation occurs exclusively in the aqueous phase and BPin hydroxyboronate formation was not observed under the reaction conditions, chemoselective oxidation is achieved since boronic acid phase transfer is significantly more favorable than BPin transfer (Scheme 6).
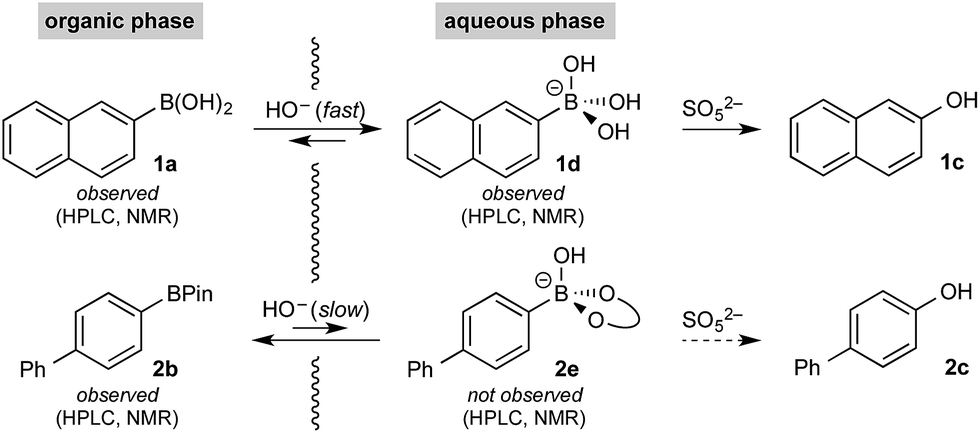 | ||
| Scheme 6 Spectroscopically observed organoboron species. | ||
Interestingly, in the absence of the active oxidant (KHSO5), protodeboronation was observed to increase proportionally with both time and temperature giving the expected product B(OH)4−.30,31 In the oxidative system this was not particularly problematic, since the rate of oxidation was rapid. However, this has clear implications for transition metal catalysis using organoboron species under basic biphasic reaction conditions (e.g., Suzuki–Miyaura cross-coupling) in which transmetallation proceeds via the neutral organoboron species15 and must engage a presumably largely organic phase-bound catalyst.
Lastly, formation of trihydroxyboronate from the boronic acid or hydroxyboronate from the BPin ester requires access to HO−. Boronic acids have typically greater aqueous solubility than the corresponding BPin.11 clogP calculations (see ESI†) indeed indicate greater aqueous solubility for boronic acids vs. BPins and this was also found for the corresponding boronate adducts. For example, boronic acid 1a has clogP = 2.64 while BPin 1b has clogP = 5.58. The corresponding boronates display the same trend with 1d cLogP = 0.50 and 1e clogP = 3.44. Accordingly, since no charged species were observed in the organic phase, we believe that selective ionization of the boronic acid occurs either at the organic/aqueous interface or in the aqueous phase following transfer of the neutral species due to its comparatively greater solubility.
Generality of the chemoselective oxidation process
With effective reaction conditions for this model system, the generality of the chemoselective oxidation was explored (Scheme 7).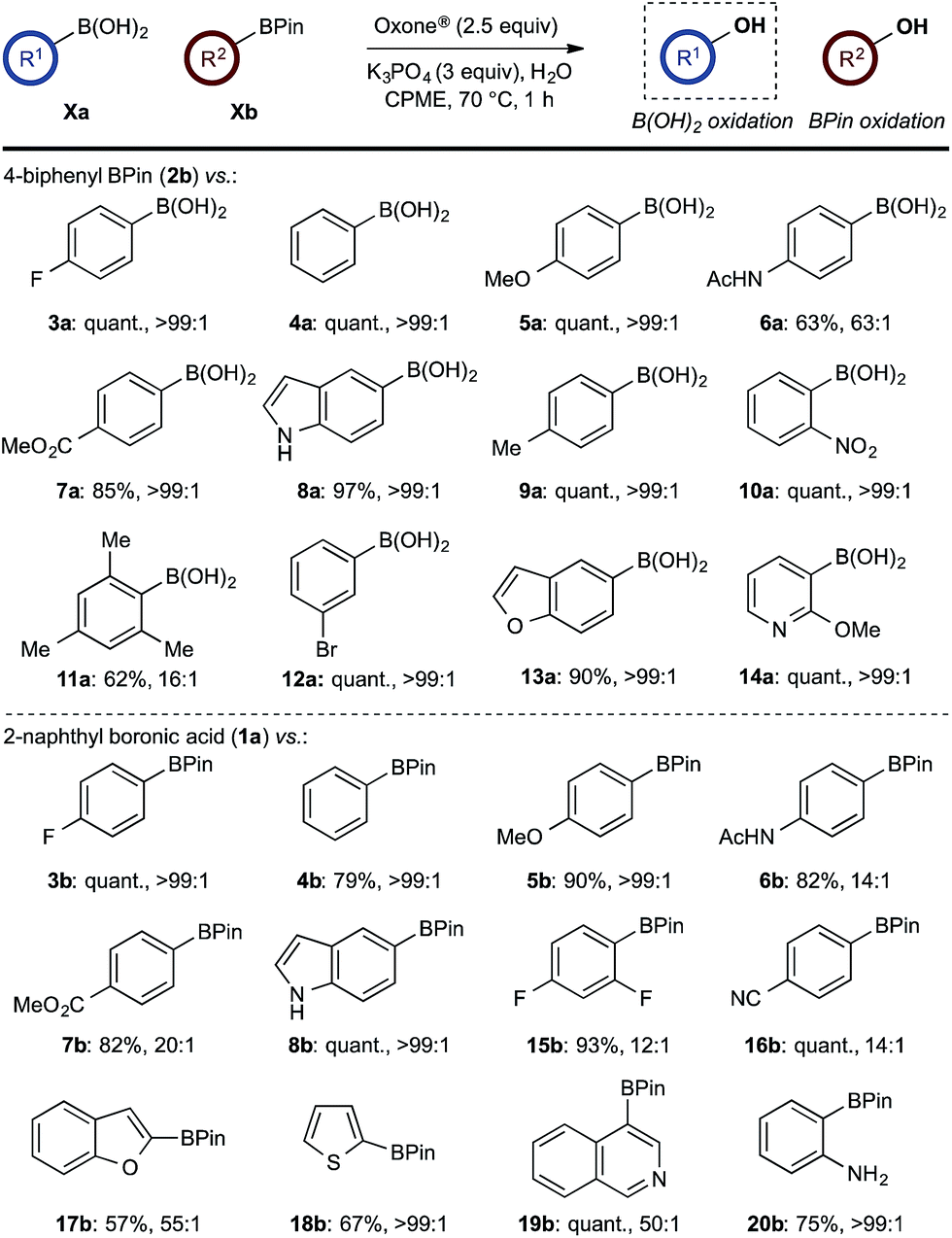 | ||
| Scheme 7 Chemoselective oxidation of aryl diboron system: B(OH)2vs. BPin substrate scope. Ratio given for oxidation of Xa:Xb. Determined by HPLC, see ESI.† | ||
The biphasic reaction conditions were found to be general across a wide variety of aryl boronic acid and BPin ester reaction partners. Conversion to products over a 1 h reaction time were generally high and the chemoselectivity for boronic acid oxidation was typically >20:1 and exclusively selective (>99:1) in many cases, regardless of functionality or regiochemistry and whether boronic acid or BPin (e.g., 3a–8avs.3b–8b). Alkenyl boronic acids were less effective substrates, giving mixtures of products.
With a framework for chemoselective oxidation of reactive diboron systems established, we sought to explore whether this biphasic protocol could challenge conventional reactivity profiles. BPin esters are typically readily oxidized in the presence of BMIDA esters.13d However, we reasoned that it might be possible to reverse this profile and selectively oxidize the BMIDA component of a BMIDA/BPin aryl diboron system via speciation-controlled hydrolysis of the BMIDA32 and oxidation of the latent boronic acid (Scheme 8).
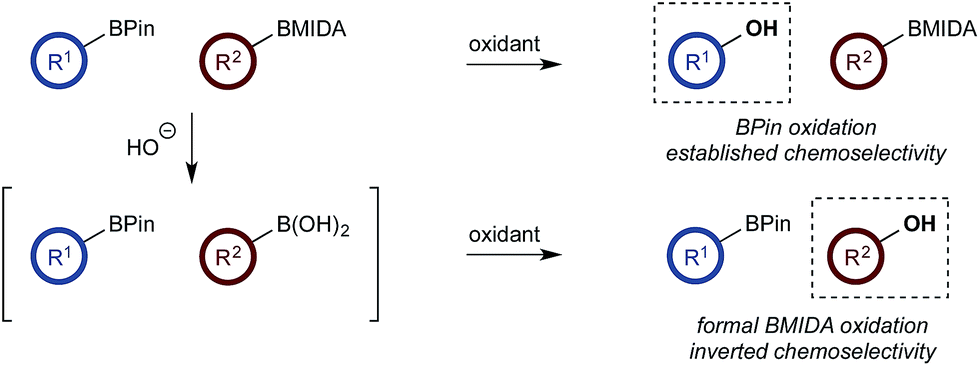 | ||
| Scheme 8 Inverting established chemoselectivity: oxidation of BMIDA in the presence of BPin. | ||
In the event, heating the reaction mixture to 80 °C for 15 min in the absence of Oxone® provided a smooth hydrolysis, which avoided any diol equilibration, and returning to 70 °C before addition of the oxidant allowed chemoselective oxidation of ArBMIDA in the presence of ArBPin (Scheme 9).
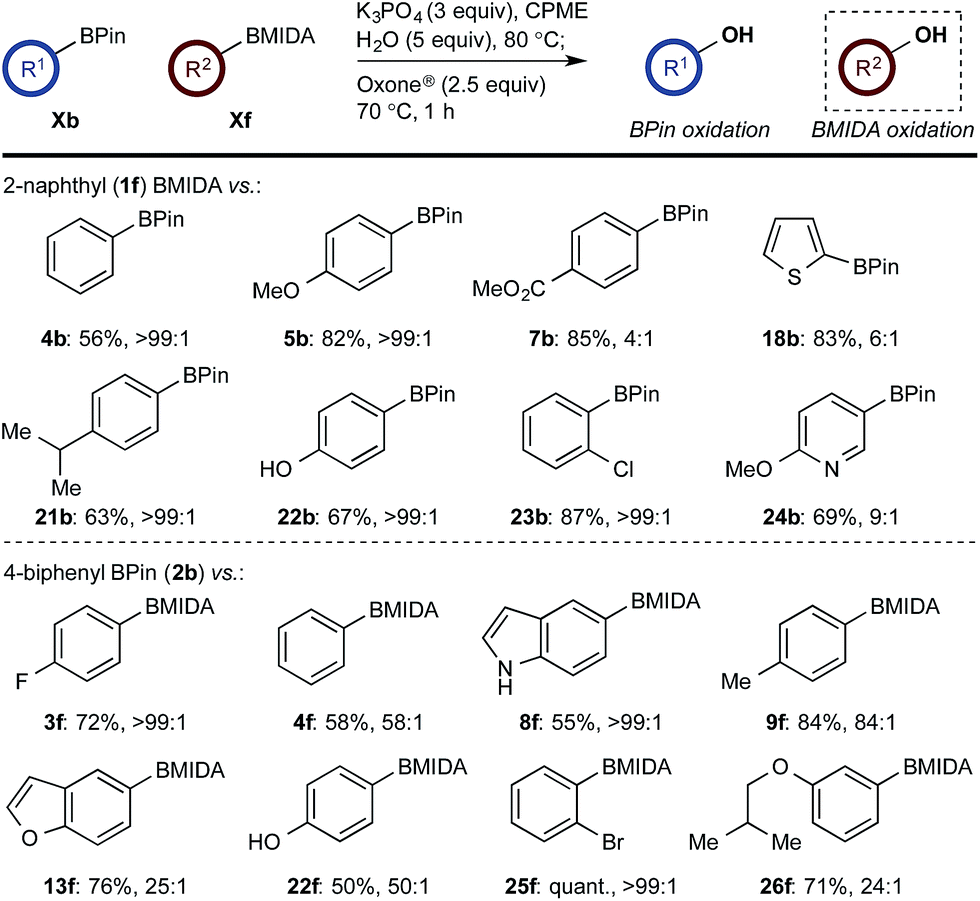 | ||
| Scheme 9 Chemoselective oxidation of aryl diboron system: BMIDA vs. BPin substrate scope. Ratio given for oxidation of Xf:Xb. Determined by HPLC, see ESI.† | ||
Once more, the efficiencies and selectivities of the process were typically excellent, with some diminished selectivity observed using specific heterocyclic derivatives (e.g., 18b, 24b). Addition of Oxone® after the hydrolysis event was necessary to avoid buffering of the basic medium. This buffering effect impeded the rate of hydrolysis providing sufficient time for equilibration and ultimately diminishing the chemoselectivity of the process.
This BMIDA oxidation process provided the opportunity to further confirm the hypothesis of the requirement to physically separate the two boron residues in order to achieve chemoselectivity. Diboron compound 27 (where both boron residues were located on the same aryl unit) was a very poor substrate that, under optimized conditions, delivered a mixture of the desired phenol 22b as well as 22a (the product of BPin oxidation and BMIDA hydrolysis), 22c (the product of global oxidation), but mainly 28 (the product of equilibration) (Scheme 10).32
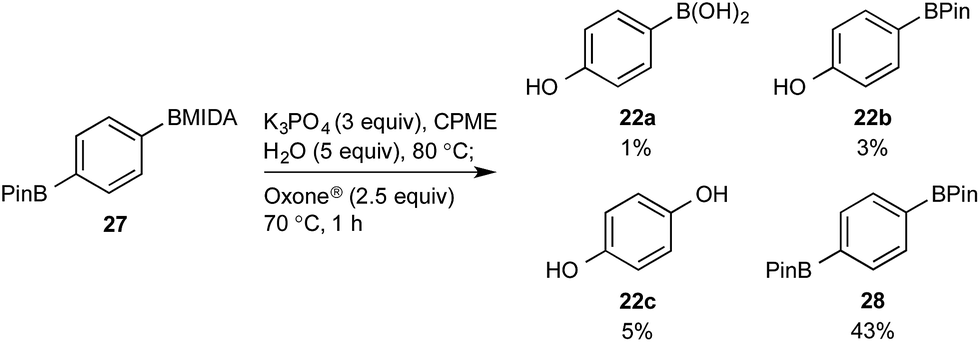 | ||
| Scheme 10 Attempted chemoselective oxidation of diboron compound 27. Determined by HPLC, see ESI.† | ||
Chemoselective oxidation of diboronic acid systems
Chemoselectivity in this system has been driven by competing boronate formation between dissimilar organoboron compounds (B(OH)2vs. BPin). However, the formation of aryl boronates, and therefore aqueous solubility, is heavily influenced by the electronics of the aryl unit.19 Specifically, substitution on the aryl unit will influence the Lewis acidity of the boronic acid and can heavily influence the aqueous solubility. Based on this, we reasoned that chemoselective discrimination within a system containing two boronic acids might be achievable by exploiting electronic effects to drive competitive boronate formation and subsequent selective phase transfer. The relative chemoselectivity might then be gauged a priori by assessing the phase separation by HPLC or NMR. This proved to be feasible and was initially evaluated using several electronically distinct phenylboronic acids vs. 2-naphthyl boronic acid 1a (Scheme 11).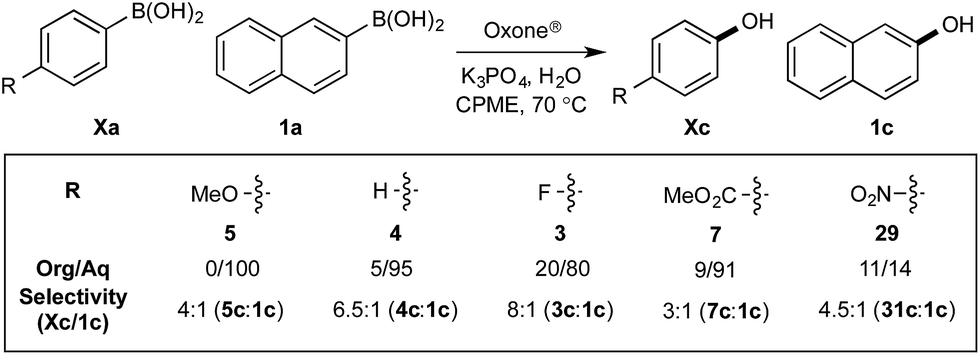 | ||
| Scheme 11 Phase distribution and chemoselective oxidation of electronically distinct aryl boronic acids vs.1a. Determined by HPLC, see ESI.† | ||
Under representative reaction conditions in the absence of oxidant, phenylboronic acids 3a, 4a, 5a, and 7a were found to preferentially distribute to the aqueous phase while 1a remained comparatively more organic phase-bound (1a org/aq average = 71/29). This translated to chemoselective oxidation of the phenyl boronic acid species over 1a in all cases. In the case of the strongly electron-deficient boronic acid 29a, protodeboronation occurred rapidly and phase distribution was less reliable as an indicator of selectivity. While the measured phase distribution allowed prediction of the favored oxidation, the exact ratio of oxidation products could not be extrapolated from this analysis. This phenomenon was also found to be transferable across a range of substrates (Table 3).
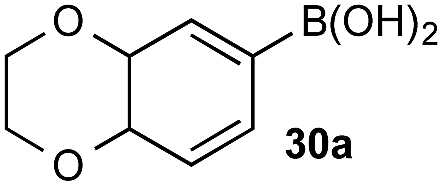
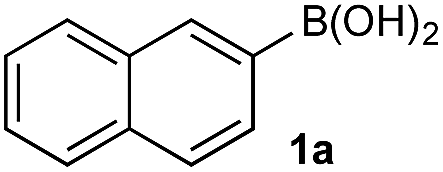
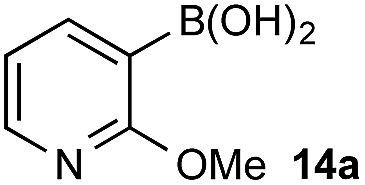
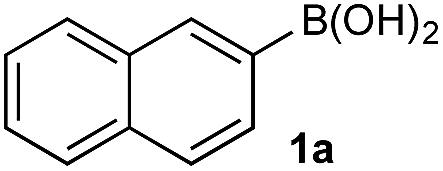
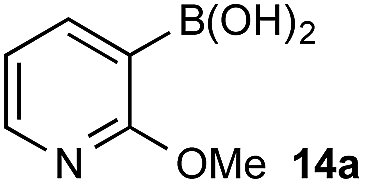
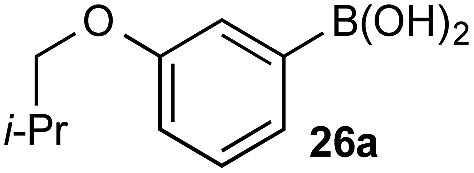
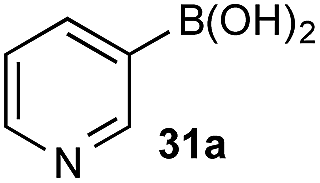
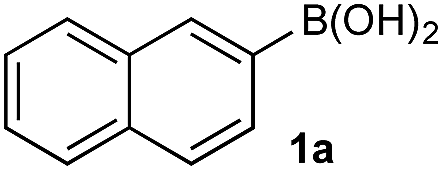
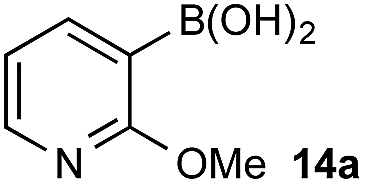
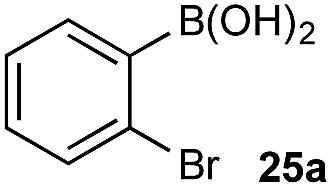
The efficiencies and chemoselectivities of the process were not as pronounced as more substantially differentiated diboron systems (e.g., B(OH)2vs. BPin in the studies above); however, this represents the first chemoselective oxidation of two ostensibly equivalent boronic acid species based on subtle differences in the substitution of the pendant aryl unit.33,34
Chemoselective oxidative nucleophile coupling
The medium-controlled chemoselective reaction manifold can potentially be leveraged to provide a number of enabling synthetic methods. As a demonstration, we have developed a chemoselective oxidative nucleophile coupling (Scheme 12).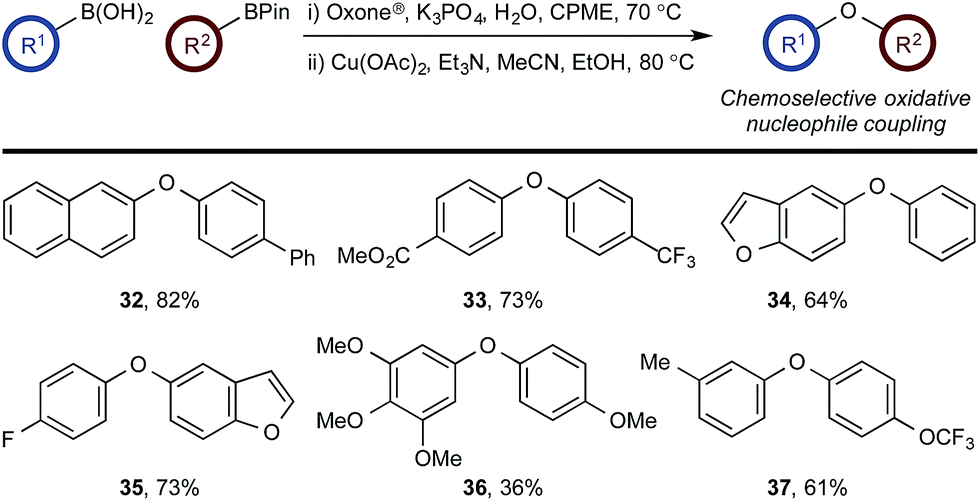 | ||
| Scheme 12 Chemoselective oxidative nucleophile coupling. | ||
Following chemoselective oxidation, a Chan–Evans–Lam etherification35,36 of the generated phenol with the remaining BPin can be effected. This process proceeds with high efficiency for the desired cross-coupled product with minimal homo-coupling detected. Biaryl ethers are prominent scaffolds in natural products, pharmaceuticals, agrochemicals, and materials,37 for example, the anticancer agents, including 36.38 Modern catalysis methods, such as Ir-catalyzed C–H activation,39 have provided convenient methods for accessing borylated arenes with substitution patterns that are not readily accessible by other methods. As such, this chemoselective oxidative nucleophile coupling process provides a novel and step-efficient synthesis of valuable chemotypes from readily accessible precursors.
Conclusions
In conclusion, chemoselective oxidation of boronic acid/BPin systems can be readily achieved in a basic biphasic reaction medium. Conventional protecting group strategies can be overturned to allow oxidation of BMIDA compounds in the presence of a normally more reactive BPin species. Spectroscopic investigations revealed that chemoselectivity is derived from a selective boronate formation and phase transfer of boronic acids to the aqueous phase. We have also shown that it is possible to chemoselectively oxidize a mixture of two boronic acids and predict the outcome of the reaction a priori by HPLC analysis of the phase distribution of the reacting partners. Lastly, the concept of chemoselectivity via medium control enabled the development of a chemoselective oxidative nucleophile coupling. The data in this study has significant ramifications for enabling chemoselectivity in non-protected organoboron systems as well as for the understanding of catalytic reactions of organoboron compounds in biphasic media.Abbreviations
| CPME | Cyclopentyl methyl ether |
| DAN | Diaminonaphthalene |
| HPLC | High performance liquid chromatography |
| MIDA | N-Methyliminodiacetic acid/N-methyliminodiacetate |
| NMR | Nuclear magnetic resonance |
| Pin | Pinacolato |
| rt | Room temperature |
Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no. [340163]. This work was supported by the EPSRC. We thank the EPSRC and GlaxoSmithKline (GSK) for PhD studentships (JJM and TAC), the EPSRC UK National Mass Spectrometry Facility at Swansea University for analyses, and Dr Vipul K. Patel and GlaxoSmithKline for financial support and chemical resources.Notes and references
- For recent reviews, see: (a) L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848–8858 RSC; (b) J. W. B. Fyfe and A. J. B. Watson, Synlett, 2015, 26, 1139–1144 CrossRef CAS; (c) L. Xu and P. Li, Synlett, 2014, 25, 1799–1802 CrossRef CAS; (d) M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2009, 48, 3565–3568 CrossRef CAS PubMed; (e) C. Wang and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 5240–5244 CrossRef CAS PubMed.
- For a classification of boron reagents, see: A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- For recent reviews, see: (a) J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed; (b) E. P. Gillis and M. D. Burke, Aldrichimica Acta, 2009, 42, 17–27 CAS.
- For the first report of this protecting group, see: H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef CAS PubMed.
- For recent reviews on the use of RBF3K, see: (a) G. A. Molander, J. Org. Chem., 2015, 80, 7837–7848 CrossRef CAS PubMed; (b) S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS PubMed.
- For examples of the use of BF3K as a protecting group, see: (a) G. Molander and D. L. Sandrock, J. Am. Chem. Soc., 2008, 130, 15792–15793 CrossRef CAS PubMed; (b) G. A. Molander and D. E. Petrillo, J. Am. Chem. Soc., 2006, 128, 9634–9635 CrossRef CAS PubMed; (c) G. A. Molander and R. Figueroa, Org. Lett., 2006, 8, 75–78 CrossRef CAS PubMed; (d) G. A. Molander and R. Figueroa, J. Org. Chem., 2006, 71, 6135–6140 CrossRef CAS PubMed; (e) G. A. Molander and N. M. Ellis, J. Org. Chem., 2006, 71, 7491–7493 CrossRef CAS PubMed.
- For selected examples of the use of protecting group strategies, see: (a) L. Xu and P. Li, Chem. Commun., 2015, 51, 5656–5659 RSC; (b) J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed; (c) L. Xu, S. Ding and P. Li, Angew. Chem., Int. Ed., 2014, 53, 1822–1826 CrossRef CAS PubMed; (d) E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484–491 CrossRef CAS PubMed; (e) J. C. H. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894–899 CrossRef CAS PubMed; (f) N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548–2549 CrossRef CAS PubMed; (g) N. Iwadate and M. Suginome, Org. Lett., 2009, 11, 1899–1902 CrossRef CAS PubMed.
- For a recent review, see: J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636–2649 CrossRef CAS PubMed.
- For examples of the use of self-protection/activation, see: (a) L. Fang, L. Yan, F. Haeffner and J. P. Morken, J. Am. Chem. Soc., 2016, 138, 2508–2511 CrossRef CAS PubMed; (b) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef CAS PubMed; (c) S. N. Mlynarski, C. H. Schuster and J. P. Morken, Nature, 2014, 505, 386–390 CrossRef CAS PubMed; (d) C. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534–6537 CrossRef CAS PubMed; (e) J. Jiao, K. Hyodo, H. Hu, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 285–295 CrossRef CAS PubMed; (f) K. Endo, T. Ohkubo, M. Hirokami and T. Shibata, J. Am. Chem. Soc., 2010, 132, 11033–11035 CrossRef CAS PubMed; (g) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS PubMed.
- For the use of additives to allow chemoselectivity in aryl/benzyl systems, see: C. M. Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo, S. Voth, M. Hutchinson, V. S. Laberge, Y. Maekawa and D. Imao, Nat. Commun., 2016, 7, 11065 CrossRef PubMed.
- D. G. Hall, Structure, Properties, and Preparation of Boronic Acid Derivatives In Boronic Acids, Preparation and Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, 2005, ch. 1, pp. 1–133 Search PubMed.
- For detailed studies on boron speciation processes, see: (a) Y. Furikado, T. Nagahata, T. Okamoto, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi, A. Odani and K. Ishihara, Chem.–Eur. J., 2014, 20, 13193–13202 CrossRef PubMed; (b) T. Okamoto, A. Tanaka, E. Watanabe, T. Miyazaki, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi, A. Odani and K. Ishihara, Eur. J. Inorg. Chem., 2014, 2389–2395 CrossRef CAS; (c) E. Watanabe, C. Miyamoto, A. Tanaka, K. Iizuka, S. Iwatsuki, M. Inamo, H. D. Takagi and K. Ishihara, Dalton Trans., 2013, 42, 8446–8453 RSC; (d) M. A. Martinez-Aguirre, R. Villamil-Ramos, J. A. Guerrero-Alvarez and A. K. Yatsmirsky, J. Org. Chem., 2013, 78, 4674–4684 CrossRef CAS PubMed; (e) C. Miyamoto, K. Suzuki, S. Iwatsuki, M. Inamo, H. D. Takagi and K. Ishihara, Inorg. Chem., 2008, 47, 1417–1419 CrossRef CAS PubMed; (f) S. Iwatsuki, S. Nakajima, M. Inamo, H. D. Takagi and K. Ishihara, Inorg. Chem., 2007, 46, 354–356 CrossRef CAS PubMed; (g) J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS; (h) J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS; (i) L. I. Bosch, T. M. Fyles and T. D. James, Tetrahedron, 2004, 60, 11175–11190 CrossRef CAS; (j) G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS; (k) R. Pizer and C. A. Tihal, Polyhedron, 1996, 15, 3411–3416 CrossRef CAS; (l) L. Babcock and R. Pizer, Inorg. Chem., 1980, 19, 56–61 CrossRef CAS.
- For examples of boron speciation in synthesis, see: (a) C. W. Muir, J. C. Vantourout, A. Isidro-Llobet, S. J. F. Macdonald and A. J. B. Watson, Org. Lett., 2015, 17, 6030–6033 CrossRef CAS PubMed; (b) C. P. Seath, J. W. B. Fyfe, J. J. Molloy and A. J. B. Watson, Angew. Chem., Int. Ed., 2015, 54, 9976–9979 CrossRef CAS PubMed; (c) J. W. B. Fyfe, E. Valverde, C. P. Seath, A. R. Kennedy, N. A. Anderson, J. M. Redmond and A. J. B. Watson, Chem.–Eur. J., 2015, 21, 8951–8964 CrossRef CAS PubMed; (d) J. J. Molloy, R. P. Law, J. W. B. Fyfe, C. P. Seath, D. J. Hirst and A. J. B. Watson, Org. Biomol. Chem., 2015, 13, 3093–3102 RSC; (e) J. W. B. Fyfe, C. P. Seath and A. J. B. Watson, Angew. Chem., Int. Ed., 2014, 53, 12077–12080 CrossRef CAS PubMed.
- H. C. Brown, Organic Synthesis via Organoboranes, Wiley Interscience, New York, 1975 Search PubMed.
- For an example in the context of Suzuki–Miyaura cross-coupling, see: B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed.
- Oxone® is poorly soluble in organic solvents, typically requiring H2O or alcoholic media. For example, see: B. M. Trost and R. Braslau, J. Org. Chem., 1988, 53, 532–537 CrossRef CAS.
- For a discussion of basic biphase in the context of organoboron chemistry, see: (a) A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed; (b) A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 7431–7441 CrossRef CAS PubMed.
- For examples of equilibration of diols in mono-boron systems, see: C. D. Roy and H. C. Brown, Monatsh. Chem., 2007, 138, 879–887 CrossRef CAS.
- Increasing the interfacial area can profoundly affect reaction rate in biphasic systems. For examples, see: F. M. Menger, J. Am. Chem. Soc., 1970, 92, 5965–5971 CrossRef CAS.
- (a) S. Mothana, J.-M. Grassot and D. G. Hall, Angew. Chem., Int. Ed., 2010, 49, 2883–2887 CrossRef CAS PubMed; (b) S. Mothana, N. Chahal, S. Vanneste and D. G. Hall, J. Comb. Chem., 2007, 9, 193–196 CrossRef CAS PubMed.
- B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2016, 55, 3909–3913 CrossRef CAS PubMed.
- Selective boronate formation has been implicated within chemoselective cross-coupling of geminal diboron compounds, see ref. 9f.
- There is some disagreement over the Lewis acidity of boronic acid pinacol esters in the literature. Their lower reactivity with respect to boronic acids has led to the conclusion that they are less Lewis acidic (see ref. 2). However, the pH depression effect suggests that boronic esters are more Lewis acidic (for example, see ref. 11j). The majority of the data supports increased Lewis acidity.
- A. A. C. Braga, N. H. Morgon, G. Ujaque and F. Maseras, J. Am. Chem. Soc., 2005, 127, 9298–9307 CrossRef CAS PubMed.
- Oxidation was too rapid to allow robust quantification of boron species in either phase.
- R. A. Bowie and O. C. Musgrave, J. Chem. Soc., Chem. Commun., 1963, 3945–3949 CAS.
- Only “HO−” boronates are shown but mixed “H/DO−” boronates are likely to have been present in these experiments.
- Temperature-proportional downfield shifting of the NMR resonances was observed and consistent with the literature. See: R. E. Hoffman, Magn. Reson. Chem., 2006, 44, 606–616 CrossRef CAS PubMed.
- NMR experiments were not agitated.
- For discussions of protodeboronation, see: (a) P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed; (b) G. Noonan and A. G. Leach, Org. Biomol. Chem., 2015, 13, 2555–2560 RSC; (c) C.-Y. Lee, S.-J. Ahn and C.-H. Cheon, J. Org. Chem., 2013, 78, 12154–12160 CrossRef CAS PubMed; (d) J. Lozada, Z. Liu and D. M. Perrin, J. Org. Chem., 2014, 79, 5365–5368 CrossRef CAS PubMed; (e) H. G. Kuivila, J. F. Reuwer Jr and J. A. Mangravi, Can. J. Chem., 1963, 41, 3081–3090 CrossRef CAS; (f) K. Nahabedian and H. G. Kuivila, J. Am. Chem. Soc., 1961, 83, 2167–2174 CrossRef CAS; (g) H. G. Kuivila and K. V. Nahabedian, J. Am. Chem. Soc., 1961, 83, 2164–2166 CrossRef CAS; (h) H. G. Kuivila and K. V. Nahabedian, J. Am. Chem. Soc., 1961, 83, 2159–2163 CrossRef CAS.
- (a) For an 11B NMR study of boric acid and the solution dynamics with its borate derivative, see: K. Ishihara, A. Nagasawa, K. Umemoto, H. Ito and K. Saito, Inorg. Chem., 1994, 33, 3811–3816 CrossRef CAS; (b) For a comprehensive analysis of BMIDA hydrolysis, see: J. A. Gonzalez, O. M. Ogba, G. F. Morehouse, N. Rosson, K. N. Houk, A. G. Leach, P. H.-Y. Cheong, M. D. Burke and G. C. Lloyd-Jones, Nat. Chem. DOI:10.1038/nchem.2571.
- 1,4-Benzene diboronic acid monopinacol ester could be prepared but not isolated cleanly. This compound was found to rapidly equilibrate leading to isolation of diboronic acid and diester. Accordingly, the BMIDA/BPin compound 27 was used to interrogate the physical separation hypothesis.
- Moderately selective counter-statistical cross-coupling can be achieved with electronically-orthogonal boronic acids. For example, see: F. Beaumard, P. Dauban and R. H. Dodd, Org. Lett., 2009, 11, 1801–1804 CrossRef CAS PubMed.
- A selective protodeboronation of regioisomeric heterocyclic boronic acids has been reported, see: L. M. Klingensmith, M. M. Bio and G. A. Moniz, Tetrahedron Lett., 2007, 48, 8242–8245 CrossRef CAS.
- (a) D. M. T. Chan, K. L. Monaco, R. P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS; (b) D. A. Evans, J. L. Katz and T. R. West, Tetrahedron Lett., 1998, 39, 2937–2940 CrossRef CAS; (c) P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef CAS.
- J. C. Vantourout, R. P. Law, A. Isidro-Llobet, S. J. Atkinson and A. J. B. Watson, J. Org. Chem., 2016, 81, 3942–3950 CrossRef CAS PubMed.
- Z. Huang and J.-P. Lumb, Angew. Chem., Int. Ed., 2016, 55, 11543–11547 CrossRef CAS PubMed.
- F. Bedos-Belval, A. Rouch, C. Vanucci-Bacqué and M. Baltas, Med. Chem. Commun., 2012, 3, 1356–1372 RSC.
- For selected examples, see: (a) M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633–8643 CrossRef CAS PubMed; (b) M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287–4299 CrossRef CAS PubMed; (c) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (d) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, assay details and spectra, characterization data for all compounds. See DOI: 10.1039/c6sc04014d |
| ‡ The following numbering key has been used throughout: R(BOH)2 = Xa; RBPin = Xb; ROH = Xc; RB(OH)3− = Xd; RBPin(OH)− = Xe; RBMIDA = Xf. |
| § Line added as a visual aid – no function has been fitted. |
| This journal is © The Royal Society of Chemistry 2017 |