DOI:
10.1039/C6SC03669D
(Edge Article)
Chem. Sci., 2017,
8, 85-90
Pyramidalization/twisting of the amide functional group via remote steric congestion triggered by metal coordination†
Received
16th August 2016
, Accepted 21st September 2016
First published on 23rd September 2016
Abstract
For decades, the planarity of the amide functional group has garnered sustained interest in organic chemistry, enticing chemists to deform its usually characteristic high-fidelity plane. As opposed to the construction of amides that are distorted by imposing rigid covalent bond assemblies, we demonstrate herein the deformation of the amide plane through increased steric bulk in the periphery of the amide moiety, which is induced by coordination to metal cations. A crystallographic analysis revealed that the thus obtained amides exhibit significant pyramidalization and twisting upon coordination to the metals, while the amide functional group remained intact. The observed deformation, which should be attributed to through-space interactions, substantially enhanced the solvolytic cleavage of the amide, providing compelling evidence that temporary crowding in the periphery of the amide functional group may be used to control the reactivity of amides.
Introduction
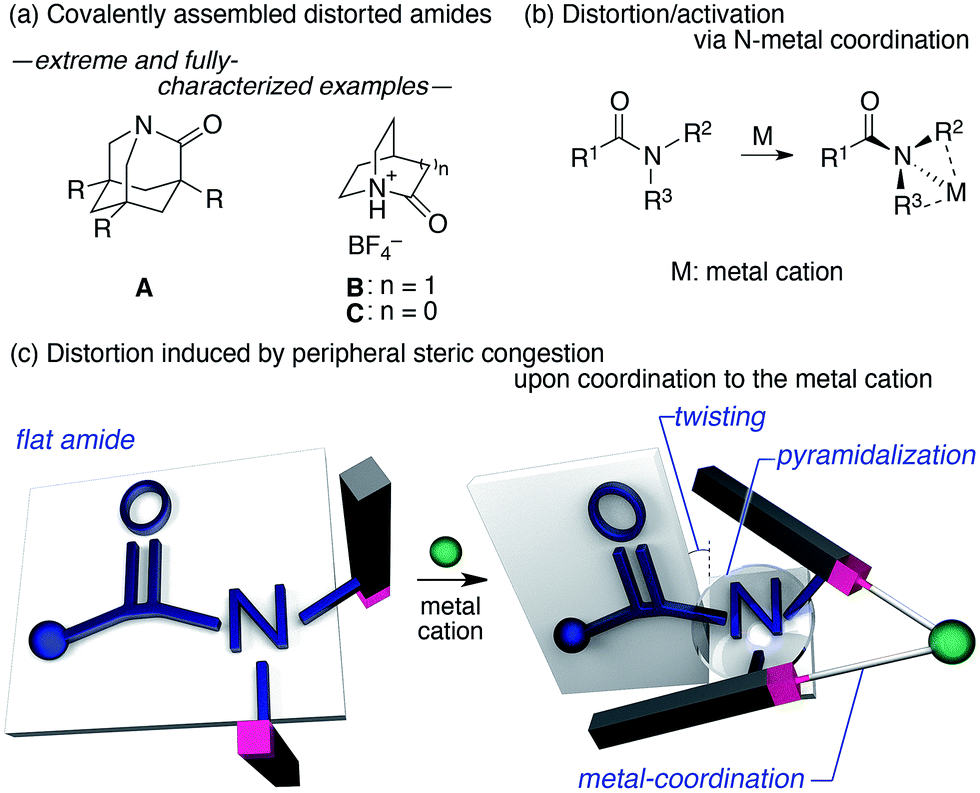
The amide bond is characterized by its thermodynamic stability and kinetic tolerance toward hydrolytic cleavage, which arises from conjugation via the planar O–C–N array.1 Under neutral conditions in aqueous solution at ambient temperature, non-activated amide bonds have a half-life of ca. 100 years.2 This high stability is usually attributed to the resonance interaction between the nN and π*C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O orbitals, which occurs most efficiently in a planar geometry with a shortened and stronger C–N bond. Therefore, amide bonds are commonly used as a robust structural motif (e.g. in synthetic polymers), and the hydrolysis of the amide bond in a practical timescale requires general harsh conditions (e.g. high or low pH at elevated temperatures). The deformation of the co-planarity of the amide bond represents an intuitive strategy to lower its robustness, which was initially proposed by Lukeš in 1938, who presented a model of strained “twisted amides” with a nitrogen atom at the bridgehead position.3 The longstanding pursuit toward twisted amides led to the identification of extreme examples of fully-characterized and highly distorted amides (A–C),4,5 which exhibited twist angles (τ) and pyramidalization (χN) values at the nitrogen, defined by Winkler and Dunitz,6 of ca. 90° and 60°, respectively (Fig. 1a). The prime importance of the resonance interaction for stabilizing the amide bond manifests in the case of B, which lacks an amide resonance, and exhibits a remarkably short half-life of <15 s in water.5d Besides these extreme examples, a number of amides that exhibit unusual τ and χN values have been reported, typically using covalently distorted bridged lactam architectures.1,7,8 These structurally intriguing bonds may not only be of fundamental academic interest, but also of significant applied importance in life science, considering that amides constitute the primary backbone of proteins. Indeed, the involvement of distorted amides has been invoked for enzymatic transformations.1,9 In this context, we were interested in distorting the amide planarity using an external trigger; more specifically, instead of constructing covalently assembled distorted amides, we aimed at inducing the amide deformation via temporary non-covalent interactions. Although several reports link the deformation and activation (e.g. hydrolysis and E/Z isomerization) of amides to the coordination of the amide nitrogen to metal cations (Fig. 1b),10,11 substantial amide deformation without direct coordination of the amide nitrogen or oxygen has, to the best of our knowledge, not yet been reported. Herein, we show that it is possible to induce significant pyramidalization and twisting of the amide functional group by remote steric congestion upon the coordination of the substituents attached to the amide nitrogen to a metal cation (Fig. 1c). A crystallographic analysis revealed that the substantial deformation of the amide planarity occurs without direct coordination of the amide. Peripheral crowding being a viable strategy to weaken the amide linkage is supported by the observed rapid solvolysis of the thus obtained distorted amides.12
O orbitals, which occurs most efficiently in a planar geometry with a shortened and stronger C–N bond. Therefore, amide bonds are commonly used as a robust structural motif (e.g. in synthetic polymers), and the hydrolysis of the amide bond in a practical timescale requires general harsh conditions (e.g. high or low pH at elevated temperatures). The deformation of the co-planarity of the amide bond represents an intuitive strategy to lower its robustness, which was initially proposed by Lukeš in 1938, who presented a model of strained “twisted amides” with a nitrogen atom at the bridgehead position.3 The longstanding pursuit toward twisted amides led to the identification of extreme examples of fully-characterized and highly distorted amides (A–C),4,5 which exhibited twist angles (τ) and pyramidalization (χN) values at the nitrogen, defined by Winkler and Dunitz,6 of ca. 90° and 60°, respectively (Fig. 1a). The prime importance of the resonance interaction for stabilizing the amide bond manifests in the case of B, which lacks an amide resonance, and exhibits a remarkably short half-life of <15 s in water.5d Besides these extreme examples, a number of amides that exhibit unusual τ and χN values have been reported, typically using covalently distorted bridged lactam architectures.1,7,8 These structurally intriguing bonds may not only be of fundamental academic interest, but also of significant applied importance in life science, considering that amides constitute the primary backbone of proteins. Indeed, the involvement of distorted amides has been invoked for enzymatic transformations.1,9 In this context, we were interested in distorting the amide planarity using an external trigger; more specifically, instead of constructing covalently assembled distorted amides, we aimed at inducing the amide deformation via temporary non-covalent interactions. Although several reports link the deformation and activation (e.g. hydrolysis and E/Z isomerization) of amides to the coordination of the amide nitrogen to metal cations (Fig. 1b),10,11 substantial amide deformation without direct coordination of the amide nitrogen or oxygen has, to the best of our knowledge, not yet been reported. Herein, we show that it is possible to induce significant pyramidalization and twisting of the amide functional group by remote steric congestion upon the coordination of the substituents attached to the amide nitrogen to a metal cation (Fig. 1c). A crystallographic analysis revealed that the substantial deformation of the amide planarity occurs without direct coordination of the amide. Peripheral crowding being a viable strategy to weaken the amide linkage is supported by the observed rapid solvolysis of the thus obtained distorted amides.12
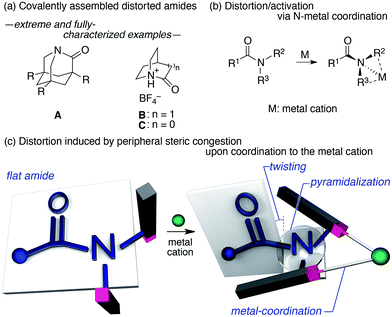 |
| | Fig. 1 (a) Extreme examples of highly distorted amides in a covalent framework, (b) deformation/activation of amides via the coordination of the amide nitrogen to metals, and (c) the distortion of the amide induced by peripheral steric constraints upon the coordination to metal cations. | |
Results and discussion
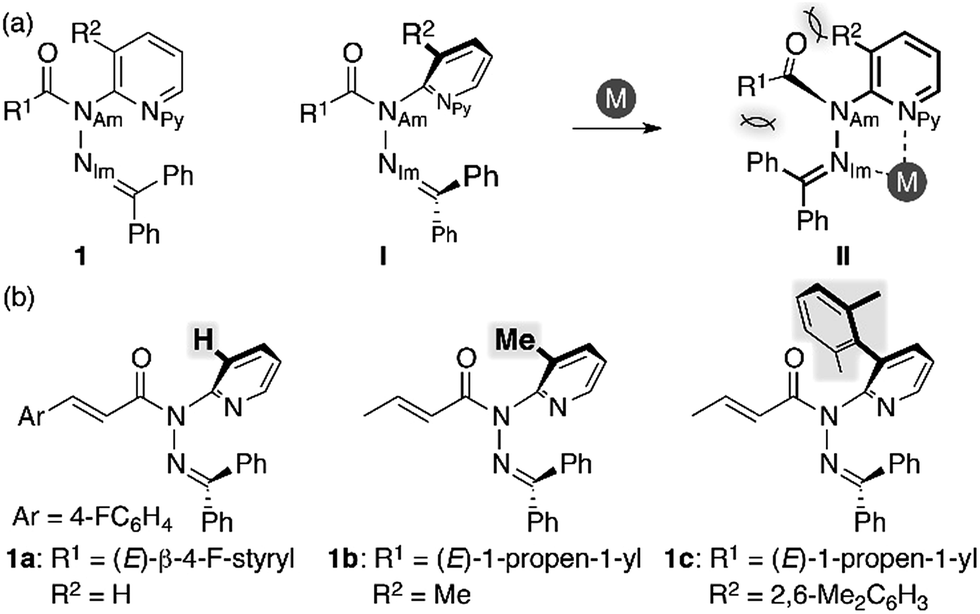
We began our study by designing a suitable amide with metal-coordination sites that may be able to create a steric bias upon the addition of appropriate metal cations. As the metal coordination should be orthogonal to the amide functional group, we selected a combination of azophilic metals and nitrogen-based bidentate coordination sites. Fig. 2a shows the generic structure of amide 1 with a 3-substituted-2-hydrazonopyridine moiety, which contains an amide (NAm) and an adjacent imine (hydrazone of benzophenone; NIm) functional group. It should be noted that 1 prefers a planar amide structure, which is achieved by tilting the pyridine ring and the imine along the C(pyridine)–NAm and NAm–NIm single bonds, respectively (Fig. 2a, I). Conversely, we anticipated that the addition of azophilic cations (M) should induce a bidentate chelation through NIm and NPy, thus affording a rigid and planar 5-membered cycle. This conformational change should provoke both the bulky benzophenone imine and the R2 group on the pyridine ring to swing close to the amide, thus compromising the amide planarity via through-space steric bias (Fig. 2a, II). With this blueprint in hand, we set out to synthesize three derivatives, which contain R2 groups of varying steric bulk: H (1a), Me (1b), and 2,6-dimethylphenyl (1c) (Fig. 2b).13 While 1b and 1c were synthesized as (E)-crotonyl amides, 1a was based on a p-fluorocinnamoyl amide in order to increase its crystallinity.
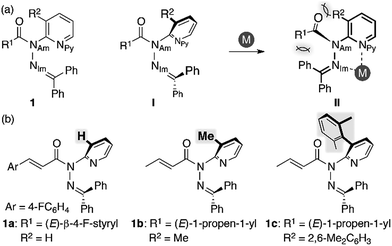 |
| | Fig. 2 (a) Amide 1 with a 3-substituted-2-hydrazonopyridine moiety as a model compound and (b) amides 1a–c with R2 substituents of different sizes. | |
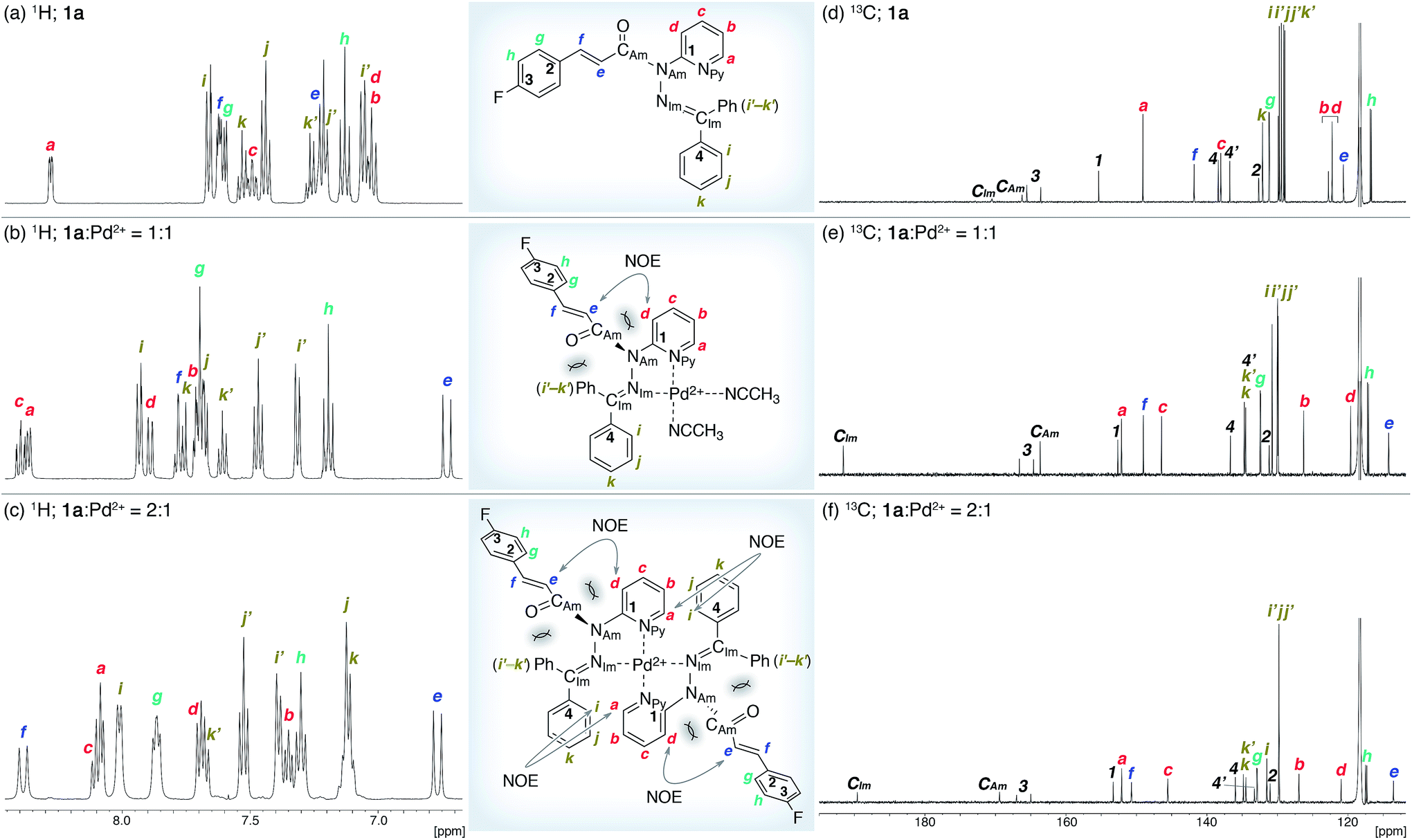
The coordination of 1a to azophilic Pd2+ cations, which favor a square-planar coordination mode, afforded in aprotic solvents the corresponding complexes. The formation of these complexes, which are thermodynamically stable under anhydrous conditions at ambient temperature, was monitored using 1H and 13C NMR spectroscopy. The NMR analysis revealed that 1a/Pd (1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd = 1:1) and (1a)2/Pd (1a:Pd = 2:1) complexes were formed depending on the ratio of 1a and [Pd(CH3CN)4](BF4)2 (Fig. 3). In CD3CN, a 1:1 mixture of 1a and [Pd(CH3CN)4(BF4)2 favored the formation of 1a/Pd. The observed NOE signals between He and Hd are consistent with the anticipated coordination mode via Npy and NIm, in which the amide nitrogen NAm is left uncoordinated (Fig. 3a and b).14 The characteristic downfield shift of the β-olefinic proton Hf upon complexation implied an increased polarization of the CO bond via distortion of the amide moiety. The formation of the homoleptic complex (1a)2/Pd from bidentate coordination via Npy and NIm induced similar spectral changes in the 1H and 13C NMR spectra, together with diagnostic NOE signals between the two 1a fragments (Ha and Hi; Fig. 3c). For (1a)2/Pd, the observed downfield shift of Hf was even more pronounced, which was tentatively ascribed to the deshielding effect of the phenyl group on the opposite 1a/Pd fragment (Fig. 3b and c and 4d; vide infra). Unfortunately, the chemical shifts in the 13C NMR spectra were not straightforward to interpret (Fig. 3d–f); in contrast to the rather subtle changes to the resonances for the amide carbonyl (CAm) moiety, the signal for the imino carbonyl (CIm) fragment experienced a substantial downfield shift. In the Ph group-rich environment of these complexes, the downfield shift of the amide carbonyl via distortion and the imino carbonyl via direct coordination might be increased and decreased, respectively, by shielding and deshielding effects from nearby multiple bonds (vide infra).
:Pd = 1:1) and (1a)2/Pd (1a:Pd = 2:1) complexes were formed depending on the ratio of 1a and [Pd(CH3CN)4](BF4)2 (Fig. 3). In CD3CN, a 1:1 mixture of 1a and [Pd(CH3CN)4(BF4)2 favored the formation of 1a/Pd. The observed NOE signals between He and Hd are consistent with the anticipated coordination mode via Npy and NIm, in which the amide nitrogen NAm is left uncoordinated (Fig. 3a and b).14 The characteristic downfield shift of the β-olefinic proton Hf upon complexation implied an increased polarization of the CO bond via distortion of the amide moiety. The formation of the homoleptic complex (1a)2/Pd from bidentate coordination via Npy and NIm induced similar spectral changes in the 1H and 13C NMR spectra, together with diagnostic NOE signals between the two 1a fragments (Ha and Hi; Fig. 3c). For (1a)2/Pd, the observed downfield shift of Hf was even more pronounced, which was tentatively ascribed to the deshielding effect of the phenyl group on the opposite 1a/Pd fragment (Fig. 3b and c and 4d; vide infra). Unfortunately, the chemical shifts in the 13C NMR spectra were not straightforward to interpret (Fig. 3d–f); in contrast to the rather subtle changes to the resonances for the amide carbonyl (CAm) moiety, the signal for the imino carbonyl (CIm) fragment experienced a substantial downfield shift. In the Ph group-rich environment of these complexes, the downfield shift of the amide carbonyl via distortion and the imino carbonyl via direct coordination might be increased and decreased, respectively, by shielding and deshielding effects from nearby multiple bonds (vide infra).
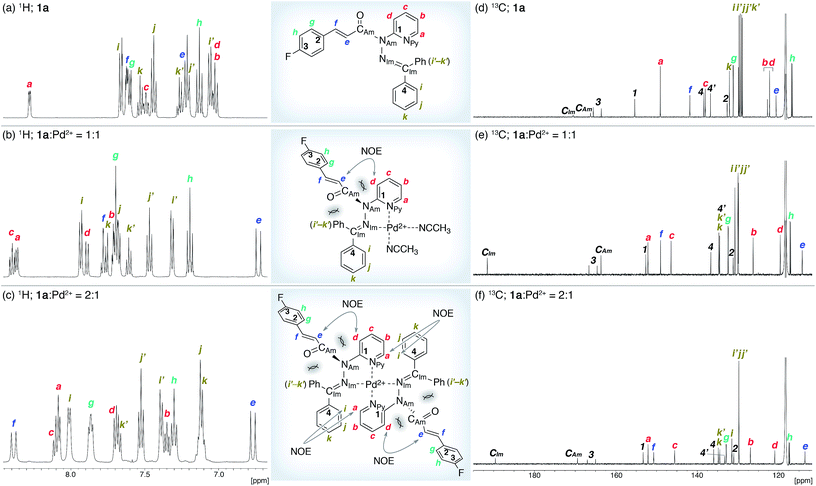 |
| | Fig. 3 NMR analysis of 1a and its Pd2+ complexes in CD3CN. (a and d) 1H and 13C NMR spectra of 1a. (b and e) 1H and 13C NMR spectra of a 1:1 mixture of 1a and [Pd(CH3CN)4](BF4)2. (c and f) 1H and 13C NMR spectra of a 2:1 mixture of 1a and [Pd(CH3CN)4](BF4)2. | |
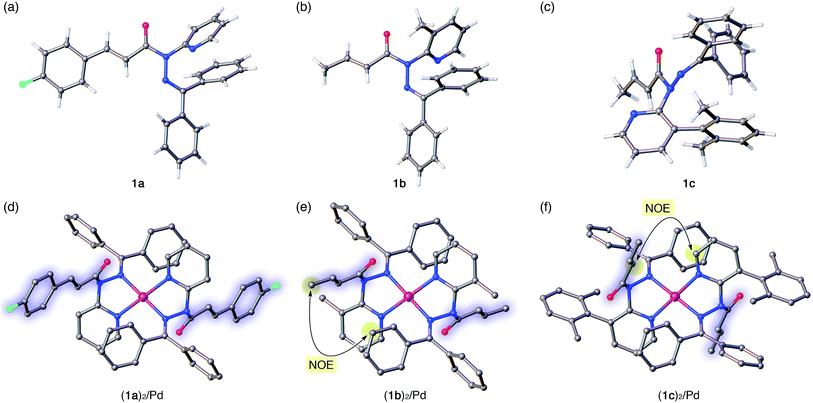 |
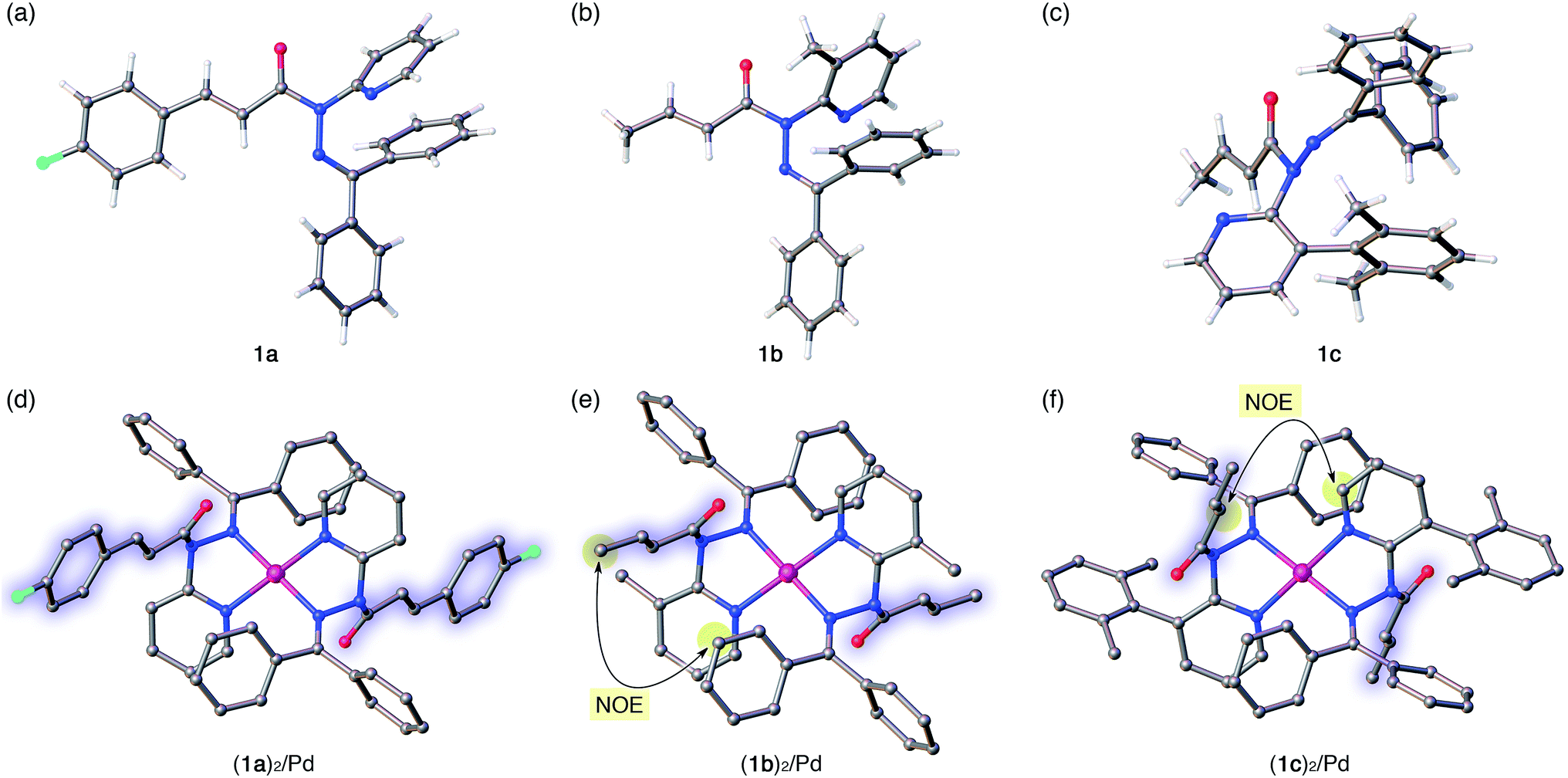
| | Fig. 4 Crystal structures of amides 1a–c (a–c) and of their Pd complexes (1a)2/Pd, (1b)2/Pd, and (1c)2/Pd (d–f). For the Pd complexes, hydrogen atoms and BF4− anions are omitted for clarity. In (d)–(f), the amide fragments are highlighted. Color code: white = hydrogen, gray = carbon, blue = nitrogen, red = oxygen, sky blue = fluorine, and pink = palladium. In (e) and (f), characteristic NOE signals between protons observed in the NMR analysis in solution are highlighted. | |
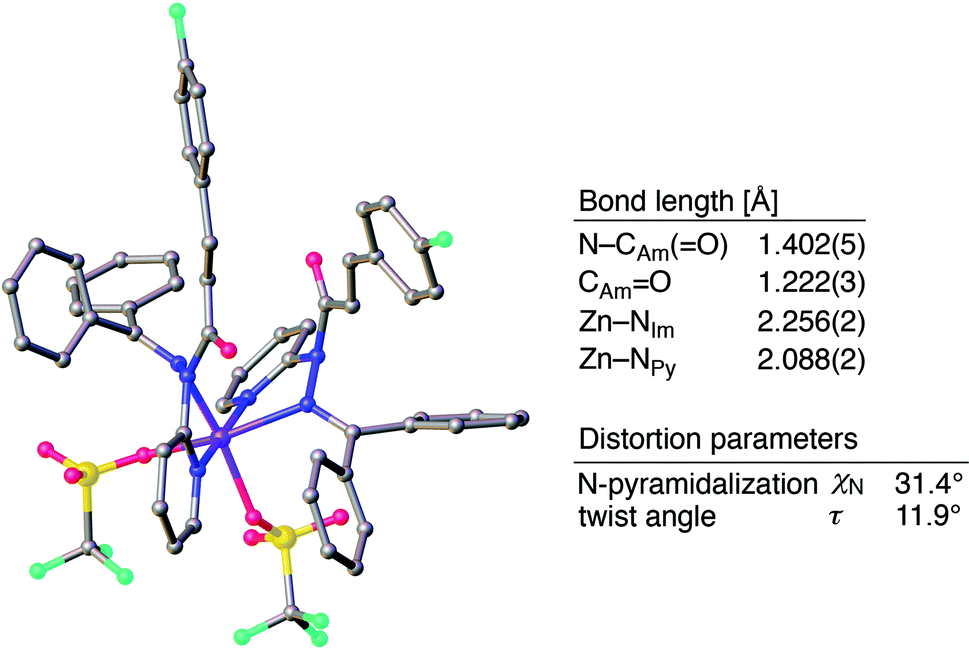
The distortion of the amide plane in 1a by peripheral crowding was further examined via single-crystal X-ray diffraction analysis (Fig. 4a and d). Single crystals of amide 1a and its Pd complex (1a)2/Pd were obtained from acetone/hexane, and their solid-state structures are shown in Fig. 4a and d, while selected bond lengths and distortion parameters are summarized in Table 1. In 1a, the pyridyl and the hydrazine group occupy the far side of the amide group in order to minimize steric repulsion. The amide group exhibited a negligible twist angle (τ = 3.0°), whereas partial pyramidalization was observed for the amide nitrogen (χN = 19.6°). In stark contrast, the complex (1a)2/Pd exhibited a significant pyramidalization (χN = 54.1°) of the amide nitrogen, and the expected bidentate coordination via Npy and NIm was confirmed. The pyramidalization of the amide nitrogen, in combination with a twisting of the amide (τ = 12.1°) resulted in a diminished amide conjugation, which is consistent with the observed decreased CAmO and increased N–CAm(O) bond lengths (Table 1). It should be noted here that the pyramidalization and twisting do not originate from the direct coordination to the amide functional group, but from the steric congestion in the periphery of the amide functional group.10b,d Similar to other covalently assembled distorted amides, the pyramidalization of the carbonyl carbon (χC) was negligible for both 1a and (1a)2/Pd. The unusual downfield shift of the imino carbonyl (CIm) resonance in the 13C NMR spectrum may arise from the deshielding effects of the amide CO bond in close proximity (Fig. 3d and f, and Table 1). Similarly, the orientation of the Ph groups in (1a)2/Pd most likely influences the chemical shift of the amide carbonyl carbon (CAm) and the β-olefinic hydrogen Hf (Fig. 3c, e and f and Table 1).
Table 1 Selected 13C NMR shifts and structural parameters for the crystal structures of 1a–c and their Pd complexes (1a)2/Pd–(1c)2/Pd
|
|
13C NMRa [ppm] |
Bond length [Å] |
Distortion parametersb |
| CAm |
CIm |
N–CAm(O) |
CAmO |
Pd–NIm |
Pd–NPy |
χ
N
|
χ
C
|
τ
|
|
Chemical shifts in CD3CN.
χ
N, χC, and τ denote the pyramidalization of the amide nitrogen atom, the carbonyl carbon atom, and the twist angle, respectively.6
|
|
1a
|
166.2 |
170.5 |
1.386(1) |
1.227(1) |
|
|
19.6° |
1.8° |
1.8° |
| (1a)2/Pd |
169.4 |
189.6 |
1.425(6) |
1.212(6) |
2.003(5) |
2.024(4) |
54.1° |
1.4° |
12.2° |
|
1b
|
168.1 |
161.9 |
1.394(3) |
1.224(3) |
|
|
28.3° |
2.7° |
4.1° |
| (1b)2/Pd |
169.0 |
189.8 |
1.421(7) |
1.216(6) |
1.996(3) |
2.022(4) |
56.3° |
3.1° |
19.3° |
|
1c
|
163.8 |
169.8 |
1.407(3) |
1.213(4) |
|
|
43.8° |
4.3° |
19.2° |
| (1c)2/Pd |
171.0 |
187.9 |
1.444(9) |
1.211(9) |
2.019(5) |
2.018(5) |
51.9° |
0.5° |
4.3° |
The crystallographic analysis of amides 1b and 1c as well as of their Pd2+ complexes revealed an identical coordination mode via Npy and NIm, as well as distortion of the amide moiety (Fig. 4b, c, e and f).15 Although the free amides 1b and 1c showed only a moderate pyramidalization of the amide nitrogen (χN), presumably due to the increased steric demand of R2 (1a: R2 = H, 1b: R2 = Me, 1c: R2 = 2,6-dimethylphenyl), the pyramidalization was significantly enhanced upon coordination to Pd2+. The elongated CAmO and compressed N–CAm(O) bonds suggest a diminished amide conjugation (Table 1). It should also be noted that all three complexes show χN values beyond 50°. Similar spectral (1H and 13C NMR) patterns were observed upon complexation of 1b and 1c. More importantly, characteristic NOE signals were observed between the two amide fragments in (1b)2/Pd and (1c)2/Pd, which provides compelling evidence for significant N-pyramidalization in solution (Fig. 4e and f).16 For 1c, some anomalies were observed: while 1a and 1b are E-amides in their free form and afford homoleptic complexes (1)2/Pd with Z-amides, the structural behavior of 1c is antipodal. Moreover, the twist angle (τ) decreased upon the coordination of 1c to Pd2+, whereas the planarity of the amide moiety was substantially disrupted by considerable pyramidalization. For amide 1c, a 1:1 Pd complex (1c/Pd) could be isolated, which exhibited an identical coordination mode with amide distortion (χN = 57.5°),17 thus supporting the structure that was assigned to 1a/Pd (Fig. 3b and e) on the basis of the NMR analysis.
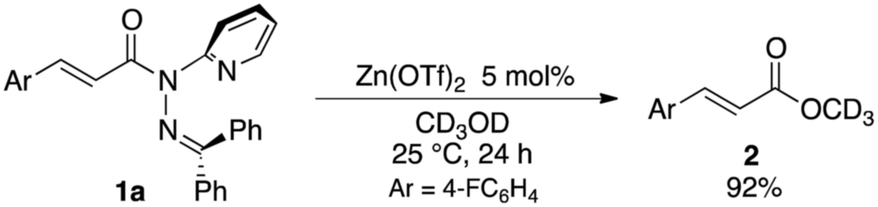
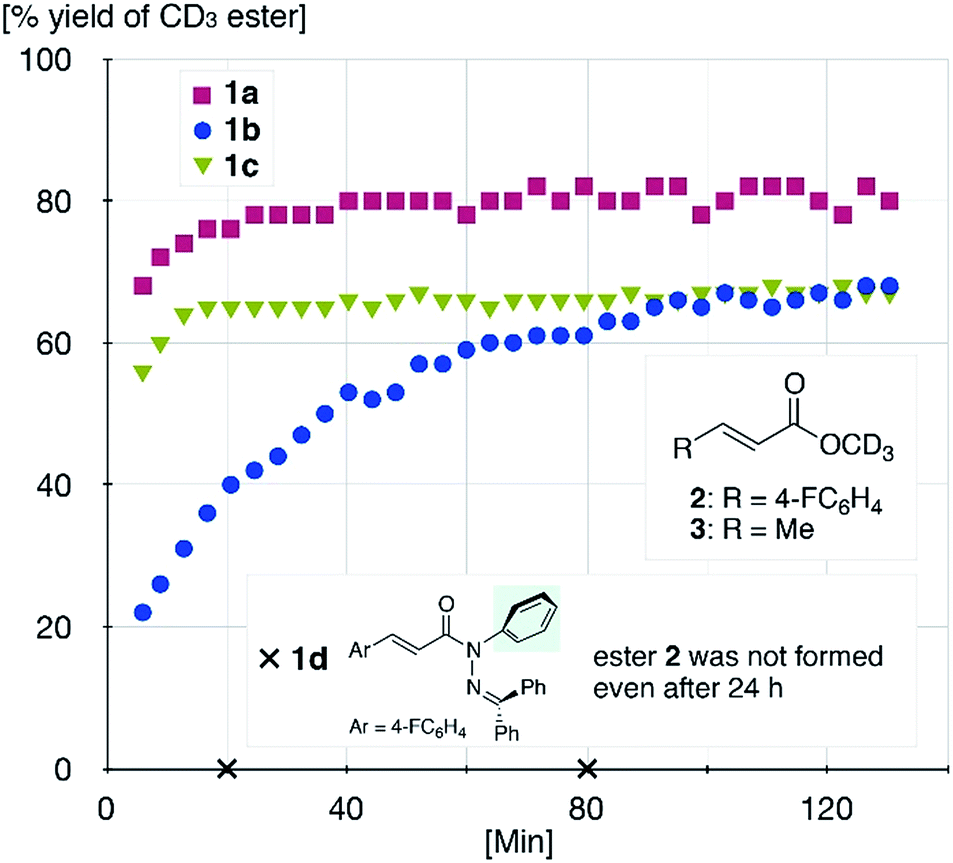
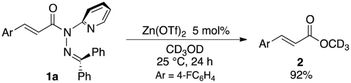
The sterically driven distortion of the amide moieties led to a substantially enhanced rate of solvolytic cleavage under neutral and ambient conditions.10a,c,d,f–i Amides 1a–c exhibited high levels of stability toward hydrolytic cleavage under such conditions, and no methanolysis was detected in CD3OD. However, in the presence of [Pd(CH3CN)4](BF4)2, 1a–c were rapidly converted to the corresponding CD3 esters 2 and 3 under otherwise identical conditions (Fig. 5). Considering the aforementioned NMR and crystallographic analyses, this reactivity should be interpreted in terms of an electrophilic activation of the amide moiety in solution, which is most likely caused by a diminished amide conjugation in response to peripheral steric congestion.18 In contrast, amide 1d, which lacks the pyridine unit for chelating complexation with Pd2+, did not produce any detectable quantity of the corresponding ester, thus further supporting the importance of the peripheral congestion. While (1c)2/Pd exhibited a small twist angle (τ = 4.3°), the methanolysis rate for 1c was significantly increased, suggesting that the pyramidalization of the amide nitrogen atom is predominantly responsible for the electrophilic activation. The chelating unit liberated by the methanolysis was found to strongly bind to Pd2+, and the slow association/dissociation kinetics of these amides and Pd2+ prevented a catalytic methanolysis.17,19 As is evident from the averaged 1H NMR spectrum, Zn2+ showed faster complexation kinetics in comparison,17 which allows the methanolysis to proceed catalytically, converting amide 1a to CD3 ester 2 in a 92% yield using 5 mol% of Zn(OTf)2 (eqn (1)).20 The crystallographic analysis of (1a)2/Zn revealed a coordination via NPy and NIm, which corroborates that a sterically driven amide activation should also be operative in the case of complexation with Zn2+ (Fig. 6).21
| |  | (1) |
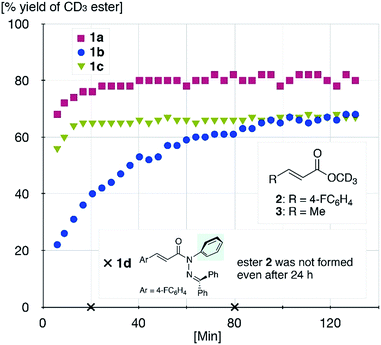 |
| | Fig. 5 Methanolysis of 1a–d in the presence of 1 equiv. of [Pd(CH3CN)4](BF4)2 at 27 °C monitored using 1H NMR spectroscopy. | |
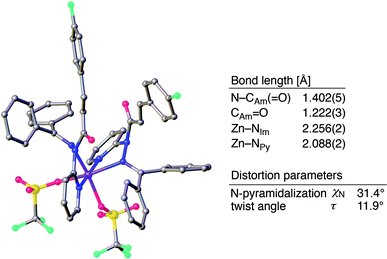 |
| | Fig. 6 Crystal structure of (1a)2/Zn with selected structural parameters. Hydrogen atoms are omitted for clarity. Color code: gray = carbon, blue = nitrogen, red = oxygen, sky blue = fluorine, yellow = sulfur, and purple = zinc. | |
Conclusions
In conclusion, we demonstrated that peripheral steric constraint may induce the pyramidalization and twisting of the amide functional group. The results summarized herein represent proof-of-concept that the distortion of the amide moiety can be induced in an indirect and temporary fashion, which stands in contrast to the previously reported permanent distortion by covalent bonds or to the temporary distortion via the direct coordination of the amide functional group. The crystallographic analysis of the Pd2+ and Zn2+ complexes of 1 revealed the presence of distorted amides in the solid state, and the observed coordination patterns, as well as the pyramidalization of the amide nitrogen were confirmed in solution via NMR analysis. The distortion of the amide moiety in solution was further supported by the enhanced solvolysis rates in the presence of metal cations. Further studies to transfer the present results to the chemoselective activation of simple amides via peripheral crowding are currently under way and will be reported in due course.
Acknowledgements
This work was financially supported by KAKENHI (No. 25713002) from JSPS (to N. K.). Dr Ryuichi Sawa, Ms Yumiko Kubota, and Dr Kiyoko Iijima are gratefully acknowledged for their assistance with the NMR analysis. The authors would also like to thank Dr Tomoyuki Kimura for assistance with the X-ray crystallography.
Notes and references
-
A. Greenberg, C. M. Breneman and J. F. Liebman, The amide linkage: selected structural aspects in chemistry, biochemistry, and materials science, Wiley-Interscience, New York, 2000 Search PubMed.
-
(a) D. Kahne and W. C. Still, J. Am. Chem. Soc., 1988, 110, 7529 CrossRef CAS;
(b) A. Radzicka and R. Wolfenden, J. Am. Chem. Soc., 1996, 118, 6105 CrossRef CAS.
- R. Lukeš, Collect. Czech. Chem. Commun., 1938, 10, 148 CrossRef.
- For computational studies, see:
(a) A. Greenberg, D. T. Moore and T. D. DuBois, J. Am. Chem. Soc., 1996, 118, 8658 CrossRef CAS;
(b) A. Creenberg and C. A. Vena, J. Am. Chem. Soc., 1993, 115, 6951 CrossRef.
-
(a) A. J. Kirby, I. V. Komarov, P. D. Wothers and N. Feeder, Angew. Chem., Int. Ed., 1998, 37, 785 CrossRef CAS;
(b) A. J. Kirby, I. V. Komarov and N. Feede, J. Am. Chem. Soc., 1998, 120, 7101 CrossRef CAS;
(c) A. J. Kirby, I. V. Komarov and N. Feeder, J. Chem. Soc., Perkin Trans. 2, 2001, 522 RSC;
(d) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731 CrossRef CAS PubMed;
(e) I. V. Komarov, S. Yanik, A. Y. Ishchenko, J. E. Davies, J. M. Goodman and A. J. Kirby, J. Am. Chem. Soc., 2015, 137, 926 CrossRef CAS PubMed;
(f) M. Liniger, D. G. VanderVelde, M. K. Takase, M. Shahgholi and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 969 CrossRef CAS PubMed.
- F. K. Winkler and J. D. Dunitz, J. Mol. Biol., 1971, 59, 169 CrossRef CAS PubMed.
- For reviews, see:
(a) H. K. Hall and A. El-Shekeil, Chem. Rev., 1983, 83, 549 CrossRef CAS;
(b)
T. G. Lease and K. J. Shea, in Advances in Theoretically Interesting Molecules, ed. R. P. Thummel, JAI Press, Greenwich, CT, 1992, vol. 2, pp. 79–112 Search PubMed;
(c) J. Clayden and W. J. Moran, Angew. Chem., Int. Ed., 2006, 45, 7118 CrossRef CAS PubMed;
(d) M. Szostak and J. Aube, Org. Biomol. Chem., 2011, 9, 27 RSC;
(e) M. Szostak and J. Aube, Chem. Rev., 2013, 113, 5701 CrossRef CAS PubMed.
- For selected examples of covalently distorted amides, see:
(a) C. Pedone, E. Benedetti, A. Immirzi and G. Allegra, J. Am. Chem. Soc., 1970, 92, 3549 CrossRef CAS;
(b) V. Somayaji and R. S. Brown, J. Org. Chem., 1986, 51, 2676 CrossRef CAS;
(c) G. V. Shustov, G. K. Kadorkina, S. V. Varlamov, A. V. Kachanov, R. G. Kostyanovsky and A. Rauk, J. Am. Chem. Soc., 1992, 114, 1616 CrossRef CAS;
(d) S. Yamada, Angew. Chem., Int. Ed., 1993, 32, 1083 CrossRef;
(e) H. Shao, X. Jiang, P. Gantzel and M. Goodman, Chem. Biol., 1994, 1, 231 CrossRef CAS PubMed;
(f) G. Yamamoto, H. Murakami, N. Tsubai and Y. Mazaki, Chem. Lett., 1997, 605 CrossRef CAS;
(g) C. F. Matta, C. C. Cow, S. Sun, J. F. Britten and P. H. M. Harrison, J. Mol. Struct., 2000, 523, 241 CrossRef CAS;
(h) R. Łysek, K. Borsuk, M. Chmielewski, Z. K. Z. Urbańczyk-Lipkowska, A. Klimek and J. Frelek, J. Org. Chem., 2002, 67, 1472 CrossRef;
(i) A. E. Gillson, S. A. Glover, D. J. Tucker and P. Turner, Org. Biomol. Chem., 2003, 1, 3430 RSC;
(j) Y. Otani, O. Nagae, Y. Naruse, S. Inagaki, M. Ohno, K. Yamaguchi, G. Yamamoto, M. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2003, 125, 15191 CrossRef CAS PubMed;
(k) S. Zaretsky, V. Rai, G. Gish, M. W. Forbes, M. Kofler, J. C. Y. Yu, J. Tan, J. L. Hickey, T. Pawson and A. K. Yudin, Org. Biomol. Chem., 2015, 13, 7384 RSC;
(l) M. Szostak, L. Yao and J. A., J. Am. Chem. Soc., 2010, 132, 2078 CrossRef CAS PubMed.
-
(a) M. K. Rosen, R. F. Standaert, A. Galat, M. Nakatsuka and S. L. Schreiber, Science, 1990, 248, 863 CAS;
(b) R. S. Brown, A. J. Bennet and H. S. Tilk, Acc. Chem. Res., 1992, 25, 481 CrossRef CAS;
(c) C. L. Perrin, Acc. Chem. Res., 1989, 22, 268 CrossRef CAS;
(d) D. G. A. Johansson, G. Wallin, A. Sandberg, B. Macao, J. Åqvist and T. Härd, J. Am. Chem. Soc., 2009, 131, 9475 CrossRef CAS PubMed;
(e) C. Cox and T. Lectka, Acc. Chem. Res., 2000, 33, 849 CrossRef CAS PubMed.
-
(a) R. P. Houghton and R. R. Puttner, J. Chem. Soc., Chem. Commun., 1970, 1270 RSC;
(b) C. Cox, D. Ferraris, N. N. Murthy and T. Lectka, J. Am. Chem. Soc., 1996, 118, 5332 CrossRef CAS;
(c) N. Niklas, F. Hampel, G. Liehr, A. Zahl and R. Alsfasser, Chem.–Eur. J., 2001, 7, 5135 CrossRef CAS PubMed;
(d) N. Niklas, F. W. Heinemann, F. Hampel and R. Alsfasser, Angew. Chem., Int. Ed., 2002, 41, 3386 CrossRef CAS;
(e) N. Niklas and R. Alsfasser, Dalton Trans., 2006, 3188, 10.1039/b516875a;
(f) M. C. Brohmer, S. Mundinger, S. Brase and W. Bannwarth, Angew. Chem., Int. Ed., 2011, 50, 6175 CrossRef PubMed;
(g) S. Mundinger, U. Jakob, P. Bichovski and W. Bannwarth, J. Org. Chem., 2012, 77, 8968 CrossRef CAS PubMed;
(h) U. Jakob, S. Mundinger and W. Bannwarth, Eur. J. Org. Chem., 2014, 2014, 6963 CrossRef CAS;
(i) S. Mundinger, U. Jakob and W. Bannwarth, Chem.–Eur. J., 2014, 20, 1258 CrossRef CAS PubMed.
- For selected examples of distorted ureas, see:
(a) R. B. Penland, S. Mizushima, C. Curran and J. V. Quagliano, J. Am. Chem. Soc., 1957, 79, 1575 CrossRef CAS;
(b) N. J. Curtis, N. E. Dixon and A. M. Sargeson, J. Am. Chem. Soc., 1983, 105, 5347 CrossRef CAS;
(c) N. E. Dixon, D. P. Fairlie, W. G. Jackson and A. M. Sargeson, Inorg. Chem., 1983, 22, 4038 CrossRef CAS;
(d) D. P. Fairlie and W. G. Jackson, Inorg. Chim. Acta, 1988, 150, 81 CrossRef CAS;
(e) D. P. Fairlie and W. G. Jackson, Inorg. Chem., 1990, 29, 3139 CrossRef CAS;
(f) P. Maslak, J. J. Sczepanski and M. Parvez, J. Am. Chem. Soc., 1991, 113, 1062 CrossRef CAS.
- For transformations of covalently crowded amides, see:
(a) F. Hu, R. Lalancette and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 5062 CrossRef CAS PubMed;
(b) G. Meng and M. Szostak, Org. Lett., 2016, 18, 796 CrossRef CAS PubMed;
(c) G. Meng and M. Szostak, Org. Biomol. Chem., 2016, 14, 5690 RSC.
- For details regarding the synthesis of 1a–c, see ESI.†.
- For full characterization details, see ESI.†.
- Some crystals contained multiple structures per unit cell, and one of these is shown in Fig. 4 and Table 1. For all structures, see ESI.†.
- Full NOE correlation confirming the structure of homoleptic (1b)2/Pd and (1c)2/Pd is detailed in ESI.†.
- See ESI† for details.
- For computational studies on the hydrolysis of distorted amides, see:
(a) X. Lopez, J. I. a. Mujika, G. M. Blackburn and M. Karplus, J. Phys. Chem. A, 2003, 107, 2304 CrossRef CAS;
(b) J. I. a. Mujika, J. M. Mercero and X. Lopez, J. Am. Chem. Soc., 2005, 127, 4445 CrossRef CAS PubMed.
- The detrimental effect of the liberated chelating unit led to incomplete conversion of the amides (Fig. 5). Indeed, in the presence of free chelating unit, methanolysis was retarded significantly.
- No methanolysis was detected for TfOH (10 equiv.) in CD3OD.
- Crystal structures with different structural parameters were found in the unit cell. For full structural data, see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1494998–1495005. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03669d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a