
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homochiral oligomers with highly flexible backbones form stable H-bonded duplexes†
Diego
Núñez-Villanueva
and
Christopher A.
Hunter
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: herchelsmith.orgchem@ch.cam.ac.uk
First published on 19th August 2016
Abstract
Two homochiral building blocks featuring a protected thiol, an alkene and a H-bond recognition unit (phenol or phosphine oxide) have been prepared. Iterative photochemical thiol–ene coupling reactions were used to synthesize oligomers containing 1-4 phosphine oxide and 1-4 phenol recognition sites. Length-complementary H-bond donor and H-bond acceptor oligomers were found to form stable duplexes in toluene. NMR titrations and thermal denaturation experiments show that the association constant and the enthalpy of duplex formation increase significantly for every additional H-bonding unit added to the chain. There is an order of magnitude increase in stability for each additional H-bonding interaction at room temperature indicating that all of the H-bonding sites are fully bound to their complements in the duplexes. The backbone of the thiol–ene duplexes is a highly flexible alkane chain, but this conformational flexibility does not have a negative impact on binding affinity. The average effective molarity for the intramolecular H-bonding interactions that zip up the duplexes is 18 mM. This value is somewhat higher than the EM of 14 mM found for a related family of duplexes, which have the same recognition units but a more rigid backbone prepared using reductive amination chemistry. The flexible thiol–ene AAAA·DDDD duplex is an order of magnitude more stable than the rigid reductive amination AAAA·DDDD duplex. The backbone of the thiol–ene system retains much of its conformational flexibility in the duplex, and these results show that highly flexible molecules can make very stable complexes, provided there is no significant restriction of degrees of freedom on complexation.
Introduction
Biomolecules encode information about supramolecular structures and function as a sequence of different monomer units assembled into a linear polymer. Of particular importance is the role of nucleic acids in storing and reproducing genetic information, through the formation of sequence-selective duplexes and self-replication via template-directed synthesis.1 These specific properties have led to the design, study and application of synthetic structures based on nucleic acids as engineering materials for nanotechnology, such as molecular switches, aptamers and multi-component nanometer-scale assemblies.2 However, the properties that allow nucleic acids to encode genetic information are not currently available in any other material.Significant effort has gone into making synthetic oligomers that can form bimolecular duplexes through different types of non-covalent interactions, such as metal–ligand coordination,3 aromatic stacking,4 salt bridges5 and H-bonding.6 The suitability of these oligomers as prototypes for synthetic information molecules is limited by the fact that the recognition sites are usually built into the backbone so that duplex formation requires precise matching of molecular geometries. The introduction of sequence variation into such systems is therefore difficult without disrupting the duplex. In contrast, the modular structure of nucleic acids allows modification of the base-pairing system independently of the backbone.7–9 We recently reported a new class of synthetic oligomers based on this blueprint (Fig. 1), which allows independent optimisation of the backbone (black), recognition sites (blue) and coupling chemistry used for synthesis of the oligomers (red).10 A variety of different oligomers have been prepared using the reductive amination chemistry illustrated in Fig. 1. These systems form stable H-bonded duplexes in toluene, and we have shown that it is possible to mix and match different backbone modules and different recognition modules without disrupting duplex formation.11,12
 | ||
| Fig. 1 A blueprint for duplex forming molecules. There are three key design elements: the coupling chemistry used for the synthesis of oligomers (red), the recognition module which controls intermolecular binding (blue) and the backbone module which links these components together (black). Two different synthesis modules are highlighted: reductive amination and thiol–ene coupling. Adapted from ref. 10. | ||
Here we explore the potential of using a different synthesis module, photochemical thiol–ene coupling (Fig. 1). Thiol–ene coupling appears to be an ideal reaction for oligomer synthesis: it is high yielding with excellent regioselectivity; the rates of reaction are fast under mild conditions; the radical reaction is compatible with a wide variety of functional groups that might be used as the recognition modules; the reaction can be carried out in the non-polar solvents required to promote H-bonding interactions between the recognition modules, and the resulting thioether linkage is non-polar and will not compete with H-bonding sites on the recognition modules.13 Moreover, thiol–ene polymerization has proven to be a useful reaction for materials synthesis,14 and nucleobase-containing homopolymers have been prepared using this reaction.15
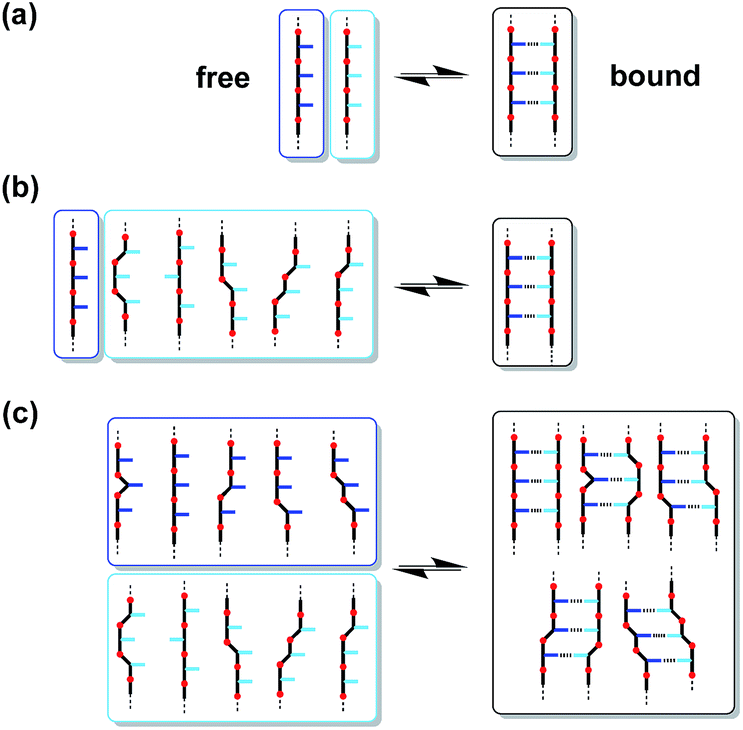
Fig. 2a shows the structure of a duplex formed between a phenol oligomer and a phosphine oxide oligomer, which we have previously prepared using reductive amination chemistry. Fig. 2b shows the design of a similar duplex accessible via thiol–ene coupling. Phenol and phosphine oxide represent an ideal recognition module, because phenol is a very good H-bond donor and a very weak acceptor (H-bond acceptor parameter β ≈ 3), and phosphine oxide is one of the best H-bond acceptors known (β ≈ 10). The H-bond between these two functional groups is exceptionally strong in toluene (K ≈ 300 M−1).16 The thiol–ene synthesis module leads to thioether linkages (Fig. 2b), which are relatively non-polar (β ≈ 3), so there will be no competition with the phosphine oxide H-bond acceptors on the recognition modules.17 The backbone module shown in the design in Fig. 2b is chiral and therefore requires enantioselective synthesis of the monomer units. The influence of monomer chirality on duplex assembly is an interesting feature of this system that could be exploited at a future date. Here we describe the synthesis of homochiral phenol and phosphine oxide monomer units, the use of these building blocks to prepare oligomers, and NMR studies of duplex formation between length-complementary oligomers. One major difference between the reductive amination oligomers in Fig. 2a and the thiol–ene oligomers in Fig. 2b is the conformational flexibility of the backbone, and the consequences for duplex stability are quantified below.
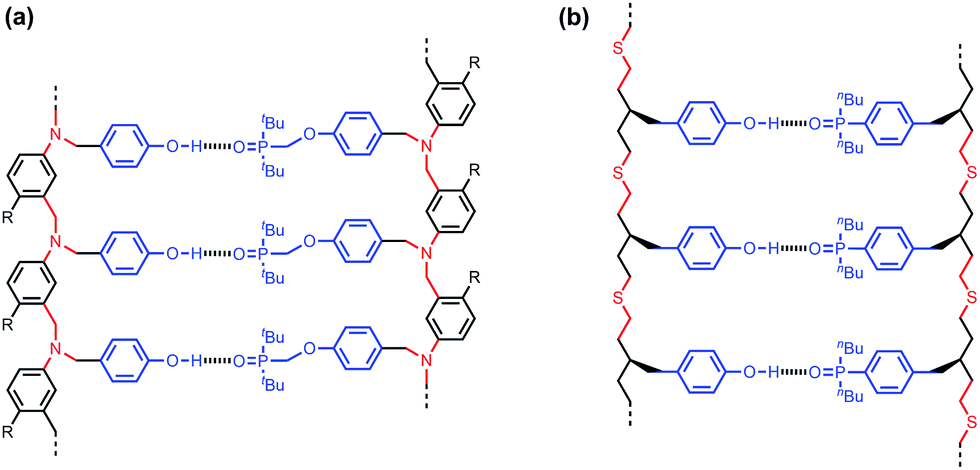 | ||
| Fig. 2 Duplex formed from a phenol oligomer and a phosphine oxide oligomer synthesized (a) using reductive amination chemistry and (b) using thiol–ene chemistry (R are 2-ethylhexoxy solubilising groups).10 | ||
Results and discussion
Synthesis
The synthesis of suitable monomers equipped with a thiol, an alkene and a recognition module is outlined in Schemes 1 and 2. The benzyl bromide derivatives of the recognition modules were prepared first (Scheme 1). The phenol group of 4-hydroxybenzaldehyde (1) was protected as a silyl ether, and the aldehyde was reduced with NaBH4. Bromination of the resulting alcohol with PBr3 yielded benzyl bromide 3a. Palladium-mediated P-arylation of 4-iodobenzaldehyde (4) with di-n-butylphosphine oxide gave 5. Reduction of 5 with NaBH4 and bromination of the resulting alcohol with N-bromosuccinimide (NBS) and triphenylphosphine provided benzyl bromide 3b. | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
Scheme 2 shows the synthetic route to the monomer required for oligomer synthesis. The first step involved formation of imide 7 from commercially available 4-bromobutyric acid 6 using published procedures for incorporation of the S-trityl group and introduction of the 2-oxazolidinone chiral auxiliary.18 Lithium bis(trimethylsilyl)amide (LiHMDS) was used as the base to form the enolate of 7, and alkylation with benzyl bromide 3a or 3b afforded 8a and 8b, respectively, as single diastereoisomers in good yields (dr > 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). Reduction of 8a and 8b removed the chiral auxiliary under mild conditions yielding alcohols 9a and 9b. Oxidation of these alcohols using Dess–Martin periodinane (DMP) followed by Wittig reactions of the aldehyde products with the ylide derived from methyltriphenylphosphonium bromide afforded 10a and 10b. The S-trityl group is not stable to radical conditions,19 so this protecting group was exchanged for an S-acetyl group: treatment of 10a or 10b with TFA in the presence of triisopropylsilane (TIPS) followed by acetylation with acetyl chloride yielded protected monomer 11a and monomer 12b, respectively. Removal of the silyl protecting group from 11a afforded monomer 12a. The synthetic route involves 6–7 steps and gives homochiral monomers 12a and 12b in overall yields of about 40%.
:5). Reduction of 8a and 8b removed the chiral auxiliary under mild conditions yielding alcohols 9a and 9b. Oxidation of these alcohols using Dess–Martin periodinane (DMP) followed by Wittig reactions of the aldehyde products with the ylide derived from methyltriphenylphosphonium bromide afforded 10a and 10b. The S-trityl group is not stable to radical conditions,19 so this protecting group was exchanged for an S-acetyl group: treatment of 10a or 10b with TFA in the presence of triisopropylsilane (TIPS) followed by acetylation with acetyl chloride yielded protected monomer 11a and monomer 12b, respectively. Removal of the silyl protecting group from 11a afforded monomer 12a. The synthetic route involves 6–7 steps and gives homochiral monomers 12a and 12b in overall yields of about 40%.
Oligomers were prepared in a stepwise manner from the monomer units using photochemically initiated thiol–ene coupling reactions (Scheme 3). Reaction of 12a or 12b with 1-hexanethiol under 365 nm irradiation in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) gave end-capped monomers 13a and 13b, respectively. Removal of the S-acetyl protecting group of 13a and 13b under basic conditions and subsequent thiol–ene coupling with monomer 12a or 12b provided 2-mers 14a (DD) and 14b (AA). The same sequential deprotection and thiol–ene coupling procedure yielded 3-mers 15a (DDD) and 15b (AAA), and 4-mer 16a (DDDD). Deprotection of the S-acetyl group of 15b resulted in the corresponding disulfide, but reduction with dithiothreitol (DTT) gave the thiol, which was coupled with 12b to give 4-mer 16b (AAAA). No epimerization was observed in any of the oligomers.
 | ||
| Scheme 3 | ||
Binding studies
31P and 1H NMR titration experiments in toluene-d8 were carried out to measure the association constants for length-complementary oligomers. The patterns of chemical shift observed for the oligomers in the free state in toluene are similar, and there is no evidence of any intramolecular folding. The donor oligomers were titrated into the acceptor oligomers, and the titration data fit well to 1:1 binding isotherms for all four systems (see ESI for details†). The association constants for the 1-mer and 2-mer complexes (A·D and AA·DD) were determined using both 1H and 31P NMR titrations, which gave the same results. The 31P signals for the 3-mer and 4-mer complexes (AAA·DDD and AAAA·DDDD) were too broad to monitor reliably, so these association constants were determined using 1H NMR titrations. The association constants for duplex formation between two oligomers with N recognition units (KN) are provided in Table 1 together with the limiting complexation-induced changes in chemical shift, Δδ. The association constant increases by an order of magnitude for every recognition module added to the oligomer, which suggests that all of the recognition modules are involved in cooperative H-bonding interactions in the duplexes. The patterns of the 31P and 1H NMR complexation-induced change in chemical shift are the same for all four duplexes suggesting that they adopt similar supramolecular motifs. In particular, the large downfield complexation-induced changes in chemical shift observed for the 31P NMR signals (+6 ppm) indicate that all of the phosphine oxide groups form H-bonds in the duplexes.20
| Complex | logKN/M−1 |
EM/mM | K EM | Δδ31P/ppm | Δδ1H/ppmb | |
|---|---|---|---|---|---|---|
| a Each titration was repeated twice and the average value is reported with errors at the 95% confidence limit. b Values for the signal due to the protons ortho to the phosphine oxide groups. | ||||||
| A·D | (12a·12b) | 2.7 ± 0.2 | — | — | 6.4 | 0.09 |
| AA·DD | (14a·14b) | 4.0 ± 0.2 | 14 ± 12 | 8 ± 5 | 6.4 | 0.08 |
| AAA·DDD | (15a·15b) | 5.4 ± 0.1 | 28 ± 16 | 16 ± 3 | 6.3 | 0.09 |
| AAAA·DDDD | (16a·16b) | 6.3 ± 0.1 | 22 ± 11 | 12 ± 2 | 5.9 | 0.10 |
Fig. 3 shows the logarithm of the association constant for duplex formation (logKN) plotted as a function of the number of H-bonding interactions (N). The data fit well to a straight line with a slope of 1.2 (eqn (1)), i.e. the association constant increases by an order of magnitude for each additional H-bond formed.
| logKN = 1.2N + 1.6 | (1) |
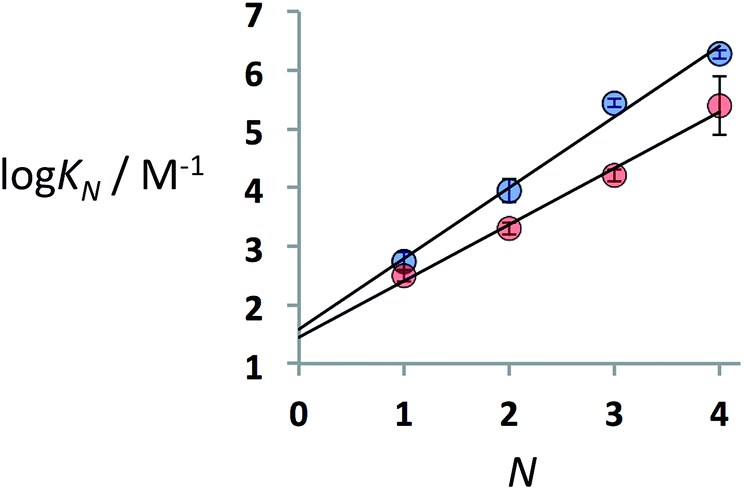 | ||
| Fig. 3 Association constants for duplex formation in toluene-d8 at 298 K (KN) plotted as a function of the number of recognition modules in the oligomer, N. Data shown are for the duplexes obtained using thiol–ene coupling in blue, and in red for the duplexes obtained using reductive amination chemistry (the best fit straight lines are shown, R2 = 0.99 and 0.99, respectively). | ||
Fig. 3 also shows the corresponding data for the duplexes obtained using reductive amination chemistry (Fig. 2a). The reductive amination oligomers have similar phosphine oxide–phenol recognition modules to the thiol–ene system, but the backbone is more rigid. Surprisingly, the thiol–ene duplexes are significantly more stable than the reductive amination duplexes. There are two factors that contribute to the overall stability of a duplex: the intrinsic strength of the H-bonding interactions, which is a property of the recognition modules, and the effective molarity (EM) for formation of intramolecular H-bonds in zipping up of the duplex, which is a property of the backbone. The association constant for formation of a duplex, which is N recognition modules long (KN), can be expressed using eqn (2).
| KN = 2KNEMN−1 | (2) |
The values of EM for the thiol–ene duplexes are reported in Table 1. The results are similar for all three duplexes that form intramolecular interactions, which implies that the backbone is sufficiently flexible to optimise the H-bond geometries in all of these oligomers. Once the first intermolecular H-bond between the two strands is formed, all subsequent intramolecular H-bonds have an average EM of 18 mM. The equilibrium constant for forming an intramolecular H-bond in a partially assembled duplex is given by the product K EM, which provides a measure of the probability of zipping up the duplex as opposed to forming intermolecular interactions that would lead to polymeric aggregates.21 For the thiol–ene duplexes, the values of K EM in Table 1 are all significantly greater than one (the average value is 12), implying that intramolecular H-bonding is highly favoured in these systems. For the reductive amination duplexes shown in Fig. 2a, the average EM is 14 mM and the average value of K EM is 5. Surprisingly, the flexible thiol–ene backbone leads to a slightly higher EM than was found for the more rigid reductive amination backbone. The differences between the properties of the thiol–ene and reductive amination duplexes are small (K = 560 and 350 M−1, and EM = 18 and 14 mM, respectively), but these subtle differences are multiplied along the length of an oligomer, so that the stability of the thiol–ene 4-mer duplex is an order of magnitude larger than the stability of the reductive amination 4-mer duplex.
Thermal denaturation studies
Thermal denaturation experiments were carried out to extract the thermodynamic parameters for duplex assembly. 31P NMR spectra of 1:1 solutions of length-complementary oligomers at 1 mM concentrations in toluene-d8 were recorded at different temperatures between 223 and 363 K. The 31P NMR signals shifted progressively downfield at low temperatures, indicative of an increase in the population of H-bonded complexes. At high temperatures, an upfield shift in the 31P NMR signals was observed consistent with disruption of the H-bonding interactions between oligomers (Fig. 4).
 | ||
| Fig. 4 Experimental 31P NMR chemical shift plotted as a function of temperature for 1:1 mixtures (1 mM) of A·D (black), AA·DD (blue), and AAA·DDD (red) in toluene-d8. The lines are the best fit to eqn (4) (total rmsd < 0.2 ppm). The horizontal bars show the transition melting temperatures, Tm,N. | ||
The data obtained for the AAAA·DDDD duplex (16a·16b) was qualitatively consistent with a duplex that melts at a higher temperature than the AAA·DDD duplex (15a·15b), but it was not possible to extract an accurate melting profile for the 4-mer because the signals were broad and overlapped. Thermodynamic parameters for the other three duplexes were obtained from the melting profiles by fitting the data to a two-state model, assuming that only duplex and denatured single strands are present, that the enthalpy and entropy changes for the formation of the N-mer duplex ( and
and  ) are temperature independent, and that the change in heat capacity between free and bound states is zero.22 The equilibrium constant for duplex formation at a given temperature T, KN(T), is given by eqn (3), which can be derived from the van't Hoff equation.10
) are temperature independent, and that the change in heat capacity between free and bound states is zero.22 The equilibrium constant for duplex formation at a given temperature T, KN(T), is given by eqn (3), which can be derived from the van't Hoff equation.10
 | (3) |
The observed chemical shift (δ) in the NMR denaturation experiment can be expressed as a function of T using eqn (4).10
 | (4) |
Fig. 4 shows the best fit of eqn (4) to the experimental melting data. The optimised values of δf and δb obtained from fitting the melting data agree with the values determined in the NMR titration experiments carried out at 298 K (δf ≈ 35–36 ppm and δb ≈ 41–42 ppm).
Table 2 shows the values of the parameters 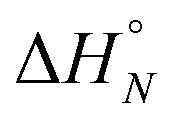 and Tm,N obtained from the thermal denaturation experiments, along with the values of logKN calculated at 298 K using eqn (3). The values of logKN at 298 K determined from the thermal denaturation experiments are similar to the corresponding values determined in the 298 K titration experiments (Table 1), indicating that the assumptions used in the treatment of the thermal denaturation data are valid. At room temperature, an increase of one order of magnitude in KN is observed for each additional H-bond formed. There is a corresponding increase in the transition melting temperature and the enthalpy change on duplex formation with increasing numbers of H-bonds. These observations are indicative of cooperative H-bonding interactions along the duplex. We also note that the values of Tm,N are consistently 10–20 degrees higher than observed for the corresponding reductive amination duplexes, confirming the enhanced stability of the more flexible thiol–ene systems.
and Tm,N obtained from the thermal denaturation experiments, along with the values of logKN calculated at 298 K using eqn (3). The values of logKN at 298 K determined from the thermal denaturation experiments are similar to the corresponding values determined in the 298 K titration experiments (Table 1), indicating that the assumptions used in the treatment of the thermal denaturation data are valid. At room temperature, an increase of one order of magnitude in KN is observed for each additional H-bond formed. There is a corresponding increase in the transition melting temperature and the enthalpy change on duplex formation with increasing numbers of H-bonds. These observations are indicative of cooperative H-bonding interactions along the duplex. We also note that the values of Tm,N are consistently 10–20 degrees higher than observed for the corresponding reductive amination duplexes, confirming the enhanced stability of the more flexible thiol–ene systems.
| Complex | T m,N/K |
|
logKN(298)/M−1 |
|
|
|
|---|---|---|---|---|---|---|
| A·D | (12a·12b) | 285 | −26 | 3.4 | −19 | −7 |
| AA·DD | (14a·14b) | 334 | −39 | 4.3 | −25 | −14 |
| AAA·DDD | (15a·15b) | 365 | −46 | 5.1 | −29 | −17 |



Molecular modelling
The structures of the duplexes were investigated using molecular mechanic calculations. The first H-bond in the duplex was fixed by constraining the distance between the phenol hydrogen and the phosphine oxide oxygen to 2 ± 1 Å. A conformational search was used to find low energy conformations compatible with this single point constraint (see ESI† for details). Fig. 5–7 show the lowest energy structures obtained from conformational searches for AA·DD, AAA·DDD and AAAA·DDDD, respectively. The fully assembled duplex was found in all cases with all of the recognition modules on one oligomer H-bonded to the complementary sites on the other oligomer. It is clear from these calculations that the thiol–ene oligomers are very flexible: the calculated structures shown in Fig. 5–7 are collapsed, unlike the idealised ChemDraw structures. Nevertheless, it appears that there is good fidelity in the pairing of the recognition modules, because complexes with out of register H-bonding interactions were not observed in the calculations: for example for the 3-mer duplex, out of register would be phenols 2 and 3 paired with phosphine oxides 3 and 2, as opposed to the in register arrangement shown in Fig. 6, which has phenols 2 and 3 paired with phosphine oxides 2 and 3.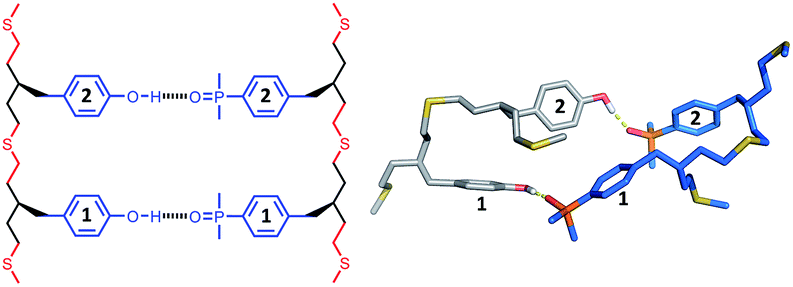 | ||
| Fig. 5 Lowest energy conformation of the 2-mer duplex AA·DD from a conformational search (MMFFs force-field and CHCl3 solvation implemented in Macromodel).23 The H-bond donor oligomer is shown in grey and the H-bond acceptor oligomer in blue. Hydrogens are not shown for clarity, and the recognition modules are numbered. | ||
 | ||
| Fig. 6 Lowest energy conformation of the 3-mer duplex AAA·DDD from a conformational search (MMFFs force-field and CHCl3 solvation implemented in Macromodel).23 The H-bond donor oligomer is shown in grey and the H-bond acceptor oligomer in blue. Hydrogens are not shown for clarity, and the recognition modules are numbered. | ||
 | ||
| Fig. 7 Lowest energy conformation of the 4-mer duplex AAAA·DDDD from a conformational search (MMFFs force-field and CHCl3 solvation implemented in Macromodel).23 The H-bond donor oligomer is shown in grey and the H-bond acceptor oligomer in blue. Hydrogens are not shown for clarity, and the recognition modules are numbered. | ||
The conformational flexibility of the thiol–ene duplexes provides a possible explanation for the unusually high stability exhibited by these systems. Fig. 8 shows an overlay of low energy duplex structures that were found for the 2-mer AA·DD: a wide range of different conformations is compatible with duplex formation. If most of the conformational states accessible to the single stranded oligomers are also available to the assembled duplex then the entropic cost restricting of degrees of freedom will be small. Fig. 9 illustrates how the population of conformations in the free and bound states contributes to the free energy of duplex formation in rigid and flexible systems. A large entropic penalty is expected for duplexes formed between a rigid and a flexible backbone, but if both of the backbones are rigid or if both of the backbones are flexible, the duplex should be relatively stable, because the change in the number of accessible conformations is relatively small. In other words, conformational flexibility is only detrimental to binding if conformational degrees of freedom are lost on complexation.
 | ||
| Fig. 8 Superposition of 11 different conformations that were found within 5 kJ mol−1 of the global minimum for the 2-mer AA·DD duplex. Two different points of views are shown. The H-bond donor oligomers are shown in grey and the H-bond acceptor oligomers in blue, and hydrogens are not shown. | ||
 | ||
| Fig. 9 The effect of conformational flexibility on duplex stability. (a) For two rigid oligomers, the duplex has similar conformational properties to the free state. (b) For a flexible oligomer (pale blue) binding to a rigid oligomer (dark blue), duplex formation significantly reduces the number of conformational states. (c) For two flexible oligomers, the duplex retains much of the conformational flexibility present in the free state. | ||
Conclusions
A new class of oligomeric molecules bearing phosphine oxide and phenol recognition modules is reported. These systems were prepared using iterative thiol–ene coupling chemistry to produce the all H-bond donor and the all H-bond acceptor oligomers up to four repeats long. The monomers for oligomer synthesis were prepared in homochiral form with a terminal alkene and a protected thiol, and no epimerisation was observed in subsequent reactions. 1H and 31P NMR titrations and thermal denaturation experiments demonstrate that length-complementary donor and acceptor oligomers form H-bonded duplexes in toluene. The association constant for duplex formation increases uniformly by an order of magnitude for each additional recognition module added to the chain, indicating that all of the phosphine oxide and phenol groups are involved in H-bonding interactions in the duplexes. Thermal denaturation experiments also show that there is an increase in the enthalpy change for duplex formation as the length of the oligomer increases, demonstrating cooperative H-bonding interactions along the duplex. The properties of the thiol–ene oligomers can be compared with a different set of oligomers that we have previously reported which were assembled using reductive amination chemistry. Both systems use phenol–phosphine oxide H-bonding interactions as the recognition motif, but they differ significantly in the flexibility of the backbone. The thiol–ene oligomers have a highly flexible alkyl chain for the backbone, whereas the reductive amination oligomers have a more rigid backbone featuring aromatic units. Surprisingly, the more flexible thiol–ene oligomers form the more stable duplexes: the thiol–ene AAAA·DDDD duplex is an order of magnitude more stable than the corresponding reductive amination duplex. Molecular modelling studies suggest that the thiol–ene duplexes can access a large number of different conformations in the bound state, and so the flexibility of the backbone is not significantly restricted on binding in this system. We have previously reported that small differences in backbone flexibility have a minimal effect on the EM for duplex formation using three different reductive amination oligomers (7–20 mM). The values of EM for duplex formation with the thiol–ene oligomers are 14–28 mM, which shows that much more dramatic increases in backbone flexibility have almost no impact on the EM for intramolecular H-bond formation. The results demonstrate that conformationally flexible molecules can form high affinity complexes provided that conformational mobility is retained in the bound state.Acknowledgements
We thank the EPSRC and ERC for funding. We thank Dr Rafel Cabot and Dr Giulia Iadevaia for helpful advice and discussions.Notes and references
- (a) J. D. Watson and F. H. Crick, Nature, 1953, 171, 964 CrossRef CAS PubMed; (b) R. R. Sinden, DNA Structure and Function, Academic Press, 1994 Search PubMed.
- (a) A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818 CrossRef CAS PubMed; (b) P. W. K. Rothemund, Nature, 2006, 440, 297 CrossRef CAS PubMed; (c) F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795 CrossRef CAS PubMed; (d) A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763 CrossRef CAS PubMed; (e) F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198 CrossRef CAS PubMed.
- (a) R. Kramer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CrossRef CAS PubMed; (b) A. Marquis, V. Smith, J. Harrowfield, J.-M. Lehn, H. Herschbach, R. Sanvito, E. Leize-Wagner and A. van Dorsselaer, Chem.–Eur. J., 2006, 12, 5632 CrossRef CAS PubMed; (c) H. L. Anderson, Inorg. Chem., 1994, 33, 972 CrossRef CAS; (d) P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538 CrossRef CAS.
- (a) V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720 CrossRef CAS PubMed; (b) V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2001, 7, 2810 CrossRef CAS PubMed.
- (a) Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., 2005, 117, 3935 CrossRef; (b) H. Ito, Y. Furusho, T. Hasegawa and E. Yashima, J. Am. Chem. Soc., 2008, 130, 14008 CrossRef CAS PubMed; (c) Y. Furusho and E. Yashima, Macromol. Rapid Commun., 2011, 32, 136 CrossRef CAS PubMed; (d) H. Yamada, Y. Furusho, H. Ito and E. Yashima, Chem. Commun., 2010, 46, 3487 RSC.
- (a) J. Sánchez-Quesada, C. Seel, P. Prados, J. de Mendoza, I. Dalcol and E. Giralt, J. Am. Chem. Soc., 1996, 118, 277 CrossRef; (b) A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856 CrossRef CAS; (c) A. P. Bisson and C. A. Hunter, Chem. Commun., 1996, 1723 RSC; (d) B. Gong, Y. Yan, H. Zeng, E. Skrzypczak-Jankunn, Y. W. Kim, J. Zhu and H. Ickes, J. Am. Chem. Soc., 1999, 121, 5607 CrossRef CAS; (e) B. Gong, Synlett, 2001, 582 CAS; (f) H. Zeng, R. S. Miller, R. A. Flowers and B. Gong, J. Am. Chem. Soc., 2000, 122, 2635 CrossRef CAS; (g) H. Zeng, H. Ickes, R. A. Flowers and B. Gong, J. Org. Chem., 2001, 66, 3574 CrossRef CAS PubMed; (h) B. Gong, Polym. Int., 2007, 56, 436 CrossRef CAS; (i) B. Gong, Acc. Chem. Res., 2012, 45, 2077 CrossRef CAS PubMed; (j) Y. Yang, Z.-Y. Yang, Y.-P. Yi, J.-F. Xiang, C.-F. Chen, L.-J. Wan and Z.-G. Shuai, J. Org. Chem., 2007, 72, 4936 CrossRef CAS PubMed; (k) W.-J. Chu, Y. Yang and C.-F. Chen, Org. Lett., 2010, 12, 3156 CrossRef CAS PubMed; (l) W.-J. Chu, J. Chen, C.-F. Chen, Y. Yang and Z. Shuai, J. Org. Chem., 2012, 77, 7815 CrossRef CAS PubMed; (m) E. A. Archer and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 5074 CrossRef CAS PubMed; (n) H. Gong and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 1719 CrossRef CAS PubMed.
- (a) J. A. Piccirilli, T. Krauch, S. E. Moroney and S. A. Benner, Nature, 1990, 343, 33 CrossRef CAS PubMed; (b) Z. Yang, D. Hutter, P. Sheng, A. M. Sismour and S. A. Benner, Nucleic Acids Res., 2006, 34, 6095 CrossRef CAS PubMed; (c) Z. Yang, F. Chen, J. B. Alvarado and S. A. Benner, J. Am. Chem. Soc., 2011, 133, 15105 CrossRef CAS PubMed; (d) S. A. Benner, Curr. Opin. Chem. Biol., 2012, 16, 581 CrossRef CAS PubMed; (e) F. Wojciechowski and C. J. Leumann, Chem. Soc. Rev., 2011, 40, 5669 RSC; (f) E. T. Kool, Acc. Chem. Res., 2002, 35, 936 CrossRef CAS PubMed; (g) H. Liu, J. Gao, S. R. Lynch, Y. D. Saito, L. Maynard and E. T. Kool, Science, 2003, 302, 868 CrossRef CAS PubMed; (h) J. N. Wilson and E. T. Kool, Org. Biomol. Chem., 2006, 4, 4265 RSC; (i) D. A. Malyshev, K. Dhami, H. T. Quach, T. Lavergne, P. Ordoukhanian, A. Torkamani and F. E. Romesberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12005 CrossRef CAS PubMed; (j) D. A. Malyshev and F. E. Romesberg, Angew. Chem., Int. Ed., 2015, 54, 11930 CrossRef CAS PubMed.
- (a) Z. Huang, K. C. Schneider and S. A. Benner, J. Org. Chem., 1991, 56, 3869 CrossRef CAS; (b) C. Richert, A. L. Roughton and S. A. Benner, J. Am. Chem. Soc., 1996, 118, 4518 CrossRef CAS; (c) S. A. Benner and D. Hutter, Bioorg. Chem., 2002, 30, 62 CrossRef CAS PubMed; (d) Z. Huang and S. A. Benner, J. Org. Chem., 2002, 67, 3996 CrossRef CAS PubMed; (e) P. Li, Z. A. Sergueeva, M. Dobrikov and B. R. Shaw, Chem. Rev., 2007, 107, 4746 CrossRef CAS PubMed; (f) H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Org. Lett., 2008, 10, 3729 CrossRef CAS PubMed; (g) P. E. Nielsen and G. Haaima, Chem. Soc. Rev., 1997, 26, 73 RSC; (h) P. E. Nielsen, Chem. Biodiversity, 2010, 7, 786 CrossRef CAS PubMed; (i) Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73 CrossRef CAS PubMed.
- (a) K.-U. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347 CrossRef; (b) A. Aerschot van, I. Verheggen, C. Hendrix and P. Herdewijn, Angew. Chem., Int. Ed., 1995, 34, 1338 CrossRef; (c) D. Renneberg and C. J. Leumann, J. Am. Chem. Soc., 2002, 124, 5993 CrossRef CAS PubMed; (d) D. A. Braasch and D. R. Corey, Chem. Biol., 2001, 8, 1 CrossRef CAS PubMed; (e) S. K. Singh, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455 RSC; (f) H. V. Nguyen, Z.-Y. Zhao, A. Sallustrau, S. L. Horswell, L. Male, A. Mulas and J. H. R. Tucker, Chem. Commun., 2012, 48, 12165 RSC; (g) L. Zhang, A. Peritz and E. Meggers, J. Am. Chem. Soc., 2005, 127, 4174 CrossRef CAS PubMed; (h) M. K. Schlegel, A. E. Peritz, K. Kittigowittana, L. Zhang and E. Meggers, ChemBioChem, 2007, 8, 927 CrossRef CAS PubMed; (i) P. Karri, V. Punna, K. Kim and R. Krishnamurthy, Angew. Chem., Int. Ed., 2013, 52, 5840 CrossRef CAS PubMed.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 94 RSC.
- G. Iadevaia, A. E. Stross, A. Neumann and C. A. Hunter, Chem. Sci., 2016, 7, 1760 RSC.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016 10.1039/c6sc01884j.
- (a) A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995 CrossRef CAS PubMed; (b) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed; (c) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed; (d) A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573 RSC; (e) W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013 RSC.
- (a) A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC; (b) M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS; (c) G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536 RSC.
- W. Xi, S. Pattanayak, C. Wang, B. Fairbanks, T. Gong, J. Wagner, C. J. Kloxin and C. N. Bowman, Angew. Chem., Int. Ed., 2015, 54, 14462 CrossRef CAS PubMed.
- H. Sun, C. A. Hunter, C. Navarro and S. Turega, J. Am. Chem. Soc., 2013, 135, 13129 CrossRef CAS PubMed.
- (a) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS PubMed; (b) C. S. Calero, J. Farwer, E. J. Gardiner, C. A. Hunter, M. Mackey, S. Scuderi, S. Thompson and J. G. Vinter, Phys. Chem. Chem. Phys., 2013, 15, 18262 RSC.
- (a) N. Qvit, H. Reuveni, S. Gazal, A. Zundelevich, G. Blum, M. Y. Niv, A. Feldstein, S. Meushar, D. E. Shalev, A. Friedler and C. Gilon, J. Comb. Chem., 2008, 10, 256 CrossRef CAS PubMed; (b) A. Hara, R. Morimoto, Y. Ishikawa and S. Nishiyama, Org. Lett., 2011, 13, 4036 CrossRef CAS PubMed.
- P. Demircioglu, M. H. Acar and Y. Yagci, J. Appl. Polym. Sci., 2003, 46, 1639–1643 CrossRef.
- (a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235 CrossRef CAS; (b) U. Mayer, W. Gerger and V. Gutmann, Monatsh. Chem., 1977, 108, 489 CrossRef CAS; (c) V. Gutmann, Pure Appl. Chem., 1979, 51, 2197 CrossRef CAS.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS PubMed.
- (a) J. Applequist and V. Damle, J. Am. Chem. Soc., 1965, 87, 1450 CrossRef CAS; (b) K. J. Breslauer, Methods Enzymol., 1995, 259, 221 CAS; (c) T. Ratilainen, A. Holmén, E. Tuite, P. E. Nielsen and B. Nordén, Biochemistry, 2000, 39, 7781 CrossRef CAS PubMed.
- MacroModel, version 9.8, Schrödinger, LLC, New York, NY, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and 1H, 13C and 31P NMR spectra of all compounds, NMR titration spectra, thermal denaturation spectra and molecular modelling methods. See DOI: 10.1039/c6sc02995g |
| This journal is © The Royal Society of Chemistry 2017 |