
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decreasing the electronic confinement in layered perovskites through intercalation†
Matthew D.
Smith
 a,
Laurent
Pedesseau
b,
Mikaël
Kepenekian
c,
Ian C.
Smith
a,
Claudine
Katan
c,
Jacky
Even
*b and
Hemamala I.
Karunadasa
*a
a,
Laurent
Pedesseau
b,
Mikaël
Kepenekian
c,
Ian C.
Smith
a,
Claudine
Katan
c,
Jacky
Even
*b and
Hemamala I.
Karunadasa
*a
aDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: hemamala@stanford.edu
bFonctions Optiques pour les Technologies de l'information, CNRS, INSA de Rennes, 35708 Rennes, France. E-mail: Jacky.Even@insa-rennes.fr
cInstitut des Sciences Chimiques de Rennes, CNRS, Université de Rennes 1, 35042 Rennes, France
First published on 10th November 2016
Abstract
We show that post-synthetic small-molecule intercalation can significantly reduce the electronic confinement of 2D hybrid perovskites. Using a combined experimental and theoretical approach, we explain structural, optical, and electronic effects of intercalating highly polarizable molecules in layered perovskites designed to stabilize the intercalants. Polarizable molecules in the organic layers substantially alter the optical and electronic properties of the inorganic layers. By calculating the spatially resolved dielectric profiles of the organic and inorganic layers within the hybrid structure, we show that the intercalants afford organic layers that are more polarizable than the inorganic layers. This strategy reduces the confinement of excitons generated in the inorganic layers and affords the lowest exciton binding energy for an n = 1 perovskite of which we are aware. We also demonstrate a method for computationally evaluating the exciton's binding energy by solving the Bethe–Salpeter equation for the exciton, which includes an ab initio determination of the material's dielectric profile across organic and inorganic layers. This new semi-empirical method goes beyond the imprecise phenomenological approximation of abrupt dielectric-constant changes at the organic–inorganic interfaces. This work shows that incorporation of polarizable molecules in the organic layers, through intercalation or covalent attachment, is a viable strategy for tuning 2D perovskites towards mimicking the reduced electronic confinement and isotropic light absorption of 3D perovskites while maintaining the greater synthetic tunability of the layered architecture.
1. Introduction
The remarkable optoelectronic properties of organic–inorganic metal–halide perovskites have recently come to light. Notably, these properties can be synthetically modulated by altering the dimensionality of the anionic inorganic lattice. Small organic cations allow for the self-assembly of three-dimensional (3D) frameworks, whereas larger organic cations partition the inorganic lattice into two-dimensional (2D) sheets.1 Recently, 3D Pb–I perovskites have been employed as low-cost absorbers in high-efficiency solar cells.2–4 Here, the weak binding energy between photogenerated electrons and holes allows for charge carriers to easily separate and migrate towards their respective current collectors. The high dielectric constant of the 3D Pb–I lattice shields the coulombic attraction between photogenerated electrons and holes.5,6 This results in free charge carriers that are ideal for solar-cell absorbers. On the other hand, typical 2D n = 1 perovskites (where n is the number of metal–halide sheets in each inorganic layer) are not suitable for standard photovoltaic devices, owing to the presence of strongly bound electron–hole pairs, or excitons. The quantum confinement of the 2D inorganic sheets increases the bandgap (Eg) and exciton binding energy (Eb) relative to the 3D materials. The Eb in these materials is further enhanced by the dielectric mismatch between organic and inorganic layers.7–12 Here, the low dielectric constant of the adjacent organic layers provides poor shielding of the electrons and holes in the inorganic layers leading to the exciton's dielectric confinement. The Eb values for n = 1 perovskites are typically above 300 meV,13 comparable to those of certain organic molecules.14 These tightly bound excitons in 2D perovskites have enabled varied applications in luminescence. The high oscillator strength of the excitons affords strong excitonic luminescence, which has been used in green phosphor15 and light-emitting-diode16 applications, and we proposed that white-light emission from 2D perovskites stems from strongly bound excitons stabilized (or self-trapped) through lattice distortions.17,18 Furthermore, perovskites with 1 < n < ∞ values provide access to intermediate Eg and Eb values,19–21 while the organic layers bring new functionality. For example, the n = 3 2D Pb–I perovskite exhibits sufficiently low Eg and Eb values to absorb sunlight and generate photocurrent in a solar cell, while hydrophobic organic layers provide enhanced moisture resistance.22,23 Owing to the greater stability and structural diversity of the layered framework compared to the 3D perovskite, we investigated how the common n = 1 Pb–I perovskites can be modified to mimic the optoelectronic properties of their 3D congeners.Herein we demonstrate that post-synthetic intercalation of highly polarizable molecules into 2D Pb–I perovskites can significantly decrease their electronic confinement and optical anisotropy, enabling new functionality in these materials. Using experimental and theoretical methods, we explain the structural, optical, and electronic consequences of intercalating I2 molecules in the perovskite's organic layers. Furthermore, using in situ powder X-ray diffraction (PXRD) and in situ optical absorption spectroscopy, we monitor the dynamics of halogen intercalation and release. We further extend halogen intercalation to halogen-mediated reactivity to access novel Pb–halide perovskites that cannot be synthesized from solution.
2. Results and discussion
We recently showed that upon exposure to halogen gas, layered perovskites containing terminal alkynes or alkenes topotactically expand by up to 36% of their original volume to form perovskites containing dihaloalkenes or dihaloalkanes, respectively.24,25 During our experiments with I2 chemisorption, we hypothesized that intercalation precedes halogenation of these nonporous solids. Here, the organic layers may stabilize I2 incorporation similar to halogen solvation in organic liquids. A paraffin-like quality has been previously ascribed to organic layers containing long-chain alkyl groups in hybrid perovskites26,27 and binding sites in the organic layers have been shown to stabilize electrochemical ion insertion in these materials.28 We therefore sought to assess if intercalation of highly polarizable molecules could provide a facile method for tuning the electronic structure of 2D perovskites.2.1. Structural effects of iodine intercalation
Weak electrostatic interactions have been previously leveraged to reversibly intercalate nonpolar aliphatic and aromatic hydrocarbons into layered perovskites containing long alkyl chains such as (C10H21NH3)2[CdCl4] and (C9H19NH3)2[PbI4].27 Stronger fluoroaryl–aryl interactions have been subsequently used to stabilize and isolate the guest-intercalated perovskites (C6H5(CH2)2NH3)2[SnI4]·(C6F6) and (C6F5(CH2)2NH3)2[SnI4]·(C6H6).29 In order to investigate the consequences of I2 intercalation in 2D perovskites, we first chose (C6H13NH3)2[PbI4]30 (hereafter denoted as (C6)2[PbI4]) in order to provide a well-defined organic bilayer with some structural flexibility (Fig. 1A, inset). Perovskite films were deposited on substrates through spin coating and then thermally annealed. Flowing air over the spinning sample improved film quality (further details are provided in the ESI†). Exposing a (C6)2[PbI4] film to a dry, inert carrier gas (N2 or Ar) containing I2 vapor results in a color change from orange to red and an expansion of the unit cell by ca. 4 Å along the c axis (perpendicular to the inorganic sheets) to yield the perovskite we formulate as (C6H13NH3)2[PbI4]·xI2 (hereafter (C6)2[PbI4]·xI2). However, (C6)2[PbI4]·xI2 completely desorbs I2 within 10 minutes of removal from an I2 atmosphere to regenerate the original material (C6)2[PbI4]. | ||
| Fig. 1 (A) Powder X-ray diffraction (PXRD) patterns of (C6)2[PbI4] (red) and (C6)2[PbI4]·xI2 (blue). Inset: X-ray crystal structure of (C6)2[PbI4].30 (B) PXRD patterns of (IC6)2[PbI4] (red) and (IC6)2[PbI4]·xI2 (blue). Inset: X-ray crystal structure of (IC6)2[PbI4]. Dark green, purple, blue, and grey spheres represent Pb, I, N, and C atoms, respectively. Disordered atoms and hydrogen atoms are omitted for clarity. | ||
To stabilize the I2-intercalated material, we then focused on the perovskite (IC6H12NH3)2[PbI4] (hereafter denoted as (IC6)2[PbI4]) containing terminal alkyl iodides (Fig. 1B, inset). The structure of (IC6)2[PbI4] at 173 K has been reported;31 however, we collected single-crystal XRD data at 298 K to gain a better match with our room-temperature PXRD patterns. The 298 K crystal structure (Fig. 2A) shows a partially interdigitated organic layer with I⋯I distances of 5.017(1) Å between organoiodines that run parallel to the inorganic sheets. Iodine–iodide interactions of 3.955(1) Å are also evident between the organoiodines and terminal iodides of the inorganic sheets. We envisioned that halogen–halogen interactions32,33 between the organoiodines and inorganic iodides could stabilize I2 intercalation in the organic layers. Exposing (IC6)2[PbI4] thin films to I2 vapor also results in a color change from orange to red. PXRD data show that I2 exposure causes the inter-layer spacing in (IC6)2[PbI4] to increase by 5.5 Å (33%) (Fig. 1B). Indeed, the iodine retention time is increased more than four-fold in (IC6)2[PbI4]·xI2 compared to (C6)2[PbI4]·xI2. Because films of (C6)2[PbI4] and (IC6)2[PbI4] are similar in both quality and thickness (250 ± 20 nm), we attribute the difference in I2 retention to stabilization of I2 molecules by the iodoalkyl groups in (IC6)2[PbI4].
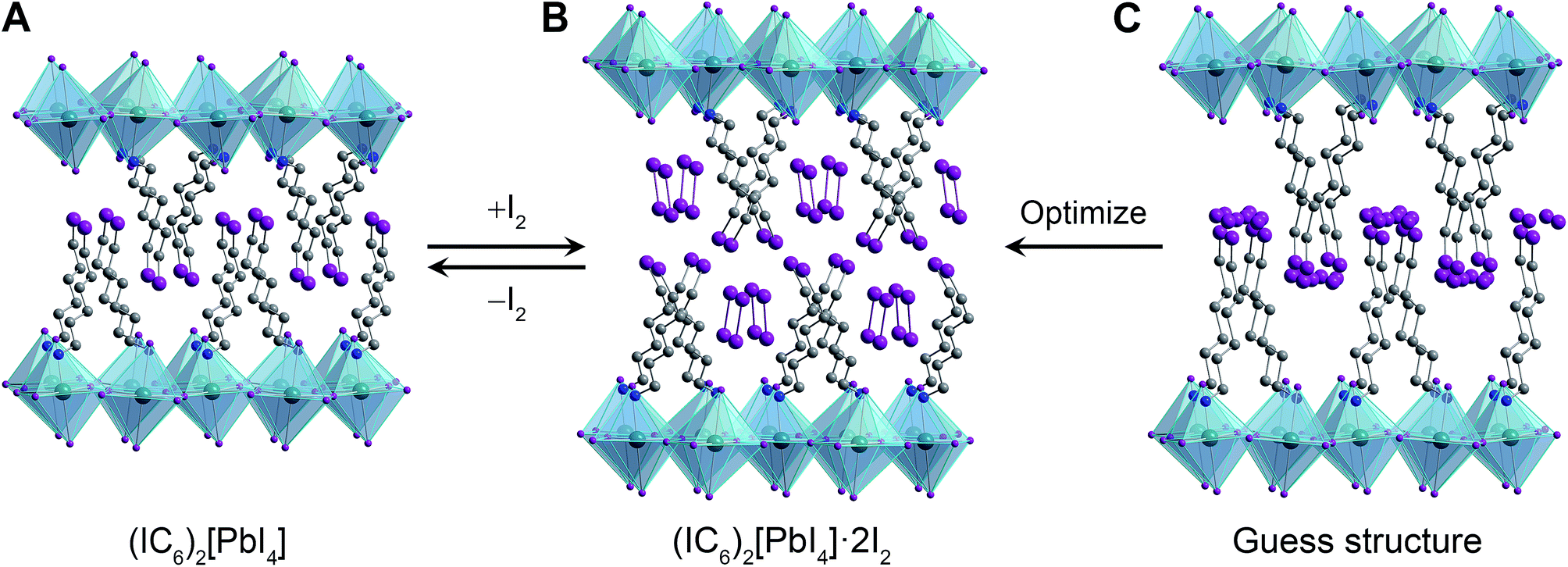 | ||
| Fig. 2 (A) Single-crystal X-ray structure of (IC6)2[PbI4]. Initial guess structure of the I2 intercalation product (C) and its geometry-optimized structure (B). Dark green, purple, blue, and grey spheres represent Pb, I, N, and C atoms, respectively. Disordered atoms and hydrogen atoms are omitted for clarity. | ||
To elucidate the structure of (IC6)2[PbI4]·xI2, we performed structural optimization at the DFT level in two steps. First, we used the local density approximation (LDA) for both (IC6)2[PbI4] and (IC6)2[PbI4]·xI2. Then, we re-optimized the LDA-optimized structure using the generalized gradient approximation (GGA) corrected to take into account van der Waals (vdW) interactions (hereafter GGA+vdW; see ESI and Table S2† for details). The alkyl chain of the IC6+ cation in (IC6)2[PbI4] is disordered across two positions. For the calculations we used only the atomic positions of the major disorder component. This final geometry-optimized structure of (IC6)2[PbI4] is consistent with the room-temperature single-crystal XRD structure, with differences ranging from ca. 1.5% to 3.8% for the a and b lattice parameters, respectively (Table S2†). Note that our structure optimizations do not include temperature effects. In layered hybrid perovskites, the evolution of unit-cell parameters with temperature is not necessarily systematic because of the rotational flexibility of the metal–halide octahedra.
In order to simulate the I2-intercalated perovskite structure, we expanded the inter-layer spacing by the experimentally observed 5.5 Å. We then positioned two intercalated I2 molecules per formula unit with their inter-nuclear axes aligned parallel to the inorganic sheets to simulate their possible configuration as the molecules enter the perovskite (Fig. 2C). During geometry optimization, no constraints were placed upon the added iodine atoms, which were allowed to move freely and independently. Upon reaching an optimized configuration at the LDA level of theory, we continued the optimization at the GGA+vdW level of theory. We find that the iodine atoms arrange as I2 molecules with their inter-nuclear axes nearly perpendicular to the inorganic sheets (Fig. 2B). The I2 molecules are flanked on either side by halogen–halogen interactions from both inorganic iodides (in the inorganic layer) as well as organoiodines (in the organic layer) with short I⋯I distances of 3.32–3.33 Å (Table S2†). The computed c lattice parameter (double the inter-layer spacing) for (IC6)2[PbI4]·2I2 compares well with that obtained from PXRD, with a difference of ca. 1% (Table S2†) of the experimental value determined at room temperature. We therefore assign the I2-intercalated perovskite as (IC6)2[PbI4]·2I2. Interestingly, the I2⋯I− fragment in (IC6)2[PbI4]·2I2 exhibits a similar geometry to the triiodide anion found in CsI3,34 indicating that the inorganic lattice may now be thought to contain axial triiodides bound to the Pb2+ centers to give the perovskite (IC6)2[PbI2(I3)2]. Therefore, we post-synthetically access a lead–iodide–triiodide perovskite, which is structurally related to the mixed-ligand 2D perovskites (H3N(CH2)8NH3)2[(AuII2)(AuIIII4)(I3)2]35 and (CH3NH3)2[PbI2(SCN)2]36 and the 1D structure (H3N(CH2)2SS(CH2)2NH3)4[Pb3I14]·I2 (ref. 37) that contains I2 molecules interacting with the lead–iodide chains.
2.2. Optical and electronic effects of iodine intercalation
The 2D Pb–I perovskites are medium-bandgap semiconductors with significant absorption in the visible range. The 298 K absorption spectrum of (IC6)2[PbI4] shows an excitonic absorption at 2.38 eV (521 nm), similar to other layered Pb–I perovskites (Fig. 3A).38 Iodine intercalation causes a redshift of this absorption band by 60 meV to 2.32 eV (535 nm), as well as an increase in absorption between 3 and 5 eV. Furthermore, the intensity ratio between the excitonic absorption peak and the above bandgap (continuum) absorption is reduced upon I2 intercalation. The high oscillator strength and temperature-induced broadening of the excitonic absorbance in lead–halide perovskites13,38 obscure the bandgap onset at room temperature. However, at 5 K we were able to resolve the bandgap in both (IC6)2[PbI4] and (IC6)2[PbI4]·2I2 as linear step-like absorption features (Fig. 3B and C and S2†) characteristic of a 2D material.39 We estimate Eg for (IC6)2[PbI4] to be ca. 2.56 eV. Iodine intercalation redshifts the Eg to ca. 2.49 eV in (IC6)2[PbI4]·2I2, a decrease of ca. 70 meV. Importantly, the perovskite's Eb substantially decreases upon I2 intercalation. Using the difference between the bandgap and the exciton peak energy in the 5 K absorption spectra, we estimate that Eb for (IC6)2[PbI4] is ca. 230 meV, which is significantly lower than typical perovskites likely owing to the polarizability of the organoiodines. This Eb value further drops in (IC6)2[PbI4]·2I2 to only 180 meV (a decrease of 50 meV). To our knowledge, this is the lowest Eb value reported for an n = 1 Pb–I perovskite. Most 2D lead–iodide perovskites have Eb values higher than 300 meV.7 The perovskite (C6H5(CH2)2NH3)2[PbI4] has been reported to have a notably low Eb of 220 meV, attributed to the polarizability of the aromatic organic cations.21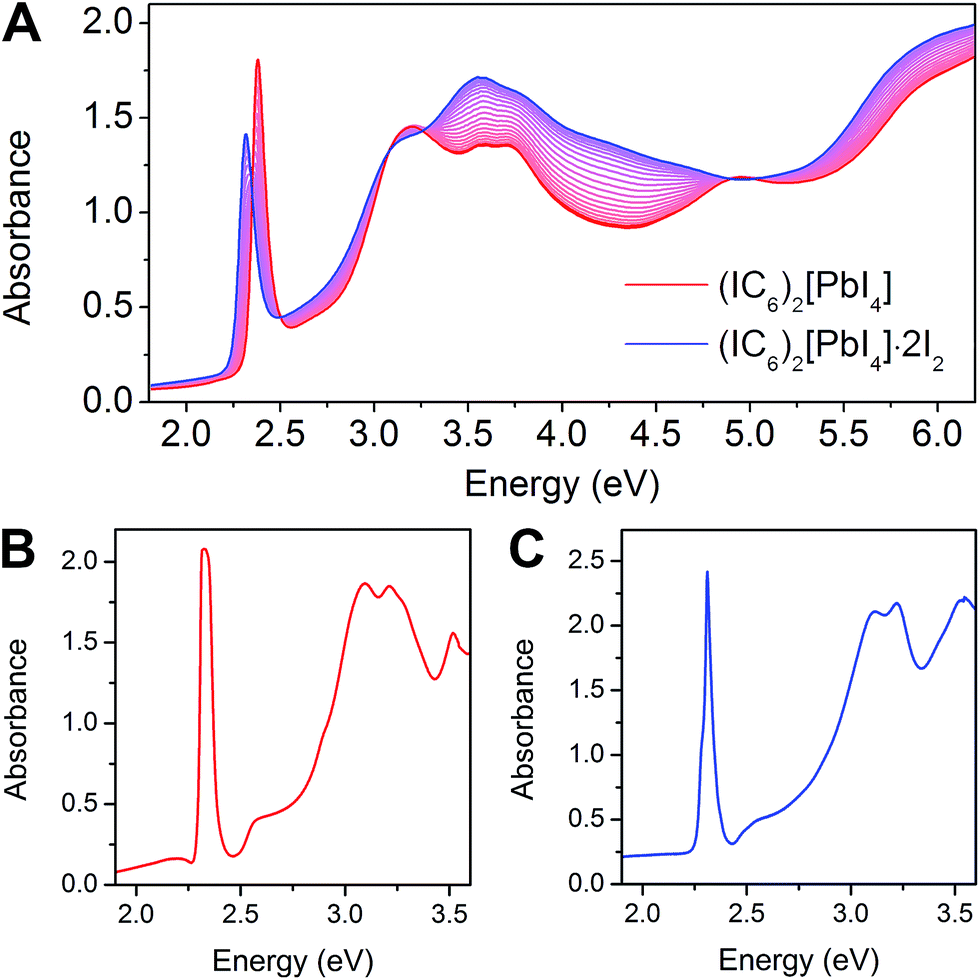 | ||
| Fig. 3 (A) Change in room-temperature absorption spectra as (IC6)2[PbI4] (red) absorbs I2 to form (IC6)2[PbI4]·2I2 (blue). Absorbance spectra collected at 5 K for (IC6)2[PbI4] (B) and (IC6)2[PbI4]·2I2 (C). | ||
Small-molecule intercalation can cause both structural and electronic changes in perovskites. As the inorganic sheets are pushed apart to accommodate the intercalants, small distortions in the metal–halide sheets can slightly alter Eg.40 However, this effect alone cannot explain the significant reduction in both Eb and Eg we observe upon I2 intercalation. In fact, the computed crystal structure reveals a sizeable increase of in-plane octahedral rotations (Table S2†) that should lead to an increase in Eg,41–43 in contradiction with our experimental findings. Therefore, intercalation-induced electronic effects should play the dominant role in the observed changes in the perovskite.
Two cooperative electronic effects greatly modify the Eb of 2D perovskites relative to the 3D analogues: (i) quantum and (ii) dielectric confinement.13,38 (i) Quantum confinement arises from the 2D structure of the lead–halide layers, causing greatly reduced band dispersion perpendicular to the inorganic sheets.38,39 In the ideal limit of a single n = 1 lead–halide layer confined by potential barriers of infinite height, purely 2D quantum confinement enhances Eb by a factor of four compared to the analogous 3D perovskite.39,44 Therefore, to parse the contribution of quantum confinement to the 2D perovskite's Eb, we first considered a hypothetical 3D perovskite with the same dielectric constant and exciton reduced mass as the 2D perovskite. Here, the 3D material's Eb can be calculated as the Rydberg energy in the hydrogenic Bohr model modified by the dielectric constant of the inorganic layers (measured at the middle of an inorganic sheet in the 2D perovskite, close to the Pb atoms) and exciton reduced mass (details in the ESI†). Using our computed values of the 3D Rydberg energies of the excitons Eb,3D, and calculating Eb,2D = 4 Eb,3D gives values of 204 and 142 meV for (IC6)2[PbI4] and (IC6)2[PbI4]·2I2, respectively. These values are lower than the corresponding experimental values (230 and 180 meV, respectively), indicating significant additional contributions from the exciton's dielectric confinement to its Eb. (ii) Dielectric confinement11 is a result of the dielectric mismatch between the inorganic (high dielectric constant)5,45 and organic (low dielectric constant)38 layers. Here, the high-frequency dielectric constant (ε∞) is an appropriate descriptor of the charge-screening ability of the layers, owing to the small exciton Bohr radius and faster timescales of electronic polarization compared to lattice vibrations.5,46 The organic layer poorly screens the electric field between the electron and hole in the inorganic layer, thereby further enhancing Eb over the conventional limit of a 2D quantum well.9,13
We reasoned that I2 intercalation could lessen the quantum confinement (the I2 molecules that orient perpendicular to the sheets could reduce its 2D nature), reduce the dielectric confinement (by increasing the polarizability of the organic layers), or both. To parse out these contributions to the exciton's confinement we turned to electronic-structure calculations.
 | ||
| Fig. 4 Electronic band structure (left) and projected density of states (pDOS, right) for (A) (IC6)2[PbI4] and (B) (IC6)2[PbI4]·2I2. The I2 pDOS was halved to bring into scale. | ||
The bulk high-frequency dielectric response (ε∞) corresponds to an average response over all layers of the perovskite and is not suited to describe the difference in dielectric constant between the organic and the inorganic layers. We therefore used a method designed to compute ε∞ profiles for nanoscale slabs and composite systems10,51 to estimate the respective contributions from the organic and inorganic layers to the bulk ε∞ value. Profiles of the high-frequency dielectric constant perpendicular to the layers (ε∞,⊥) in (IC6)2[PbI4] and (IC6)2[PbI4]·2I2 are shown in Fig. 5. The behavior of ε∞,⊥ changes substantially when I2 molecules intercalate. The average ε∞,⊥ dielectric profile stemming from the inorganic layers increases from 5.4 to 7.0 when including I2. For the ε∞,⊥ dielectric profile associated with the organic layers, we see a dramatic three-fold increase from 3.7 to 11.1 upon I2 intercalation. Therefore, I2 intercalation significantly decreases the dielectric confinement of excitons in the inorganic layers by better screening electric field lines in the organic layer.
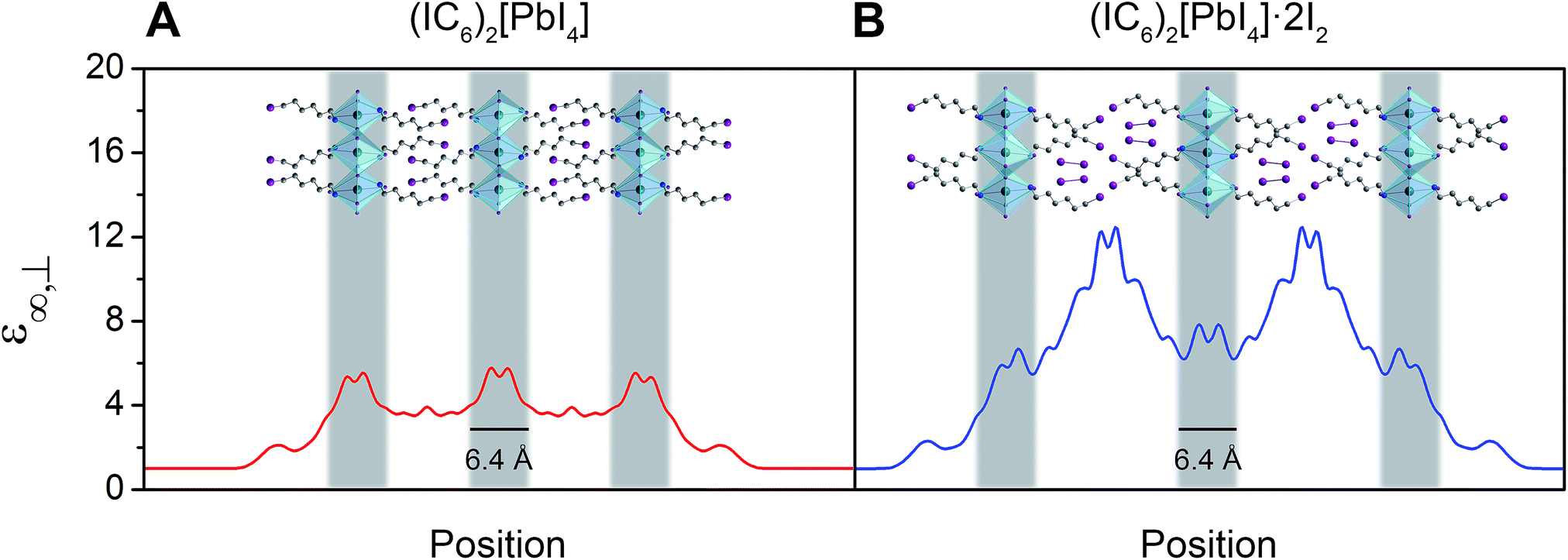 | ||
| Fig. 5 Slabs of (IC6)2[PbI4] (A) and (IC6)2[PbI4]·2I2 (B) and their corresponding calculated high-frequency dielectric profiles ε∞,⊥. Here, ε∞,⊥ is the high-frequency dielectric constant perpendicular to the direction of layer propagation. Dark green, purple, blue, and grey spheres represent Pb, I, N, and C atoms, respectively. Hydrogen atoms omitted for clarity. | ||
Layered perovskites have been considered as quantum-well-like structures where the more polarizable (higher dielectric constant) inorganic sheets form the “wells” and the less polarizable organic layers (low dielectric constant) form the “barriers”.8,38 Notably, I2 intercalation completely inverts this dielectric profile to yield the first example of a 2D perovskite containing organic layers with a higher dielectric constant than the inorganic layers. The exciton's dielectric confinement is thus substantially reduced, though not completely eliminated because the exciton is likely screened only by the immediately adjacent portion of the organic layer.
The resulting Eb values for (IC6)2[PbI4] and (IC6)2[PbI4]·2I2 are 288 and 171 meV, respectively. These numbers compare well with the experimental Eb values of 230 and 180 meV for (IC6)2[PbI4] and (IC6)2[PbI4]·2I2, respectively (determined using the exciton peak positions and bandgaps from the 5 K absorption spectra in Fig. 3B and C). Similar to our experimental absorption spectrum, the calculated spectrum for (IC6)2[PbI4]·2I2 (Fig. S7†) also shows a reduction in the ratio between the excitonic absorption peak and the above bandgap continuum compared to that of (IC6)2[PbI4], which is a good indicator of decreased dielectric confinement upon I2 intercalation (Fig. 3 and 5).55 The difference between the exciton peak positions in the computed absorption spectra for (IC6)2[PbI4] and (IC6)2[PbI4]·2I2 relative to experiment result from the difference between experimental and computed Eg and Eb values.
The experimentally observed reduction in the exciton's confinement upon I2 intercalation is therefore well supported by theory. Our calculations further indicate that the dominant contribution to this effect is the decrease in the exciton's dielectric confinement upon inclusion of the polarizable I2 molecules in the organic layers, which results in an inversion of the pseudo-quantum-well dielectric profile. The intercalating I2 molecules extend perpendicular to the inorganic sheets and further mitigate the 2D nature of the material's optical properties leading to more isotropic light absorption.
2.3. Dynamics of halogen intercalation
The foregoing calculations assumed that we experimentally accessed a single product through I2 intercalation: (IC6)2[PbI4]·2I2, with no intermediate species with varying I2 content. To test this assumption, we studied the dynamics of I2 intercalation. Because reaction with I2 vapor occurs in seconds, we designed in situ PXRD and electronic absorption spectroscopy cells for use with halogen gas to track potential reaction intermediates (see ESI for details and Fig. S8†).We first studied the reaction between the alkyl perovskite (C6)2[PbI4] and I2. Here, we see evidence for reaction intermediates in both PXRD and optical absorption spectroscopy (see ESI† for details). The excitonic absorption band continuously redshifts upon I2 intercalation and it continuously blueshifts upon I2 release, indicating an evolving structure (Fig. S9 and S10†). In contrast, we did not observe any crystalline intermediates in the PXRD patterns during I2 absorption or desorption from the iodoalkyl perovskite (IC6)2[PbI4]·2I2 (Fig. 6, inset). The absence of intermediates is further supported by optical absorption spectroscopy. When we plot the exciton's spectral progression during I2 intercalation and deintercalation for (IC6)2[PbI4] and (IC6)2[PbI4]·2I2, respectively, we do not observe a continuous shift of the excitonic absorption energy (Fig. 6 and S11†). Instead, at intermediate scan times, the excitonic absorbance contains only contributions from the excitonic bands of (IC6)2[PbI4] and (IC6)2[PbI4]·2I2. The absence of intermediates in both the PXRD and absorption data suggests that I2 molecules that enter the perovskite rapidly localize to the binding sites provided by the organoiodines and inorganic iodides. Compared to (C6)2[PbI4], the organic layers in (IC6)2[PbI4] should interact more strongly with the inorganic sheets. Upon I2 loss from (IC6)2[PbI4]·2I2, the organic layers may more rapidly find their equilibrium positions in (IC6)2[PbI4] aided by iodine–iodine interactions between the organic and inorganic layers. Such halogen–halogen interactions have been previously invoked as structure-directing agents during the assembly of layered lead–halide perovskites.41 To further test if I2 molecules intercalate between the organic bilayers, we synthesized the alkyldiammonium perovskite (H3NC6H12NH3)[PbI4],56 which contains an organic monolayer that forms hydrogen bonds at both ends with adjacent inorganic sheets. We did not observe any new phases in the PXRD patterns during I2 exposure, indicating that an organic bilayer is necessary for I2 intercalation (Fig. S12†).
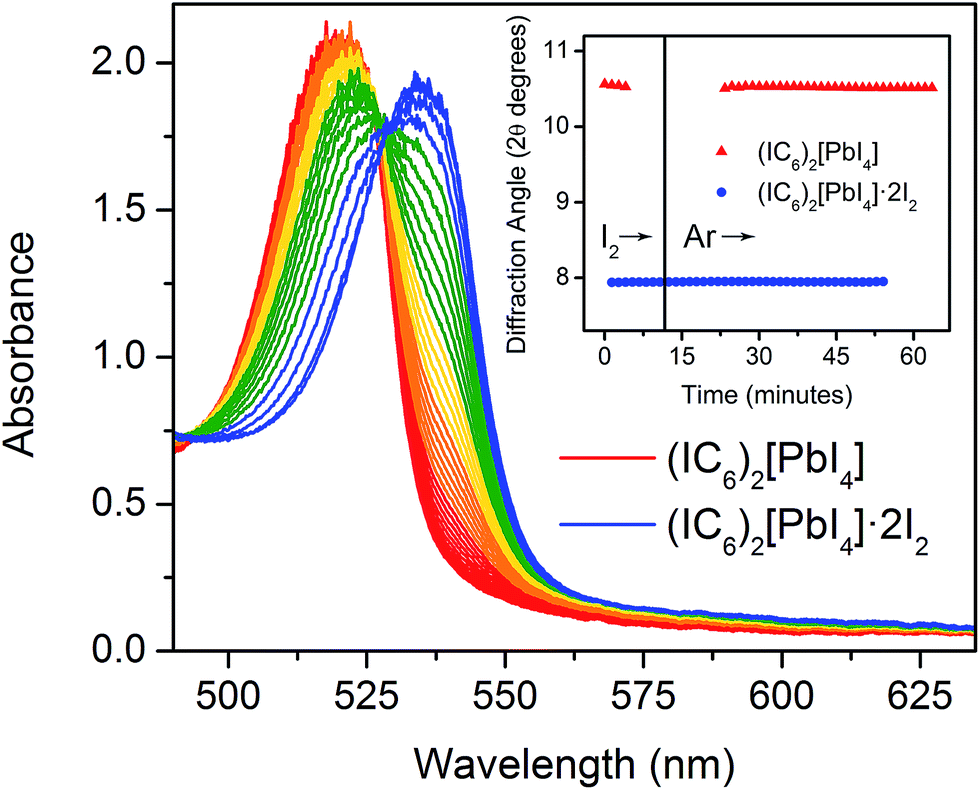 | ||
| Fig. 6 In situ optical absorbance spectra acquired as (IC6)2[PbI4]·2I2 (blue) desorbs I2 to yield (IC6)2[PbI4] (red). Inset: position of the (004) reflection in the powder X-ray diffraction pattern as (IC6)2[PbI4] absorbs and then desorbs I2 vapor. | ||
2.4. Halogen exchange
We finally turned our attention to reactivity that may follow halogen intercalation. As we reported previously, Br2 exposure converts Pb–I perovskites to Pb–Br perovskites through redox-mediated halogen exchange.25 To further extend this reactivity, we investigated the reaction product of (IC6)2[PbI4] with Br2. In addition to inorganic halide substitution (95% conversion), analysis of the digested product by 1H NMR shows that the organoiodines have also been converted (90% conversion) to organobromines to yield the new perovskite: (BrC6H12NH3)2[PbBr4] (Fig. 7C, S13 and S17†). While atypical, a few examples of solution-state halogen-mediated organohalogen substitution reactions are known.57,58 To further parse the reactivity of the organic and inorganic components, we then studied the reaction between a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of (IC6)2[PbI4] and Br2. The major product of this reaction (95% yield) was (IC6)2[PbBr4] (identified through 1H NMR, inductively coupled plasma mass spectrometry, and PXRD), where the organoiodines remain unsubstituted (Fig. 7A, S14 and S18†). This indicates that inorganic halide substitution precedes organohalogen exchange in the organic layer. Notably, we could not form this perovskite through solution-state reactions. When we combined IC6+ salts and PbBr2 in solution, Br− ions partially displaced the organoiodines to form organobromines (Fig. S16†).
:2 molar ratio of (IC6)2[PbI4] and Br2. The major product of this reaction (95% yield) was (IC6)2[PbBr4] (identified through 1H NMR, inductively coupled plasma mass spectrometry, and PXRD), where the organoiodines remain unsubstituted (Fig. 7A, S14 and S18†). This indicates that inorganic halide substitution precedes organohalogen exchange in the organic layer. Notably, we could not form this perovskite through solution-state reactions. When we combined IC6+ salts and PbBr2 in solution, Br− ions partially displaced the organoiodines to form organobromines (Fig. S16†).
 | ||
| Fig. 7 Schematic of products obtained through reaction of (IC6)2[PbI4] (B) with a stoichiometric amount of Br2 (A) and with excess Br2 (C). Turquoise and brown polyhedra represent Pb–I and Pb–Br octahedra, respectively. Dark green, purple, brown, blue, and grey spheres represent Pb, I, Br, N, and C atoms, respectively. Hydrogen atoms omitted for clarity. | ||
3. Conclusions
We demonstrate that small molecules can be stabilized in binding pockets designed into 2D perovskites. Notably, placing highly polarizable molecules in the organic layers can cause large changes in the electronic and optical properties of the inorganic sheets. Here we show that I2 intercalation results in more polarizable organic layers compared to the inorganic layers, which considerably decreases the dielectric confinement of excitons generated in the inorganic layers. Control over the exciton binding energy in 2D perovskites can enable their application in a broad range of optoelectronic technologies. Although the I2-intercalated perovskite studied here is metastable, our studies show that incorporation of polarizable functionalities through intercalation or covalent attachment in the organic layers is a viable approach for substantially decreasing the electronic confinement of these layered materials.We further extend halogen intercalation to halogen-mediated reactivity where our studies show that inorganic halide exchange precedes organohalogen exchange. This gas–solid reaction allows us to synthesize perovskites that cannot be formed through traditional solution-state routes.
Acknowledgements
This work was funded by the National Science Foundation (NSF) CAREER award (DMR-1351538), the Global Climate and Energy Project (GCEP), and the Alfred P. Sloan Fellowship. M. D. S. is supported by an NSF Graduate Research Fellowship (DGE-114747). Single-crystal and powder X-ray diffraction experiments were performed at the Stanford Nanocharacterization Laboratory (SNL) and at beamline 11.3.1 at the Advanced Light Source (ALS). The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. J. E. and L. P. acknowledge the HPC resources of CINES under the allocations 2016-[x2016096724] and 2015-[x2015096724] made by GENCI (Grand Equipement National de Calcul Intensif). The theoretical work was supported by the University of Rennes 1 (Action Incitative, Défis Scientifiques Emergents 2015). J. E.'s work is also supported by the Fondation d'entreprises banque Populaire de l'Ouest under Grant PEROPHOT 2015.Notes and references
- D. B. Mitzi, in Prog. Inorg. Chem., ed. K. D. Karlin, John Wiley & Sons Inc, New York, 1999, vol. 48, pp. 1–121 Search PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- S. Kazim, M. K. Nazeeruddin, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2014, 53, 2812–2824 CrossRef CAS PubMed.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- Q. Q. Lin, A. Armin, R. C. R. Nagiri, P. L. Burn and P. Meredith, Nat. Photonics, 2015, 9, 106–112 CrossRef CAS.
- J. Even, L. Pedesseau and C. Katan, J. Phys. Chem. C, 2014, 118, 11566–11572 CAS.
- E. A. Muljarov, S. G. Tikhodeev, N. A. Gippius and T. Ishihara, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 14370–14378 CrossRef CAS.
- J. Even, L. Pedesseau and C. Katan, ChemPhysChem, 2014, 15, 3733–3741 CrossRef CAS PubMed.
- L. V. Keldysh, Pis'ma Zh. Eksp. Teor. Fiz., 1979, 29, 716–719 Search PubMed.
- D. Sapori, M. Kepenekian, L. Pedesseau, C. Katan and J. Even, Nanoscale, 2016, 8, 6369–6378 RSC.
- E. Hanamura, N. Nagaosa, M. Kumagai and T. Takagahara, Mater. Sci. Eng., B, 1988, 1, 255–258 CrossRef.
- J. Even, L. Pedesseau, M.-A. Dupertuis, J.-M. Jancu and C. Katan, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 205301 CrossRef.
- H. Takagi, H. Kunugita and K. Ema, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 125421 CrossRef.
- M. Knupfer, Appl. Phys. A: Mater. Sci. Process., 2003, 77, 623–626 CrossRef CAS.
- K. Gauthron, J.-S. Lauret, L. Doyennette, G. Lanty, A. Al Choueiry, S. J. Zhang, A. Brehier, L. Largeau, O. Mauguin, J. Bloch and E. Deleporte, Opt. Express, 2010, 18, 5912–5919 CrossRef CAS PubMed.
- M. Era, S. Morimoto, T. Tsutsui and S. Saito, Appl. Phys. Lett., 1994, 65, 676–678 CrossRef CAS.
- E. R. Dohner, E. T. Hoke and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 1718–1721 CrossRef CAS PubMed.
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS PubMed.
- J. Calabrese, N. L. Jones, R. L. Harlow, N. Herron, D. L. Thorn and Y. Wang, J. Am. Chem. Soc., 1991, 113, 2328–2330 CrossRef CAS.
- D. B. Mitzi, S. Wang, C. A. Feild, C. A. Chess and A. M. Guloy, Science, 1995, 267, 1473–1476 CrossRef CAS PubMed.
- X. Hong, T. Ishihara and A. V. Nurmikko, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 6961–6964 CrossRef CAS.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- A. H. Slavney, R. W. Smaha, I. C. Smith, A. Jaffe, D. Umeyama and H. I. Karunadasa, Inorg. Chem., 2016 DOI:10.1021/acs.inorgchem.6b01336.
- D. Solis-Ibarra and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 1039–1042 CrossRef CAS PubMed.
- D. Solis-Ibarra, I. C. Smith and H. I. Karunadasa, Chem. Sci., 2015, 6, 4054–4059 RSC.
- G. F. Needham, R. D. Willett and H. F. Franzen, J. Phys. Chem., 1984, 88, 674–680 CrossRef CAS.
- Y. I. Dolzhenko, T. Inabe and Y. Maruyama, Bull. Chem. Soc. Jpn., 1986, 59, 563–567 CrossRef CAS.
- A. Jaffe and H. I. Karunadasa, Inorg. Chem., 2014, 53, 6494–6496 CrossRef CAS PubMed.
- D. B. Mitzi, D. R. Medeiros and P. R. L. Malenfant, Inorg. Chem., 2002, 41, 2134–2145 CrossRef CAS PubMed.
- D. G. Billing and A. Lemmerer, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 735–747 CAS.
- A. Lemmerer and D. G. Billing, CrystEngComm, 2010, 12, 1290–1301 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- H. A. Tasman and K. H. Boswijk, Acta Crystallogr., 1955, 8, 59–60 CrossRef CAS.
- L. M. Castro-Castro and A. M. Guloy, Angew. Chem., Int. Ed., 2003, 42, 2771–2774 CrossRef CAS PubMed.
- M. Daub and H. Hillebrecht, Angew. Chem., Int. Ed., 2015, 54, 11016–11017 CrossRef CAS PubMed.
- N. Louvain, W. Bi, N. Mercier, J.-Y. Buzaré, C. Legein and G. Corbel, Dalton Trans., 2007, 965–970 RSC.
- T. Ishihara, J. Lumin., 1994, 60–1, 269–274 CrossRef.
- X. Hong, T. Ishihara and A. V. Nurmikko, Solid State Commun., 1992, 84, 657–661 CrossRef CAS.
- J. L. Knutson, J. D. Martin and D. B. Mitzi, Inorg. Chem., 2005, 44, 4699–4705 CrossRef CAS PubMed.
- S. Sourisseau, N. Louvain, W. Bi, N. Mercier, D. Rondeau, F. Boucher, J.-Y. Buzaré and C. Legein, Chem. Mater., 2007, 19, 600–607 CrossRef CAS.
- C. Katan, L. Pedesseau, M. Kepenekian, A. Rolland and J. Even, J. Mater. Chem. A, 2015, 3, 9232–9240 CAS.
- D. B. Mitzi, C. D. Dimitrakopoulos and L. L. Kosbar, Chem. Mater., 2001, 13, 3728–3740 CrossRef CAS.
- M. Shinada and S. Sugano, J. Phys. Soc. Jpn., 1966, 21, 1936–1946 CrossRef CAS.
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef CAS.
- K. Tanaka, T. Takahashi, T. Kondo, K. Umeda, K. Ema, T. Umebayashi, K. Asai, K. Uchida and N. Miura, Jpn. J. Appl. Phys., Part 1, 2005, 44, 5923–5932 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Ángyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- T. Umebayashi, K. Asai, T. Kondo and A. Nakao, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 155405 CrossRef.
- J. Even, L. Pedesseau and M. Kepenekian, Phys. Chem. Chem. Phys., 2014, 16, 25182–25190 RSC.
- R. R. Guseinov, Phys. Status Solidi B, 1984, 125, 237–243 CrossRef CAS.
- D. B. T. Thoai, R. Zimmermann, M. Grundmann and D. Bimberg, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5906–5909 CrossRef.
- J. Even, L. Pedesseau, J.-M. Jancu and C. Katan, J. Phys. Chem. Lett., 2013, 4, 2999–3005 CrossRef CAS.
- A. V. Rodina and A. L. Efros, J. Exp. Theor. Phys., 2016, 122, 554–566 CrossRef CAS.
- G. A. Mousdis, G. C. Papavassiliou, C. P. Raptopoulou and A. Terzis, J. Mater. Chem., 2000, 10, 515–518 RSC.
- C. Friedel, Justus Liebigs Ann. Chem., 1865, 135, 203–207 CrossRef.
- E. J. Corey and W. J. Wechter, J. Am. Chem. Soc., 1954, 76, 6040–6042 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1487885. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02848a |
| This journal is © The Royal Society of Chemistry 2017 |