
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical optogenetic modulation of inflammation and immunity†
Bibudha
Parasar
 a and
Pamela V.
Chang
*b
a and
Pamela V.
Chang
*b
aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA
bDepartment of Microbiology and Immunology, Cornell University, 930 Campus Road, VMC C4-185, Ithaca, NY 14853, USA. E-mail: pamela.chang@cornell.edu
First published on 21st October 2016
Abstract
The immune system is an essential component of host defense against pathogens and is largely mediated by inflammatory molecules produced by immune cells, such as macrophages. These inflammatory mediators are regulated at the transcriptional level by chromatin-modifying enzymes including histone deacetylases (HDACs). Here we describe a strategy to regulate inflammation and immunity with photocontrolled HDAC inhibitors, which can be selectively delivered to target cells by UV irradiation to minimize off-target effects. We strategically photocaged the active moiety of an HDAC inhibitor and showed that mild UV irradiation leads to the selective release of the inhibitor in a spatiotemporal manner. This methodology was used to decrease the amount of pro-inflammatory mediators produced by a subpopulation of macrophages. Our approach could ultimately be used to control inflammation in vivo as a therapeutic for inflammatory diseases, while minimizing off-target effects to healthy tissues.
Introduction
Inflammation is a complex biological process that evolved as a protective response to eliminate the presence of noxious stimuli and initiate tissue repair.1 This physiological process is part of the host immune response, which serves as our main defense against infection by pathogens. The mammalian immune system is a complex network of specialized cell types that orchestrates the immune response through the secretion of many factors, including cytokines and chemokines.2 These proteins can generally be thought of as pro- or anti-inflammatory, and their levels in the local milieu greatly influence the inflammatory state of the tissue.Given the essential role of immune cells in regulating inflammation, dysregulation of immune responses can disrupt the delicate balance of pro- and anti-inflammatory signals that are necessary to maintain tissue homeostasis.3 This imbalance can lead to chronic inflammation and many inflammatory disorders, including autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, and type 1 diabetes, as well as allergy, cancer, and metabolic syndrome. These conditions often arise from an unnecessary increase in immune cell activation and inflammation in the absence of tissue injury or infection.
Macrophages are ubiquitous immune cells that play major roles in innate immunity, the host's first-line defense against infection.4 These cells act as sentinels that patrol tissues in search of foreign microorganisms, which in turn stimulate the macrophages to initiate innate immune responses. These responses include activation of phagocytosis and antigen presentation. In addition, macrophages greatly influence inflammation through their secretion of cytokines and chemokines, which results in the activation and recruitment of additional immune cells, including those from the adaptive immune system. Thus, the ability to modulate the inflammatory activities of these cells in a spatiotemporal manner would represent a powerful strategy to control unnecessary immune responses.
Gene regulation is mediated by chromatin-modifying enzymes, including histone acetyltransferases and histone deacetylases (HDACs).5 These writers and erasers, respectively, are thought to regulate gene transcription through the reversible chemical modification of histone lysine residues with post-translational modifications including acetylation. Previously, we have shown that inhibition of HDACs modulates macrophage activity through the downregulation of pro-inflammatory mediators, including microbicidal reactive nitrogen species (RNS) and various cytokines.6 This regulation was shown to be at the level of gene transcription, suggesting the potential therapeutic use of HDAC inhibitors for controlling immunity as epigenetic modulators of gene expression.
HDAC inhibitors are currently used both preclinically and clinically to treat many diseases, most notably neurodegenerative diseases and cancer.7 These therapeutics are typically delivered systemically to humans or mouse models of human disease; however, many HDAC inhibitors exhibit toxic side effects, including cardiotoxicity.8 In addition, HDAC inhibitors have pleiotropic effects and affect numerous cell types, so the need for technologies to selectively deliver them with precision is in high demand.9–11 Therefore, the ability to release HDAC inhibitors in a controlled, spatiotemporal manner would enable the local delivery of therapeutics to tissues of interest while minimizing off-target effects.
Chemical optogenetics is an emerging paradigm involving the use of light and small molecule probes to manipulate and study biological processes with spatiotemporal control.12 Photocaged approaches have been used to activate immune cells using UV light to uncage Toll-like receptor agonists.13,14 Here, we describe a photocontrolled chemical strategy to selectively deliver HDAC inhibitors to macrophages to downregulate local inflammation. Using this approach, the inhibitor is released in the vicinity of or within the target cell and can subsequently bind to HDACs within activated macrophages to decrease inflammation through changes in gene expression (Fig. 1). Similar methods have been used previously to photocage HDAC inhibitors;15,16 however, our approach is used to inhibit HDACs within live cells and to modulate cellular behavior and function. We envision that our strategy can be used for the therapeutic treatment of many inflammatory diseases, while avoiding unwanted side effects such as immunosuppression of other tissues, which could lead to infection by opportunistic pathogens.
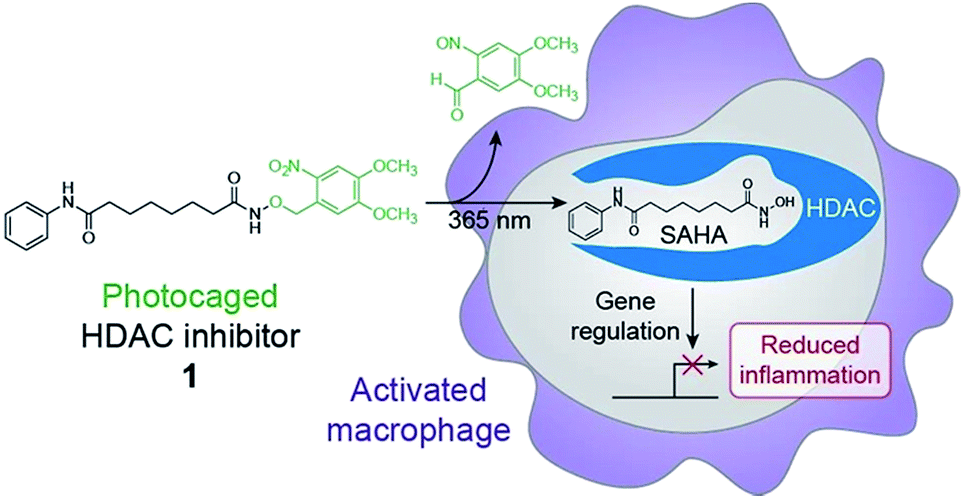 | ||
| Fig. 1 Schematic of chemical optogenetic approach for modulating inflammatory state of macrophages. UV light irradiation of photocaged histone deacetylase (HDAC) inhibitor 1 releases SAHA, which inhibits HDAC activity and affects macrophage function by reducing inflammation through changes in gene expression. | ||
Results and discussion
Suberoylanilide hydroxamic acid (SAHA) is a well-studied competitive inhibitor of class I and II Zn2+-dependent HDACs that binds within the enzyme active site.17 Importantly, SAHA is cell-permeable and blocks the effects of these enzymes at nanomolar concentrations in vitro.18 SAHA has been shown to have anti-inflammatory effects on immune cells,19 and we found that treatment of lipopolysaccharide (LPS)-stimulated murine bone marrow derived macrophages (BMDM) with SAHA led to a decrease in the production of the RNS nitric oxide (NO) (Fig. S1†), a free radical that has potent and broad microbial killing activity. SAHA also decreased the amounts of secreted pro-inflammatory cytokines IL-6 and IL-12 (Fig. S1†), which are involved in the activation of other cell types and instruction of adaptive immunity. These findings are in agreement with our previous work showing that pan-HDAC inhibition leads to downregulation of these pro-inflammatory mediators through alterations in gene transcription.6To control the release of SAHA in a spatiotemporal manner, we chose to cage its activity as an HDAC inhibitor with a photoremovable protecting group. In this design, a photolabile group is used to mask the hydroxamic acid moiety,15,16 which tightly binds to the Zn2+ cofactor required for catalytic activity within the HDAC active site (Fig. S2†).17 The photoremovable protecting group 6-nitroveratryl (NV) was selected because it is a well-characterized group that is released relatively quickly upon irradiation with UV light wavelengths that are compatible with biological systems.20 Compared to alternative photocages, the NV group enables a facile, one-step synthesis to obtain the photocaged HDAC inhibitor.15,16 Toward this goal, we have synthesized a photo-activatable HDAC inhibitor consisting of SAHA conjugated to NV via its hydroxamic acid (1, Fig. 1). We prepared 1 in a one-step displacement reaction from SAHA and 6-nitroveratryl bromide (ESI Scheme 1†).
We first assessed whether SAHA could be uncaged in vitro upon irradiation of 1 with 365 nm UV light. This treatment resulted in the rapid disappearance of 1 (k = 3.51 × 10−4 s−1; Fig. S3†) and release of SAHA, as analyzed by reverse-phase HPLC and mass spectrometry (Fig. 2A). The major product from this reaction was SAHA, though minor photodecomposition products were also observed, including the expected nitroso aldehyde and the amide analog of SAHA, which results from N–O bond cleavage upon UV irradiation (Fig. 2B and S4†).16,21 Importantly, the byproducts did not affect the amounts of IL-6, IL-12, and NO produced by LPS-stimulated BMDM (Fig. S4†). Furthermore, 1 was stable to decomposition in aqueous buffer at 37 °C for at least 24 h (Fig. S5†).
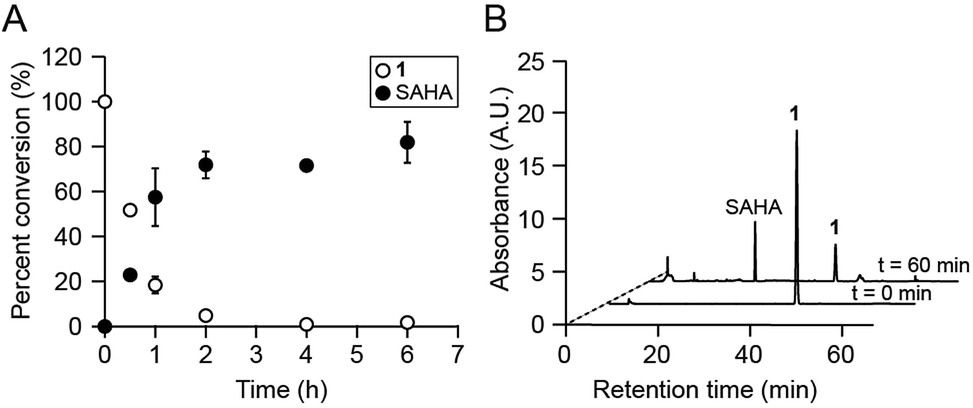 | ||
| Fig. 2 Compound 1 is photo-uncaged by UV light. (A) Percent conversion of 1 to SAHA after irradiation with 365 nm light. (B) HPLC analyses of photo-uncaging reactions. Traces shown were monitored by UV absorbance at 254 nm. A.U. = arbitrary units. | ||
We next verified that 1 does not inhibit HDACs until it is irradiated with UV light to release SAHA. In these studies, we used whole cell lysates from BMDM as a source of HDACs. We found that samples treated with SAHA and irradiated 1 inhibited HDAC activity. Importantly, non-irradiated 1 did not inhibit enzymatic activity (Fig. S6†).
We then determined whether photo-uncaging of 1 affects the inflammatory state of macrophages after UV irradiation. BMDM were stimulated with LPS to activate inflammatory pathways that lead to the production of pro-inflammatory factors, followed by treatment with 1 and UV irradiation. Similar to treatment with SAHA, photo-uncaging of 1 led to a decrease in the production of IL-6, IL-12, and NO, which was specific to exposure to UV light (Fig. 3). Importantly, treatment of cells with 1 or UV light did not result in increased cytotoxicity, as measured by the MTT cell viability assay (Fig. S7†). We also verified that UV light irradiation did not affect the production of these pro-inflammatory mediators by LPS-stimulated BMDM (Fig. S8†).
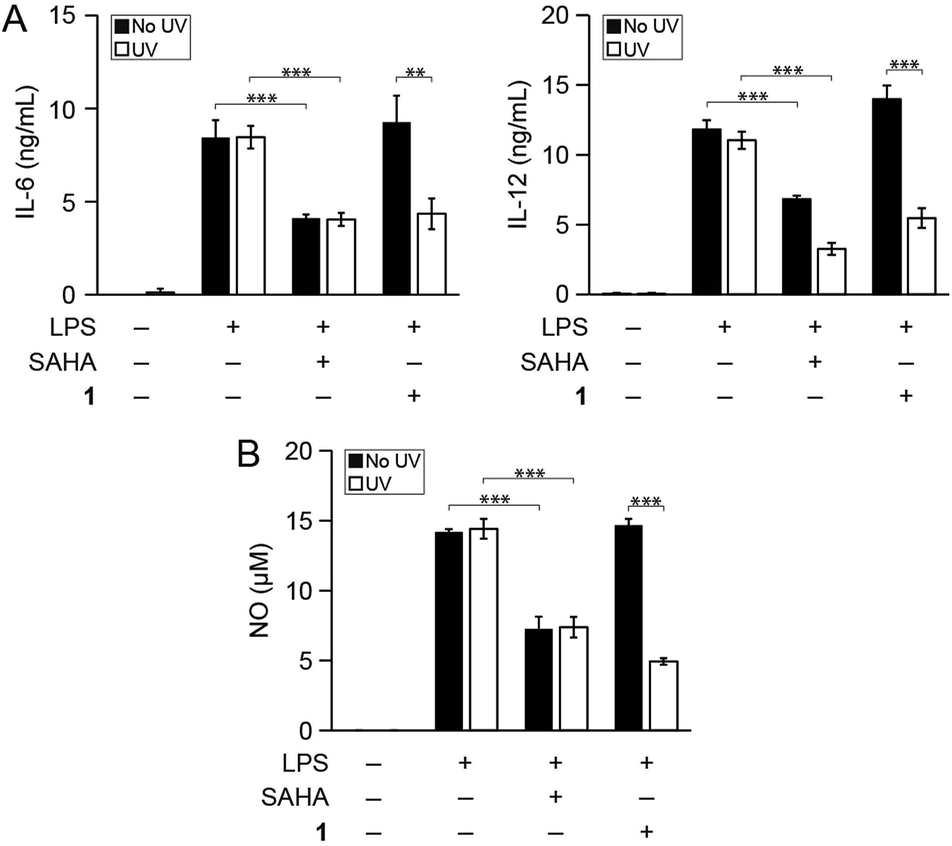 | ||
| Fig. 3 Photo-uncaging of 1 leads to a decrease in inflammatory mediator production by macrophages. Bone marrow derived macrophages (BMDM) from mice were treated with the indicated combinations of lipopolysaccharide (LPS, 100 ng mL−1), SAHA (500 nM), and/or 1 (3 μM). Cells were then irradiated with UV light (365 nm, 10 min, white bars) or kept in the dark (black bars), and secretion of (A) IL-6 and IL-12 and (B) NO were measured by ELISA and the Griess assay, respectively, after 24 h. **p < 0.01, ***p < 0.001. | ||
Finally, to showcase the high level of spatial control afforded by our technique, we used fluorescence microscopy to uncage 1 in subpopulations of BMDM and subsequently evaluate levels of cytokine production in these cells. For these studies, we retrovirally transduced BMDM with a photoswitchable fluorescent protein, mEos3.2, to mark the irradiated cells.22 We pre-treated BMDM with 1 overnight to pre-load the cells with the probe and then irradiated a subpopulation of cells with UV light to selectively uncage 1 within only these cells after removing excess un-internalized probe. Exclusive uncaging of internalized 1 is designed to prevent off-target effects on surrounding cell populations. We estimate that the effective concentration of SAHA that is released within the cells upon UV irradiation is approximately 500 pM (Fig. S9†). Next, we stimulated the BMDM with LPS to activate the cells. After allowing for cytokine production over several hours in the presence of a protein secretion inhibitor, we performed immunofluorescence staining for intracellular IL-12. Confocal imaging revealed that within the same dish, only the irradiated region of BMDM (exhibiting red mEos3.2-derived fluorescence) exhibited lower levels of cytokine, similar to SAHA-treated cells, while the peripheral, non-irradiated cells (exhibiting only green mEos3.2-derived fluorescence) were more activated, producing similar levels of IL-12 to LPS-activated BMDM (Fig. 4). The striking spatial resolution of this approach can be appreciated by examining adjacent irradiated and non-irradiated cell populations (Fig. 5, dotted line denotes the border region of illumination).
 | ||
| Fig. 4 Photo-uncaging of 1 is spatially selective and leads to a decrease in IL-12 production by a subpopulation of macrophages. BMDM expressing mEos3.2 were pre-treated with 1 (3 μM), and the center of the dish was irradiated with UV light (epifluorescence using DAPI filter, 30 s). The BMDM were then activated with the indicated combinations of LPS (100 ng mL−1) and/or SAHA (500 nM). After 6 h, the cells were fixed, and immunofluorescence labeling for IL-12 was performed. (A) Schematic of experimental setup. (B) Maximum intensity z-projection images of cells in the periphery (top three rows, no UV) and center (bottom three rows, UV) of the same dish, imaged by confocal microscopy. Green indicates non-photoconverted state of mEos3.2 and red indicates photoconverted state of mEos3.2. Scale bar: 50 μm. (C) Quantification of data from (B), where % activated cells represents the percentage of IL-12-positive cells. n.s. = not significant, *p < 0.05. | ||
 | ||
| Fig. 5 Photo-uncaging of 1 is spatially selective and leads to a decrease in IL-12 production in only the irradiated macrophages. BMDM were pre-treated with 1 (3 μM), and the center of the dish was irradiated with UV light (epifluorescence using DAPI filter, 30 s). The BMDM were then activated with LPS (100 ng mL−1). After 6 h, the cells were fixed, and immunofluorescence labeling for IL-12 was performed, followed by imaging with confocal microscopy. Shown are maximum intensity z-projection images of cells at the border of UV irradiation. Dotted line indicates the border region of illumination. First three panels represent monochrome images of non-photoconverted mEos3.2, photoconverted mEos3.2, and IL-12, respectively. Far right panel represents merged image of photoconverted mEos3.2 (red) and IL-12 (cyan). Scale bar: 50 μm. | ||
Conclusions
We have developed a strategy for modulating inflammation and immunity in a spatiotemporal manner using a photo-activatable HDAC inhibitor. Given that inflammation is linked to many disorders, including autoimmune diseases and cancer, this approach has promise for the selective delivery of therapeutics to limit off-target effects.23 HDAC inhibitors such as SAHA are already FDA-approved for the clinical treatment of cutaneous T cell lymphoma in humans and have the potential to modulate epigenetic processes in many diseases.8 Furthermore, HDAC inhibitors have pleiotropic effects that can act synergistically within the target tissue, and pan-HDAC inhibitors such as SAHA have anti-inflammatory effects on multiple immune cell types.24 Thus, the selective delivery of photo-activatable HDAC inhibitors could elicit different desirable physiological outcomes, depending on the target tissue and the disease state of the organism.Acknowledgements
This work was supported by a PCCW Affinito-Stewart Grant. We thank Timothy Bumpus, Jessica Daughtry, Gael Nicolas, and Samantha Scott for technical assistance. We also thank Xiaoyu Zhang, Prof. Hening Lin, Prof. Helene Marquis, Prof. David Russell, and the Weill Institute for Cell and Molecular Biology for resources and reagents.Notes and references
- R. Medzhitov, Nature, 2008, 454, 428–435 CrossRef CAS PubMed.
- A. Miyajima, T. Kitamura, N. Harada, T. Yokota and K. Arai, Annu. Rev. Immunol., 1992, 10, 295–331 CrossRef CAS PubMed.
- W. S. Garrett, J. I. Gordon and L. H. Glimcher, Cell, 2010, 140, 859–870 CrossRef CAS PubMed.
- C. K. Glass and G. Natoli, Nat. Immunol., 2015, 17, 26–33 CrossRef PubMed.
- V. W. Zhou, A. Goren and B. E. Bernstein, Nat. Rev. Genet., 2010, 12, 7–18 CrossRef PubMed.
- P. V. Chang, L. Hao, S. Offermanns and R. Medzhitov, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2247–2252 CrossRef CAS PubMed.
- K. J. Falkenberg and R. W. Johnstone, Nat. Rev. Immunol., 2014, 13, 673–691 CAS.
- A. C. West and R. W. Johnstone, J. Clin. Invest., 2014, 124, 30–39 CrossRef CAS PubMed.
- A. R. Maolanon, A. S. Madsen and C. A. Olsen, Cell Chem. Biol., 2016, 23, 759–768 CrossRef CAS PubMed.
- A. V. Bieliauskas and M. K. H. Pflum, Chem. Soc. Rev., 2008, 37, 1402–1413 RSC.
- S. A. Reis, B. Ghosh, J. A. Hendricks, D. M. Szantai-Kis, L. Törk, K. N. Ross, J. Lamb, W. Read-Button, B. Zheng, H. Wang, C. Salthouse, S. J. Haggarty and R. Mazitschek, Nat. Chem. Biol., 2016, 12, 317–323 Search PubMed.
- T. Fehrentz, M. Schönberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156–12182 CrossRef CAS PubMed.
- K. A. Ryu, L. Stutts, J. K. Tom, R. J. Mancini and A. P. Esser-Kahn, J. Am. Chem. Soc., 2014, 136, 10823–10825 CrossRef CAS PubMed.
- J. M. Govan, D. D. Young, M. O. Lively and A. Deiters, Tetrahedron Lett., 2015, 56, 3639–3642 CrossRef CAS PubMed.
- N. Ieda, S. Yamada, M. Kawaguchi, N. Miyata and H. Nakagawa, Bioorg. Med. Chem., 2016, 24, 2789–2793 CrossRef CAS PubMed.
- A. Leonidova, C. Mari, C. Aebersold and G. Gasser, Organometallics, 2016, 35, 851–854 CrossRef CAS.
- B. E. L. Lauffer, R. Mintzer, R. Fong, S. Mukund, C. Tam, I. Zilberleyb, B. Flicke, A. Ritscher, G. Fedorowicz, R. Vallero, D. F. Ortwine, J. Gunzner, Z. Modrusan, L. Neumann, C. M. Koth, P. J. Lupardus, J. S. Kaminker, C. E. Heise and P. Steiner, J. Biol. Chem., 2013, 288, 26926–26943 CrossRef CAS PubMed.
- J. E. Bradner, N. West, M. L. Grachan, E. F. Greenberg, S. J. Haggarty, T. Warnow and R. Mazitschek, Nat. Chem. Biol., 2010, 6, 238–243 CrossRef CAS PubMed.
- T. Roger, J. Lugrin, D. Le Roy, G. Goy, M. Mombelli, T. Koessler, X. C. Ding, A. L. Chanson, M. K. Reymond, I. Miconnet, J. Schrenzel, P. Francois and T. Calandra, Blood, 2011, 117, 1205–1217 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- W. R. Grither, J. Korang, J. P. Sauer, M. P. Sherman, P. L. Vanegas, M. Zhang and R. D. McCulla, J. Photochem. Photobiol., A, 2012, 227, 1–10 CrossRef CAS.
- M. Zhang, H. Chang, Y. Zhang, J. Yu, L. Wu, W. Ji, J. Chen, B. Liu, J. Lu, Y. Liu, J. Zhang, P. Xu and T. Xu, Nat. Methods, 2012, 9, 727–729 CrossRef CAS PubMed.
- B. E. Gryder, Q. H. Sodji and A. K. Oyelere, Future Med. Chem., 2012, 4, 505–524 CrossRef CAS PubMed.
- R. Tao, E. F. de Zoeten, E. Özkaynak, C. Chen, L. Wang, P. M. Porrett, B. Li, L. A. Turka, E. N. Olson, M. I. Greene, A. D. Wells and W. W. Hancock, Nat. Med., 2007, 13, 1299–1307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and supplemental figures. See DOI: 10.1039/c6sc03702j |
| This journal is © The Royal Society of Chemistry 2017 |