
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic circular dichroism studies of iron(II) binding to human calprotectin†
Tessa M.
Baker
a,
Toshiki G.
Nakashige
b,
Elizabeth M.
Nolan
*b and
Michael L.
Neidig
*a
aDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: neidig@chem.rochester.edu
bDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: lnolan@mit.edu
First published on 11th October 2016
Abstract
Calprotectin (CP) is an abundant metal-chelating protein involved in host defense, and the ability of human CP to bind Fe(II) in a calcium-dependent manner was recently discovered. In the present study, near-infrared magnetic circular dichroism spectroscopy is employed to investigate the nature of Fe(II) coordination at the two transition-metal-binding sites of CP that are a His3Asp motif (site 1) and a His6 motif (site 2). Upon the addition of sub-stoichiometric Fe(II), a six-coordinate (6C) Fe(II) center associated with site 2 is preferentially formed in the presence of excess Ca(II). This site exhibits an exceptionally large ligand field (10Dq = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 045 cm−1) for a non-heme Fe(II) protein. Analysis of CP variants lacking residues of the His6 motif supports that CP coordinates Fe(II) at site 2 by employing six His ligands. In the presence of greater than one equiv. of Fe(II) or upon mutation of the His6 motif, the metal ion also binds at site 1 of CP to form a five-coordinate (5C) Fe(II)–His3Asp motif that was previously unidentified in this system. Notably, the introduction of His-to-Ala mutations at the His6 motif results in a mixture of 6C (site 2) and 5C (site 1) signals in the presence of sub-stoichiometric Fe(II). These results are consistent with a reduced Fe(II)-binding affinity of site 2 as more weakly coordinating water-derived ligands complete the 6C site. In the absence of Ca(II), both sites 1 and 2 are occupied upon addition of sub-stoichiometric Fe(II), and a stronger ligand field is observed for the 5C site. These spectroscopic studies provide further evaluation of a unique non-heme Fe(II)–His6 site for metalloproteins and support the notion that Ca(II) ions influence the Fe(II)-binding properties of CP.
045 cm−1) for a non-heme Fe(II) protein. Analysis of CP variants lacking residues of the His6 motif supports that CP coordinates Fe(II) at site 2 by employing six His ligands. In the presence of greater than one equiv. of Fe(II) or upon mutation of the His6 motif, the metal ion also binds at site 1 of CP to form a five-coordinate (5C) Fe(II)–His3Asp motif that was previously unidentified in this system. Notably, the introduction of His-to-Ala mutations at the His6 motif results in a mixture of 6C (site 2) and 5C (site 1) signals in the presence of sub-stoichiometric Fe(II). These results are consistent with a reduced Fe(II)-binding affinity of site 2 as more weakly coordinating water-derived ligands complete the 6C site. In the absence of Ca(II), both sites 1 and 2 are occupied upon addition of sub-stoichiometric Fe(II), and a stronger ligand field is observed for the 5C site. These spectroscopic studies provide further evaluation of a unique non-heme Fe(II)–His6 site for metalloproteins and support the notion that Ca(II) ions influence the Fe(II)-binding properties of CP.
Introduction
Calprotectin (CP, S100A8/S100A9 oligomer, MRP8/MRP14 oligomer) is an abundant metal-chelating protein that contributes to host defense.1–4 CP is released from neutrophils in high concentrations at sites of infection and functions as a metal-scavenging antimicrobial protein during the host innate immune response.3–7 Its antimicrobial mechanism of action is attributed to its capacity to withhold nutrient transition metals from microbial pathogens.1–4,8 Indeed, human CP sequesters a number of divalent first-row transition metal ions, including Zn(II),3 Mn(II),4 and Fe(II).8Human CP is a heterooligomer of the proteins S100A8 (α, 10.8 kDa) and S100A9 (β, 13.2 kDa) that each have two EF-hand domains that coordinate Ca(II).9 In the presence of excess Ca(II), CP transforms from the αβ heterodimer to the α2β2 heterotetramer and exhibits enhanced transition-metal-binding affinity.8,10,11 Two transition-metal-binding sites are formed at the S100A8/S100A9 dimer interface (Fig. 1).10–16 Site 1 is a His3Asp motif and is most celebrated for coordinating Zn(II) with high affinity.10,13 This site has also been shown to bind Mn(II) and Co(II), albeit with lower affinity.10,11,14 Site 2 is a His6 motif that chelates a variety of first-row transition metal ions, including Mn(II), Fe(II) and Zn(II), with high affinity.8,13–15,17 The His6 motif is formed by four His residues at the S100A8/S100A9 interface and two His residues of the S100A9 C-terminal tail. Both sites coordinate divalent first-row transition metals in the absence of Ca(II) and exhibit enhanced metal-ion affinities in the presence of excess Ca(II).8,10,15–17
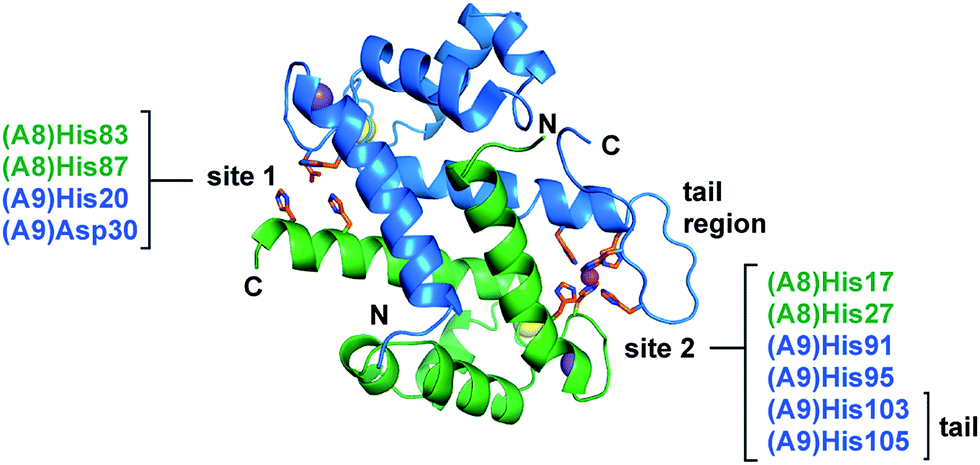 | ||
| Fig. 1 The two transition-metal-binding sites of CP. The Mn(II)-, Ca(II)-, and Na(I)-bound CP dimer unit is shown (PDB 4XJK).16 Site 1 comprises His83 and His87 of S100A8 and His20 and Asp30 of S100A9. Site 2 comprises His17 and His27 of S100A8 and His91, His95, His103, and His105 of S100A9. The S100A8 subunit is green, and the S100A9 subunit is blue. Na(I), Ca(II), and Mn(II) ions are depicted as purple, yellow, and magenta spheres, respectively. The N- and C-termini of each subunit and the S100A9 C-terminal tail region are labeled. | ||
To the best of our knowledge, the hexahistidine Fe(II) site of CP is unique amongst known iron-coordinating proteins.8 Fe(II) is a relatively labile metal ion, and several Fe(II)-binding proteins that have been characterized, including the transcriptional regulator Fur of Escherichia coli18 and the dioxygenase TfdA of Ralstonia eutropha,19 exhibit micromolar affinities for this divalent metal ion.20 Moreover, octahedral Fe(II)-binding motifs found in biological systems typically have ligands that are not protein-based, such as water or other molecules.21,22 Mononuclear non-heme iron enzymes that employ α-ketoglutarate (α-KG), for example, must accommodate this cofactor at the metal center.23 On the other hand, CP chelates Fe(II) with remarkably high affinity (apparent Kd,Fe(II) < 2.2 pM, +Ca),8 and all of the Fe(II)-coordinating ligands of CP are amino acid residues. Unlike other proteins in which their function is regulated by the reversible binding of Fe(II) (e.g., transcription factors), or enzymes that employ Fe(II) at the catalytic site where cofactors, substrates, and solvent water molecules must come in close proximity to the metal center (e.g., dioxygenase, hydroxylase), CP employs an effective metal-sequestering mechanism in which formation of a coordination complex results in entrapment of Fe(II) and other metals. On the basis of our structural and spectroscopic studies of Mn(II) coordination, we hypothesize that coordinative saturation of a metal ion at the hexahistidine site, coupled with encapsulation of the metal ion by the S100A9 C-terminal tail, precludes the access of solvent water molecules, and thereby overcomes the kinetic lability of the metal center.16 In the context of metal sequestration by the host, this binding mode enables CP to be a versatile metal-chelating host-defense protein, and prevents microbial acquisition of multiple nutrient metals.
In the current work, we further investigate the electronic and structural properties of the high-affinity Fe(II) site of CP. One method to interrogate Fe(II) coordination is near-infrared (NIR) magnetic circular dichroism (MCD) spectroscopy. This technique allows examination of the electronic transitions of high-spin Fe(II) complexes that are often inaccessible by other spectroscopic methods and gives insight into the geometry and ligand field of the metal center.24 We present a systematic study of Fe(II)-bound CP and CP variants by NIR MCD spectroscopy (Table 1). We show that the MCD spectrum of the Fe(II)–His6 site affords the largest ligand field of 6-coordinate (6C) Fe(II) proteins that have been studied to date. Analysis of CP variants that lack residues of this site supports the notion that the native His6 motif impedes solvent water molecules from accessing the metal ion. Furthermore, our findings support that Ca(II) binding enhances the ability of CP to coordinate Fe(II) at site 2. Lastly, we provide the first spectroscopic evidence for Fe(II) coordination at the His3Asp site in both the absence and presence of Ca(II). The His3Asp site forms a five-coordinate (5C) Fe(II) motif and coordinates Fe(II) with lower affinity than the His6 site. This observation is consistent with our expectation that both transition-metal-binding sites of CP are able to accommodate metal ions between Mn and Zn on the Periodic Table.
| Protein | S100A8 mutation(s) | S100A9 mutation(s) | Referencea |
|---|---|---|---|
| a The reference where each variant was purified and biochemically characterized is given. | |||
| CP | N/A | N/A | 10 |
| CP-Ser | C42S | C3S | 10 |
| CP-Ser ΔHis3Asp | C42S, H83A, H87A | C3S, H20A, D30A | 10 |
| CP-Ser ΔHis4 | C42S, H17A, H27A | C3S, H91A, H95A | 10 |
| CP-Ser(H103A) | C42S | C3S, H103A | 15 |
| CP-Ser-AHA | C42S | C3S, H103A, H105A | 15 |
| CP-Ser-AAA | C42S | C3S, H103A, H104A, H105A | 15 |
| CP-Ser ΔHis3Asp(H103A) | C42S, H83A, H87A | C3S, H20A, D30A, H103A | 17 |
Experimental
Preparation of MCD samples
All samples were prepared using D2O (99.9% D, Cambridge Isotope Laboratories, Inc.). Buffers were prepared using Ultrol grade HEPES (Calbiochem), TraceSELECT NaCl (Sigma), and NaOD (99.9% D, Sigma). Concentrated Fe(II) (100 mM) and Ca(II) (1.0 M) stocks were prepared in an anaerobic N2 atmosphere glove box (Vacuum Atmospheres Company Omni-Lab or MBraun) by dissolving (NH4)2Fe(SO4)2·6H2O (trace metals basis, Sigma) and CaCl2 (trace metals basis, Sigma) in deoxygenated D2O. All buffers and metal stock solutions were prepared in acid-washed volumetric glassware and stored in polypropylene containers.Protein was purified as previously described10 and stored in the apo form in 20 mM HEPES, 100 mM NaCl, pH 8.0 at −80 °C. Characterization of the human CP variants (Table 1) is reported elsewhere.10,15,17 CP-Ser is the heterooligomer of S100A8(C42S) and S100A9(C3S), and all variants are based on this protein.10 Concentrated protein solutions (up to ≈3.0 mM) were prepared by buffer exchanging into deuterated buffer (150 mM HEPES, 200 mM NaCl, pD 7.4) using 10k MWCO Amicon spin concentrators (EMD Millipore). Glycerol samples were prepared by adding 60% (v/v) glycerol-d8 (99%, Cambridge Isotope Laboratories, Inc.) to the concentrated protein samples and gently mixing by pipetting. Saturated sucrose buffer was prepared by dissolving BioUltra grade sucrose (≥99.5%, Sigma) into deuterated buffer solution (where ≈2.0 g sucrose were dissolved for every ≈1.0 mL buffer), gently heating at 37 °C, and vortexing. The sucrose samples were prepared by buffer exchanging protein solutions into the saturated sucrose buffer. The final concentration of protein was determined based on the calculated extinction coefficient for CP-Ser and variants (ε280 = 18450 M−1 cm−1) from the online ExPASy ProtParam tool using a Take3 plate and BioTek Synergy HT plate reader or a Thermo Scientific NanoDrop 1000 spectrophotometer.
All samples for MCD spectroscopy were prepared in a nitrogen atmosphere glove box equipped with a liquid nitrogen fill port to allow sample freezing to 77 K within the inert atmosphere. Frozen-solution MCD samples were prepared in copper cells fitted with quartz disks and a 3 mm gasket. Protein solutions with glycerol or sucrose (≈300 μL) were degassed by gently purging with N2, transferred to the glove box, and allowed to equilibrate in the N2 atmosphere for at least 30 min. Fe(II) and Ca(II) were added to each protein solution in the glove box from the concentrated stock solutions, and the resulting solutions were mixed by pipetting and allowed to equilibrate for at least 1 h. Protein solutions that were prepared at the University of Rochester were injected directly to MCD cells and flash frozen in liquid N2 in the glove box. Protein solutions that were prepared at MIT were transferred out of the glove box in microcentrifuge tubes, flash frozen immediately in liquid N2, shipped overnight in a dry shipping dewar cooled with liquid N2, transferred to the University of Rochester glove box, and thawed for injection into MCD cells. Cells containing ≈250 μL of sample were flash frozen in liquid N2 and stored in liquid N2 until data collection. The protein and metal concentrations for each sample are detailed in Table S1.† An [Fe(H2O)6]2+ MCD sample (1.5 mM) was prepared from diluting the (NH4)2Fe(SO4)2 stock solution in deuterated buffer (150 mM HEPES, 200 mM NaCl, pD 7.4) with 60% glycerol-d8 (v/v) added to form an optical glass at low temperature.
Magnetic circular dichroism spectroscopy
NIR MCD experiments were conducted using a Jasco J-730 spectropolarimeter and a liquid nitrogen-cooled InSb detector. The spectral range accessible with this NIR MCD setup is 2000–600 nm. A modified sample compartment was utilized incorporating focusing optics and an Oxford Instruments SM4000-7T superconducting magnet/cryostat. This setup enabled MCD measurements from 1.6 to 290 K with magnetic fields of up to 7 T. A calibrated Cernox sensor directly inserted into the copper sample holder was used to measure the temperature of the sample to ±0.001 K. All MCD spectra were baseline-corrected against zero-field scans. Saturation magnetization data were analysed using previously reported fitting procedures.25,26 For saturation magnetization data fitting, both negative and positive zero-field splitting models were evaluated. Reported errors for spin Hamiltonian parameters were determined via evaluation of the effects of systematic variations of the fit parameters on the quality of the overall fit.Results and analysis
In the NIR MCD spectroscopic studies performed herein, the energies and splitting pattern of observed MCD bands are related to the geometric and electronic structures of Fe(II) bound to CP-Ser and its variants. A distorted octahedral Fe(II) site is typically split by 10Dq ≈ 10000 cm−1 due to a doubly degenerate 5Eg ligand field (LF) excited state as well as a triply degenerate 5T2g LF ground state.24 Moreover, the lower symmetry of protein metal-binding sites causes the 5Eg state to further split allowing for two LF transitions centred at ≈10000 cm−1, split by ≈2000 cm−1. Two different 5C sites lead to slightly different splitting of these bands. Square pyramidal sites show transitions at ≈10000 cm−1 and ≈5000 cm−1, whereas trigonal bipyramidal sites show these transitions at <10000 cm−1 and <5000 cm−1. Distorted four-coordinate (4C) sites have smaller 10Dq values, which results in low-energy LF transitions in the 4000–7000 cm−1 region. Table 2 summarizes the LF transitions observed for CP-Ser and its variants, along with the corresponding Δ5Eg and 10Dq values and assignments of binding sites.
| Name | Transition energy (cm−1) | Δ5Eg (cm−1) | 10Dq (cm−1) |
|---|---|---|---|
| a Same values for sucrose and glycerol samples. b 6C only in glycerol; both 5C and 6C components observed in sucrose. c 5C and 6C components present in both sucrose and glycerol but in different ratios. d Parameters from sucrose samples. | |||
| CP-Ser/Ca(II)/Fe(II)a | 6C: 10520, 11570 |
1050 | 11045 |
| H103A/Ca(II)/Fe(II)b | 6C: 9920, 11500 |
1580 | 10710 |
| 5C: <5000, 8345 | — | — | |
| AHA/Ca(II)/Fe(II)c | 6C: 9670, 11160 |
1490 | 10415 |
| 5C: <5000, 8345 | — | — | |
| AAA/Ca(II)/Fe(II)c | 6C: 9570, 11100 |
1530 | 10335 |
| 5C: <5000, 8345 | — | — | |
| ΔHis4/Ca(II)/Fe(II)a | 5C: <5000, 8345 | — | — |
| ΔHis3Asp/Ca(II)/Fe(II)a | 6C: 10520, 11570 |
1050 | 11045 |
| CP-Ser/Fe(II)d | 6C: 10520, 11570 |
1050 | 11045 |
| 5C: <5000, 8710 | — | — | |
| ΔHis3Asp/Fe(II)d | 6C: 10520, 11570 |
1050 | 11045 |
Circular dichroism (CD) spectroscopy is often used to ensure glycerol effects, although uncommon, are not present as a result of using glycerol for the preparation of low-temperature glasses required for spectroscopic studies; however, insufficient concentrations and, hence, CD intensities were accessible with the CP-Ser and variant samples. Therefore, MCD samples containing either glycerol or sucrose as the glassing agent were prepared and analyzed to probe for the presence of any glycerol effects. Furthermore, all observed transitions in the protein samples were compared to those of [Fe(H2O)6]2+ to confirm the identity of transitions resulting from protein-bound Fe(II). The NIR MCD results for CP-Ser and its variants are presented below, including evaluation of (i) CP-Ser, (ii) variants at the His6 site, (iii) a variant at the His3Asp site, and (iv) the effect of Ca(II) on Fe(II) binding.
Fe(II) binding to CP-Ser
The 5 K, 7 T NIR MCD of CP-Ser + 0.9 equiv. Fe(II) in the presence of excess Ca(II) (CP-Ser/Ca(II)/Fe(II)) in both glycerol (Fig. 2D) and sucrose (Fig. 2E) contains two LF transitions at 10520 cm−1 and 11570 cm−1, indicative of a distorted six-coordinate (6C) octahedral Fe(II)-binding site with Δ5Eg = 1050 cm−1 and 10Dq = 11045 cm−1. The distorted 6C Fe(II) site with a large 10Dq value is consistent with Fe(II) binding to the His6 site, where six His ligands comprise the coordination environment of Fe(II). It is noteworthy that the observed 10Dq value for the 6C Fe(II) site in CP-Ser/Ca(II)/Fe(II) is significantly larger than the 10Dq values observed for sites derived from facial triad Fe(II)-binding sites and is, to the best of our knowledge, the largest 10Dq value for any characterized 6C Fe(II) site in a non-heme protein environment. Saturation magnetization data were collected for both the glycerol (Fig. 2E) and sucrose (Fig. 2F) samples at 10310 cm−1. In both cases, the saturation magnetization data are well described by an S = 2 positive zero-field split (+ZFS) non-Kramers doublet model with D = 11 ± 2 cm−1 and E = 1 ± 0.5.
 | ||
| Fig. 2 NIR MCD studies of Fe(II) binding to CP-Ser. The 5 K, 7 T NIR MCD spectra of CP-Ser/Ca(II)/Fe(II) in (A) glycerol and (B) sucrose glasses. (C) The 5 K, 7 T NIR MCD spectra for the titration of apo CP-Ser with 0.3 equiv. (orange), 0.6 equiv. (green), 0.9 equiv. (blue) and 1.2 equiv. (red) of Fe(II). Saturation magnetization data (dots) and best fit (lines) for CP-Ser/Ca(II)/Fe(II) in (D) glycerol and (E) sucrose at 10310 cm−1. | ||
CP has two transition-metal-binding sites at the S100A8/S100A9 interface,10,11,13 and in order to evaluate whether a second Fe(II)-binding site may be present, a titration study of Fe(II) addition to CP-Ser in the presence of Ca(II) was performed in sucrose (Fig. 2C). When sub-stoichiometric Fe(II) is added, only the distorted 6C site previously described is observed by MCD. Upon addition of excess Fe(II), two new ligand–field transitions are observed in addition to the 6C site bands: a low-energy transition at <5000 cm−1 (observed as a tail in Fig. 2C) and a band at 8345 cm−1. Together, these latter two transitions indicate that a new distorted 5C Fe(II) site is formed in the presence of >1 equiv. of Fe(II). At 1.2 equiv. of added Fe(II) to CP-Ser, ≈90% of Fe(II) is bound to the 6C site and ≈10% is bound to the 5C site utilizing the Δε values for pure 6C and 5C sites (vide infra) determined in this study. Note that the Δε value for the 5C site far exceeds that for the 6C site as expected since the lower symmetry of the 5C site allows higher energy intense transitions to mix into parity forbidden d–d transitions.27,28 The 5C Fe(II) site is assigned to Fe(II) binding to site 1 (His3Asp site) of CP-Ser, consistent with previous studies indicating that site 1 in CP-Ser binds metals (i.e. Mn(II), Co(II), Zn(II))10,11,13 and supported by NIR MCD studies of CP-Ser variants (vide infra). The observation of the highest energy LF transition for the 5C site at 8345 cm−1 is most consistent with a distorted trigonal bipyramidal geometry.24 This work represents the first evidence for Fe(II) binding at site 1 in CP-Ser.
Fe(II) binding to site 2 variants
Studies of CP-Ser variants lacking residues of the His6 site were performed to provide further support for the His6 coordination of Fe(II) at site 2. Four variants of the His6 site were investigated (Table 1): (i) H103A, where a single His residue of the S100A9 C-terminal tail is mutated to a non-coordinating Ala residue; (ii) AHA, where both His103 and His105 of the His6 site are mutated to Ala residues; (iii) AAA, where His103, His104 and His105 are mutated to three Ala residues; and (iv) ΔHis4, where the four interfacial His residues of the His6 site are mutated to Ala residues. The NIR MCD data for these His6 site variants are given in Fig. 3 and 4.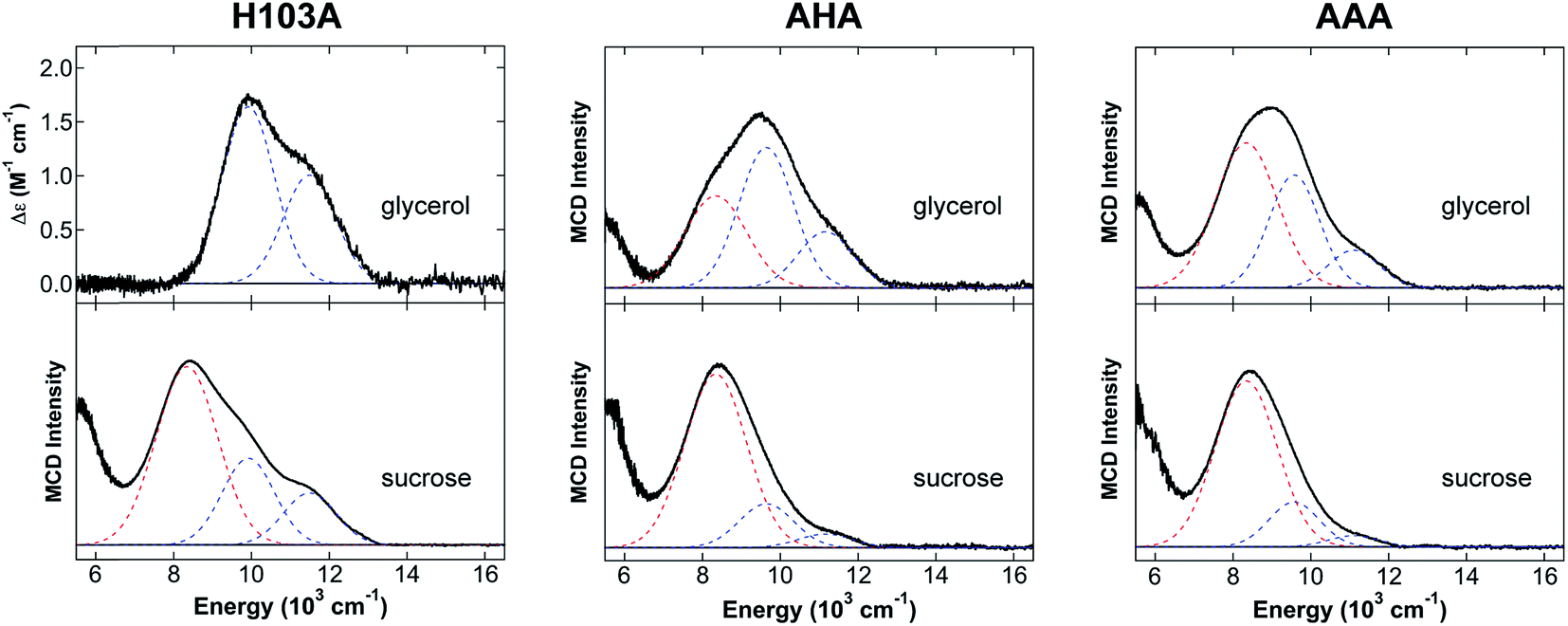 | ||
| Fig. 3 NIR MCD studies of site 2 variants of CP-Ser. The 5 K, 7 T NIR MCD spectra of H103A/Ca(II)/Fe(II), AHA/Ca(II)/Fe(II) and AAA/Ca(II)/Fe(II) in glycerol and sucrose. Best fits are shown in dashed lines with blue denoting 6C site LF transitions and red denoting the highest-energy 5C site LF transition (the low energy tail was not fit). | ||
 | ||
| Fig. 4 NIR MCD studies of ΔHis4. The 5 K, 7 T NIR MCD spectra of ΔHis4/Ca(II)/Fe(II) in (A) glycerol and (B) sucrose glasses. Saturation magnetization data (dots) and best fit (lines) for ΔHis4/Ca(II)/Fe(II) in glycerol collected at (C) 8905 cm−1, (D) 7810 cm−1 and (E) 5880 cm−1. The low-energy tail transitions were not included in the fits as only part of the transition was observable. | ||
The 5 K, 7 T NIR MCD spectrum of H103A/Ca(II)/Fe(II) (Fig. 3) in glycerol contains two LF bands at 9920 cm−1 and 11500 cm−1, indicating a distorted 6C octahedral site with Δ5Eg = 1580 cm−1 and 10Dq = 10710 cm−1. Notably, the energies of both LF transitions as well as the observed 10Dq value for the distorted 6C Fe(II) site in the H103A variant are reduced compared to the values for the 6C site of CP-Ser/Ca(II)/Fe(II). This result is consistent with a change in the ligand environment of the metal center. We propose that a His ligand is replaced by a water-derived ligand in H103A/Ca(II)/Fe(II). In sucrose (Fig. 3), an identical 6C site with Δ5Eg = 1580 cm−1 and 10Dq = 10710 cm−1 is observed. Moreover, additional ligand field features consistent with the distorted 5C site observed in CP-Ser/Ca(II)/Fe(II) in the presence of excess Fe(II) are also observed for the sucrose sample. The 6C site comprises ≈85% of Fe(II) in the H103A/Ca(II)/Fe(II) sample in sucrose, and the 5C site comprises ≈15% of Fe(II) in the sample. The differences observed between the glycerol and sucrose samples in the H103A variant are consistent with a glycerol effect in this sample, where the increased coordination number (i.e. all 6C) is similar to the glycerol effect previous observed in wild-type soybean lipoxygenase NIR MCD (all 6C Fe(II) in glycerol whereas a mix of 5C and 6C Fe(II) is present in sucrose).29 It should be noted that in previous studies of non-heme iron proteins including soybean lipoxygenase29 and factor inhibiting hypoxia-inducible factor30 where glycerol effects have been observed, the Fe(II) distribution in sucrose was found to be the accurate distribution in the proteins.
For the AHA and AAA variants, both 5C and 6C Fe(II) LF transitions are observed with small differences in the ratios (but not the transition energies) present as a function of the glassing solvent, where more 6C Fe(II) is present in glycerol than sucrose. For example, the 5C site comprises ≈10% of Fe(II) in the AHA glycerol sample, whereas this site comprises ≈30% of the Fe(II) in the corresponding sucrose sample. The same trend occurs for the AAA variant, where ≈15% of Fe(II) is in the 5C site and ≈25% of Fe(II) is in the 5C site for the glycerol and sucrose samples, respectively. Both datasets are given in Fig. 3, and the following analysis will focus on the sucrose samples. As the number of His residues mutated to Ala residues at the His6 site increases, a mixture of 5C and 6C Fe(II) species is observed where the transition energies and 10Dq values of the 6C Fe(II) component decreases with an increasing number of alanines. The 5 K, 7 T NIR MCD spectrum of AHA/Ca(II)/Fe(II) (Fig. 3) contains LF transitions at 9670 cm−1 and 11160 cm−1 consistent with a distorted 6C site (Δ5Eg = 1490 cm−1 and 10Dq = 10415 cm−1) where the reduced ligand field is assigned to a site with mixed His and water-derived ligands. In addition, transitions assigned to the distorted 5C site observed throughout these studies are also present. In AAA/Ca(II)/Fe(II), the 5 K, 7 T NIR MCD spectrum indicates the presence of a further reduction of the ligand field of the distorted 6C site, with LF transitions observed at 9570 cm−1 and 11100 cm−1 (Fig. 3) corresponding to Δ5Eg = 1530 cm−1 and 10Dq = 10335 cm−1. Similar to the H103A variant LF transitions for the distorted 5C Fe(II) site are also observed. The reduced ligand field for the distorted 6C Fe(II) site is consistent with the presence of additional water-derived ligands and/or weakened Fe(II)–His interactions compared to AHA.
In the ΔHis4 variant, the four interfacial His ligands of site 2 are mutated to knock out this Fe(II)-binding site. The 5 K, 7 T NIR MCD spectra of ΔHis4/Ca(II)/Fe(II) in either glycerol (Fig. 4A) or sucrose (Fig. 4B) are analogous, containing only a LF transition at 8345 cm−1 and the presence of a low-energy tail consistent with the presence of the distorted 5C Fe(II) site. No transitions corresponding to a distorted 6C component are observed, indicating the mutation of four His ligands at the His6 site results in selective Fe(II) binding at the distorted 5C Fe(II) site, attributed to the His3Asp site (vide infra). Saturation magnetization data collected at 8905 cm−1, 7810 cm−1 and 5880 cm−1 (Fig. 4C–E, respectively) on ΔHis4/Ca(II)/Fe(II) in glycerol are all well described by an S = 2 –ZFS non-Kramers doublet model with ground-state spin-Hamiltonian parameters of δ = 2.4 ± 0.2 cm−1 and g∥ = 8.8 ± 0.2 and only differ with respect to transition polarizations. Thus, in addition to providing further confirmation that the distorted 6C Fe(II) site in CP-Ser/Ca(II)/Fe(II) corresponds to the coordination of six His ligands at site 2, studies of the variants further support that the distorted 5C site observed in the presence of excess Fe(II) in the CP-Ser studies (vide supra) results from Fe(II) binding to the His3Asp site.
Fe(II) binding to His3Asp site variants
Since the MCD studies described above indicate binding of Fe(II) to site 1, this site was deleted (ΔHis3Asp) in order to confirm that the distorted 5C Fe(II) is associated with site 1. The 5 K, 7 T NIR MCD spectrum of ΔHis3Asp/Ca(II)/Fe(II) displays two LF transitions at energies identical to those observed for the 6C Fe(II) site of CP-Ser/Ca(II)/Fe(II) (Fig. 5A). Consistent with this assignment, saturation magnetization data collected at 10310 cm−1 are well fit to identical ground-state parameters as the distorted 6C His6 site of CP-Ser/Ca(II)/Fe(II) (see ESI†).
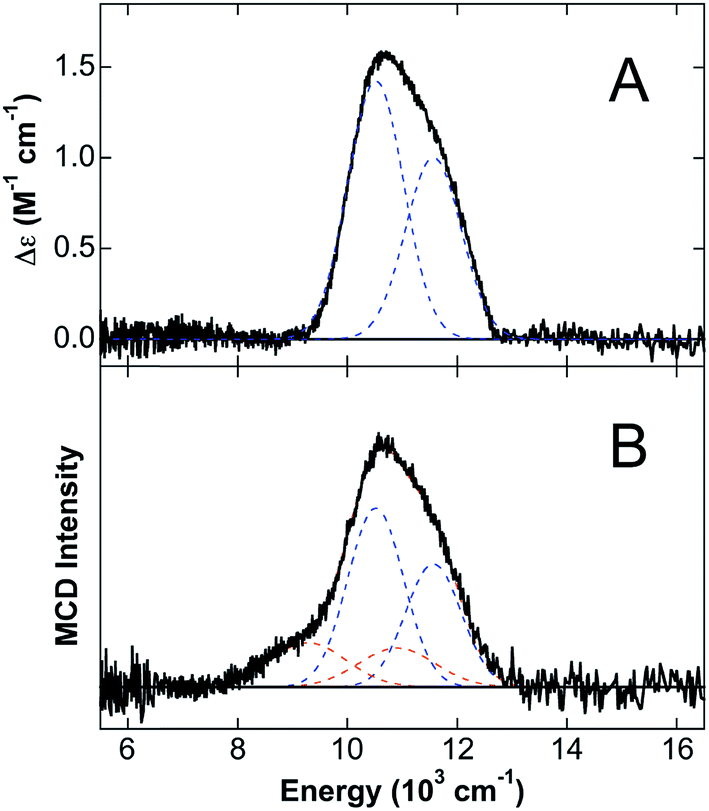 | ||
| Fig. 5 NIR MCD studies of ΔHis3Asp. The 5 K, 7 T NIR MCD spectra of (A) ΔHis3Asp/Ca(II)/Fe(II) and (B) ΔHis3Asp/Ca(II) + 1.5 equiv. Fe(II) in sucrose. Best fits are shown in dashed lines with blue denoting 6C His6 site LF transitions and orange denoting LF transitions due to [Fe(H2O)6]2+. | ||
The observation of a distorted 5C Fe(II) site upon (i) the addition of excess Fe(II) to CP-Ser/Ca(II) and (ii) mutation of metal-binding His residues at the His6 site suggested that the 5C Fe(II) site is likely associated with Fe(II) binding at site 1 of CP-Ser/Ca(II)/Fe(II). To test this hypothesis further, excess Fe(II) (1.5 equiv. with respect to protein) was added to the ΔHis3Asp variant in order to determine if the same 5C Fe(II) site is formed in the absence of the His3Asp site. From the 5 K, 7 T NIR MCD spectrum of ΔHis3Asp/Ca(II) with 1.5 equiv. Fe(II) (Fig. 5B), only LF transitions consistent with the His6 6C site as well as an additional 6C Fe(II) species assigned as free [Fe(H2O)6]2+ are observed. No 5C Fe(II) is present. Thus, this result supports that the observed distorted 5C site results from Fe(II) binding to the His3Asp site. Consistent with the distorted 5C Fe(II) site being attributed to Fe(II) binding to site 1 of CP-Ser/Ca(II)/Fe(II), the 5 K, 7 T NIR MCD spectrum of a ΔHis3Asp(H103A) variant in sucrose (see ESI†) contains only the distorted 6C LF transitions for Fe(II) at site 2. Thus, the 5C component observed in the H103A variant in sucrose is absent upon deletion of site 1, further confirming that the 5C site is due to Fe(II) binding to the His3Asp motif at site 1.
Effect of calcium on Fe(II) binding
The NIR MCD studies presented thus far were performed in the presence of excess Ca(II) because CP-Ser exhibits enhanced metal-binding affinity for Fe(II) and other first-row transition metals in the presence of Ca(II).8,10,11 Nevertheless, CP-Ser also binds Fe(II) in the absence of Ca(II).8 To evaluate whether the absence of Ca(II) affects the nature of Fe(II) binding to CP-Ser, NIR MCD was employed to investigate Fe(II) binding to CP-Ser and the ΔHis3Asp variant in the absence of Ca(II). The 5 K, 7 T NIR MCD spectrum of CP-Ser/Fe(II) (i.e. without Ca(II)) (Fig. 6A) displays multiple LF features, including higher energy bands associated with the 6C Fe(II) His6 site and an intense transition at 8710 cm−1 assigned to the 5C Fe(II)-binding site. Notably, the 5C LF transition is ≈300 cm−1 higher in energy than in the presence of Ca(II). Therefore, the absence of Ca(II) not only leads to the presence of both 6C and 5C Fe(II) sites (in contrast to only the 6C Fe(II) His6 site in the presence of Ca(II)), but also results in a change in the LF strength of the 5C Fe(II) site whereas no change in the LF strength is observed at the His6 site. In contrast, the 5 K, 7 T NIR MCD spectrum of ΔHis3Asp/Fe(II) contains only the 6C Fe(II) His6 site transitions, indicating that in the absence of a 5C Fe(II) site (i) all of the Fe(II) binds to the His6 site (i.e. there is no free Fe(II) in solution) and (ii) the 6C Fe(II) His6 site is unperturbed (i.e. the same transition energies and LF are observed). The lack of perturbation of the His6 6C site in ΔHis3Asp/Fe(II) is consistent with the unperturbed 6C Fe(II) site in CP-Ser/Fe(II), and indicates that the presence or absence of Ca(II) has no observable effect on the electronic structure of the Fe(II)–His6 coordination sphere.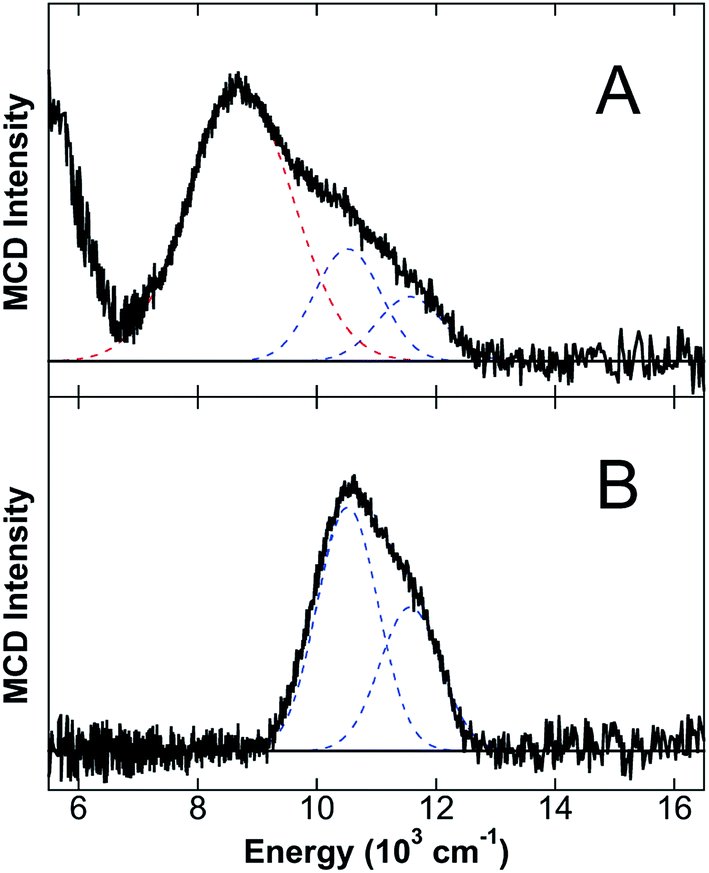 | ||
| Fig. 6 NIR MCD studies of CP-Ser and ΔHis3Asp in the absence of Ca(II). The 5 K, 7 T NIR MCD spectra of (A) CP-Ser/Fe(II) and (B) ΔHis3Asp/Fe(II) in sucrose, where no Ca(II) is present for either sample. Best fits are shown in dashed lines with blue denoting 6C His6 site LF transitions and red denoting LF transitions due to a 5C Fe(II) site. | ||
Discussion
CP has two transition-metal-binding sites, a His3Asp motif (site 1) and a His6 motif (site 2), and the protein coordinates a variety of first-row transition metals in a Ca(II)-dependent manner.8,10,11,13,15 NIR MCD spectroscopy provides a powerful method for probing the coordination number, geometry, and LF parameters of transition metals such as high-spin Fe(II) bound to metalloproteins. In the present study, NIR MCD was utilized to evaluate Fe(II) binding to CP-Ser, including studies of variants where mutations at the His6 and His3Asp sites were incorporated.Studies of Fe(II) (0.9 equiv.) binding to CP-Ser in the presence of Ca(II) indicate the generation of a single 6C Fe(II) site for CP-Ser/Ca(II)/Fe(II). This site is characterized by a very large ligand field for high-spin Fe(II) in a protein environment (10Dq = 11045 cm−1). To the best of our knowledge, this represents the largest LF for a 6C non-heme Fe(II) site in a metalloprotein; it is larger than those reported for a variety of facial triad (2 His, 1 carboxylate) Fe(II) sites as well as the 3 His containing Fe(II) site in Dke1 (Table 3). The large LF of the 6C site in CP-Ser is consistent with the coordination of six His ligands, and analogous studies of site 2 variants where His residues are mutated to non-coordinating Ala residues further support this conclusion. In the H103A variant, the 6C site undergoes a reduction in LF (from 10Dq = 11045 cm−1 to 10710 cm−1). Such a reduction in ligand field is consistent with replacement of a strong His ligand with a more weakly coordinating water-derived ligand in order to maintain a 6C site. In fact, as more His residues of the His6 site are mutated to non-coordinating Ala residues, further reductions in 10Dq for the 6C site are observed. Consistent with prior work,8 Fe(II) coordination at site 2 is lost in the ΔHis4 variant. Combined, the studies on CP-Ser and the site 2 variants definitively demonstrate that, in the presence of excess Ca(II), the Fe(II) preferentially binds to the His6 motif to form a 6C Fe(II) site.
| Protein | Ligands | 10Dq (cm−1) | Δ5Eg (cm−1) |
|---|---|---|---|
| Naphthalene dioxygenase28 | 2 His, 1 Asp, 3H2O | 9300 | 2750 |
| Tyrosine dioxygenase31 | 2 His, 1 Glu, 3H2O | 9735 | 2030 |
| Taurine hydroxylase32 | 2 His, 1 Asp, 3H2O | 9800 | 1800 |
| Factor inhibiting hypoxia-inducible factor30 | 2 His, 1 Glu, 3H2O | 10000 |
1800 |
| Clavaminate synthase 233 | 2 His, 1 Glu, 3H2O | 10055 |
1690 |
| Dke134 | 3 His, 3H2O | 10100 |
1600 |
| CP (this work) | 6 His | 11045 |
1050 |
The current work also provides the first evidence of Fe(II) coordination at site 1. Indeed, the NIR MCD studies also demonstrate that the His3Asp site can form a 5C Fe(II) site. For example, when excess Fe(II) is added to CP-Ser in the presence of Ca(II) such that the His6 site is fully occupied, this 5C site is observed to form. If the His6 site is deleted (i.e. in the ΔHis4 variant), only the 5C Fe(II) His3Asp site is observed in the presence of sub-stoichiometric Fe(II). Furthermore, the addition of excess Fe(II) to the ΔHis3Asp variant results in only the 6C site and free [Fe(H2O)6]2+, confirming that the 5C Fe(II) site is associated with Fe(II) binding to the His3Asp site and not simply adventitious binding to the protein. Although the 5C Fe(II) site likely contains either bidentate Asp coordination or an additional water-derived ligand, these possibilities cannot be unambiguously resolved in the present study. In prior biochemical studies, no evidence for Fe(II) binding at site 1 was observed in (i) Fe(II) competition experiments with a Fe(II)-chelating small molecule and (ii) size-exclusion chromatography experiments where only one equiv. of Fe(II) was retained for CP-Ser in the absence of Ca(II).8 Taken together with the current insights, we conclude that the Fe(II) affinity of site 1 is too low for Fe(II) binding to be detected by the prior methods. Indeed, further studies of Fe(II) coordination at site 1 and investigating whether the Fe(II)–His3Asp site forms under physiological conditions are warranted. Lastly, we observed in variants of the His6 site that both the 6C His6-derived site as well at the 5C His3Asp site coordinate Fe(II) when sub-stoichiometric Fe(II) was added. This result suggests that as the His ligands are replaced by water-derived ligands at the His6 site, the relative binding affinities of the variant His6 (6C) and His3Asp (5C) sites becomes more similar under these conditions, enabling Fe(II) binding to both sites.
Previous studies demonstrated that CP binds Fe(II) with relatively low affinity at the His6 site in the absence of Ca(II).8 To determine whether the absence of Ca(II) influences the coordination number and/or electronic structure of Fe(II) at this site, NIR MCD studies were also performed on samples where Ca(II) was omitted from the buffer. In the absence of Ca(II), the addition of 0.9 equiv. of Fe(II) to CP-Ser resulted in both 5C (His3Asp) and 6C (His6) Fe(II) sites being occupied. The observed transition energies and LF for the 6C His6 site in the absence of Ca(II) are identical to those found in the presence of Ca(II) (Table 2). This observation is reminiscent of prior studies of Mn(II) binding to the His6 site, where advanced EPR spectroscopy indicated no change to the nature of the Mn(II)–His6 site when Ca(II) was omitted from the sample.16 Taken together, these observations indicate that the enhanced metal-binding affinities at the His6 site that occur in the presence of Ca(II) cannot be explained by a change in the primary coordination sphere that results from Ca(II) binding to the EF-hands of the S100A8/S100A9 subunits. In contrast, the absence of Ca(II) resulted in an increase in the highest energy LF transition of the 5C His3Asp site (by ≈300 cm−1), indicating a perturbation of the Fe(II) His3Asp site in the absence of Ca(II). Overall, these results suggest that the presence or absence of Ca(II) plays a significant role in modulating the Fe(II)-binding affinities as well as determining the Fe(II) distribution between the His6 and His3Asp sites in CP. This result is also consistent with prior spectroscopic studies of Mn(II)-CP species, which revealed that sub-stoichiometric Mn(II) binds exclusively to the His6 site in the presence of excess Ca(II) and to the His6 site as well as one or more other sites in the absence of Ca(II).16
Conclusion
The NIR MCD spectroscopic studies presented in this work provide insights into the Fe(II)-binding properties of the metal-sequestering host-defense protein human CP. These studies demonstrate that (i) Fe(II) preferentially binds to the His6 site of CP-Ser to form a hexahistidine Fe(II) site in the presence of excess Ca(II), (ii) Fe(II) binds to the His3Asp site to generate a 5C Fe(II) species upon mutation of His ligands that compose the His6 site or upon addition of excess Fe(II) to CP-Ser in the presence of Ca(II), and (iii) the absence of Ca(II) results in both 5C (His3Asp) and 6C (His6) Fe(II) sites in the presence of sub-stoichiometric Fe(II) as well as a perturbed LF at the 5C site. These studies provide further support for the notion that Ca(II) ions modulate the transition-metal-binding properties and biological function of human CP.Acknowledgements
We thank the Alfred P. Sloan Foundation (M. L. N. and E. M. N.) and the MIT Center for Environmental Health Sciences (NIH Grant P30-ES002109) for financial support. T. M. B. and T. G. N. are recipients of the NSF Graduate Research Fellowship. We thank Prof. K. L. Bren for the use of an analytical balance and microcentrifuge in her laboratory and Prof. G. M. Culver for the use of the spectrophotometer in her laboratory.References
- M. I. Hood and E. P. Skaar, Nat. Rev. Microbiol., 2012, 10, 525–537 CrossRef CAS PubMed.
- V. E. Diaz-Ochoa, S. Jellbauer, S. Klaus and M. Raffatellu, Front. Cell. Infect. Microbiol., 2014, 4, 2 CrossRef PubMed.
- P. G. Sohnle, C. Collins-Lech and J. H. Wiessner, J. Infect. Dis., 1991, 164, 137–142 CrossRef CAS PubMed.
- B. D. Corbin, E. H. Seeley, A. Raab, J. Feldmann, M. R. Miller, V. J. Torres, K. L. Anderson, B. M. Dattilo, P. M. Dunman, R. Gerads, R. M. Caprioli, W. Nacken, W. J. Chazin and E. P. Skaar, Science, 2008, 319, 962–965 CrossRef CAS PubMed.
- P. A. Clohessy and B. E. Golden, Scand. J. Immunol., 1995, 42, 551–556 CrossRef CAS PubMed.
- B. Johne, M. K. Fagerhol, T. Lyberg, H. Prydz, P. Brandtzaeg, C. F. Naess-Andresen and I. Dale, J. Clin. Pathol.: Mol. Pathol., 1997, 50, 113–123 CrossRef CAS.
- H. J. Loomans, B. L. Hahn, Q.-Q. Li, S. H. Phadnis and P. G. Sohnle, J. Infect. Dis., 1998, 177, 812–814 CrossRef CAS PubMed.
- T. G. Nakashige, B. Zhang, C. Krebs and E. M. Nolan, Nat. Chem. Biol., 2015, 11, 765–771 CrossRef CAS PubMed.
- T. Vogl, N. Leukert, K. Barczyk, K. Strupat and J. Roth, Biochim. Biophys. Acta, 2006, 1763, 1298–1306 CrossRef CAS PubMed.
- M. B. Brophy, J. A. Hayden and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18089–18100 CrossRef CAS PubMed.
- J. A. Hayden, M. B. Brophy, L. S. Cunden and E. M. Nolan, J. Am. Chem. Soc., 2013, 135, 775–787 CrossRef CAS PubMed.
- I. P. Korndörfer, F. Brueckner and A. Skerra, J. Mol. Biol., 2007, 370, 887–898 CrossRef PubMed.
- T. E. Kehl-Fie, S. Chitayat, M. I. Hood, S. Damo, N. Restrepo, C. Garcia, K. A. Munro, W. J. Chazin and E. P. Skaar, Cell Host Microbe, 2011, 10, 158–164 CAS.
- S. M. Damo, T. E. Kehl-Fie, N. Sugitani, M. E. Holt, S. Rathi, W. J. Murphy, Y. Zhang, C. Betz, L. Hench, G. Fritz, E. P. Skaar and W. J. Chazin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3841–3846 CrossRef CAS PubMed.
- M. B. Brophy, T. G. Nakashige, A. Gaillard and E. M. Nolan, J. Am. Chem. Soc., 2013, 135, 17804–17817 CrossRef PubMed.
- D. M. Gagnon, M. B. Brophy, S. E. J. Bowman, T. A. Stich, C. L. Drennan, R. D. Britt and E. M. Nolan, J. Am. Chem. Soc., 2015, 137, 3004–3016 CrossRef CAS PubMed.
- T. G. Nakashige, J. R. Stephan, L. S. Cunden, M. B. Brophy, A. J. Wommack, B. C. Keegan, J. M. Shearer and E. M. Nolan, J. Am. Chem. Soc., 2016, 138, 12243–12251 CrossRef CAS PubMed.
- S. A. Mills and M. A. Marletta, Biochemistry, 2005, 44, 13553–13559 CrossRef CAS PubMed.
- J. C. Dunning Hotopp, T. A. Auchtung, D. A. Hogan and R. P. Hausinger, J. Inorg. Biochem., 2003, 93, 66–70 CrossRef CAS PubMed.
- J. J. A. Cotruvo and J. Stubbe, Metallomics, 2012, 4, 1020–1036 RSC.
- K. D. Koehntop, J. P. Emerson and L. Que, J. Biol. Inorg. Chem., 2005, 10, 87–93 CrossRef CAS PubMed.
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed.
- M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS PubMed.
- M. L. Neidig and E. I. Solomon, Chem. Commun., 2005, 5843–5863, 10.1039/b510233m.
- E. G. Pavel, N. Kitajima and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 3949–3962 CrossRef CAS.
- F. Neese and E. I. Solomon, Inorg. Chem., 1999, 38, 1847–1865 CrossRef CAS PubMed.
- E. I. Solomon and M. A. Hanson, in Inorganic Electronic Structure and Spectroscopy, ed. E. I. Solomon and A. B. P. Lever, Wiley-Interscience, New York, 1999, vol. II, pp. 1–129 Search PubMed.
- T. Ohta, S. Chakrabarty, J. D. Lipscomb and E. I. Solomon, J. Am. Chem. Soc., 2008, 130, 1601–1610 CrossRef CAS PubMed.
- M. A. Pavlosky and E. I. Solomon, J. Am. Chem. Soc., 1994, 116, 11610–11611 CrossRef CAS.
- K. M. Light, J. A. Hangasky, M. J. Knapp and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 9665–9674 CrossRef CAS PubMed.
- M. S. Chow, B. E. Eser, S. A. Wilson, K. O. Hodgson, B. Hedman, P. F. Fitzpatrick and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 7685–7698 CrossRef CAS PubMed.
- M. L. Neidig, C. D. Brown, K. M. Light, D. G. Fujimori, E. M. Nolan, J. C. Price, E. W. Barr, J. M. Bollinger, C. Krebs, C. T. Walsh and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 14224–14231 CrossRef CAS PubMed.
- E. G. Pavel, J. Zhou, R. W. Busby, M. Gunsior, C. A. Townsend and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 743–753 CrossRef CAS.
- G. D. Straganz, A. R. Diebold, S. Egger, B. Nidetzky and E. I. Solomon, Biochemistry, 2010, 49, 996–1004 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sample details and supplemental magnetic circular dichroism spectra. See DOI: 10.1039/c6sc03487j |
| This journal is © The Royal Society of Chemistry 2017 |