
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThioester reduction and aldehyde transamination are universal steps in actinobacterial polyketide alkaloid biosynthesis†
U. R.
Awodi‡
,
J. L.
Ronan‡
,
J.
Masschelein‡
,
E. L. C.
de los Santos
 and
G. L.
Challis
*
and
G. L.
Challis
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: g.l.challis@warwick.ac.uk
First published on 22nd August 2016
Abstract
Actinobacteria produce a variety of polyketide alkaloids with unusual structures. Recently, it was shown that a type I modular polyketide synthase (PKS) is involved in the assembly of coelimycin P1, a polyketide alkaloid produced by Streptomyces coelicolor M145. However, the mechanisms for converting the product of the PKS to coelimycin P1 remain to be elucidated. Here we show that the C-terminal thioester reductase (TR) domain of the PKS and an ω-transaminase are responsible for release of the polyketide chain as an aldehyde and its subsequent reductive amination. Bioinformatics analyses identified numerous gene clusters in actinobacterial genomes that encode modular PKSs with a C-terminal TR domain and a homolog of the ω-transaminase. These are predicted to direct the biosynthesis of both known and novel polyketide alkaloids, suggesting that reductive chain release and transamination constitutes a conserved mechanism for the biosynthesis of such metabolites.
Introduction
An astonishing array of structurally-diverse polyketide metabolites with numerous applications in medicine, animal health and agriculture are produced by actinobacteria.1 Remarkable molecular machines known as type I modular polyketide synthases (PKSs) are responsible for the biosynthesis of the overwhelming majority of these natural products.2 Modular PKSs employ several distinct strategies for the creation of structural diversity, including the utilization of a variety of starter and extender units for assembly of the polyketide chain,3 diverse mechanisms for release of the fully-assembled chain from the PKS,4 and a wide range of on- and post-PKS tailoring reactions. Among these, the mechanism of chain release is arguably of prime importance in defining which structural class a metabolite belongs to. Thus, in macrolide biosynthesis chain release proceeds via macrolactonization,4 whereas in ansamycin biosynthesis the chain is released via macrolactamization;4 polyether biosynthesis employs hydrolytic chain release;4 (spiro)tetronates are assembled by condensation of the polyketide chain with a glyceryl thioester;5 and chain release in prodiginine biosynthesis proceeds via nucleophilic attack of an amino acid α-carbanion equivalent (see ESI† for further details).6Incorporation experiments with isotope-labeled precursors have shown that several mono-, di-, tri- and tetracyclic alkaloids produced by actinobacteria, including latumcidin 1 (also known as abikoviromycin),7 nigrifactin 2,8 pyrindicin 3,9 cyclizidine 4,10 and streptazolin 5 (ref. 11) (Fig. 1) are derived from polyketide precursors. Several other actinobacterial alkaloids, such as streptazone E 6,12 4-hydroxy(2-penta-1,3-dienyl)piperidine 7,13 and indolizomycin 8 (ref. 14) can similarly be hypothesized to have a polyketide origin. In 2012, we reported the identification of the unusual alkaloid coelimycin P1 9 (Fig. 1) as the metabolic product of a cryptic polyketide (cpk) biosynthetic gene cluster in Streptomyces coelicolor M145.15 This provided the first biochemical insights into the molecular mechanisms underlying actinobacterial polyketide alkaloid biosynthesis.
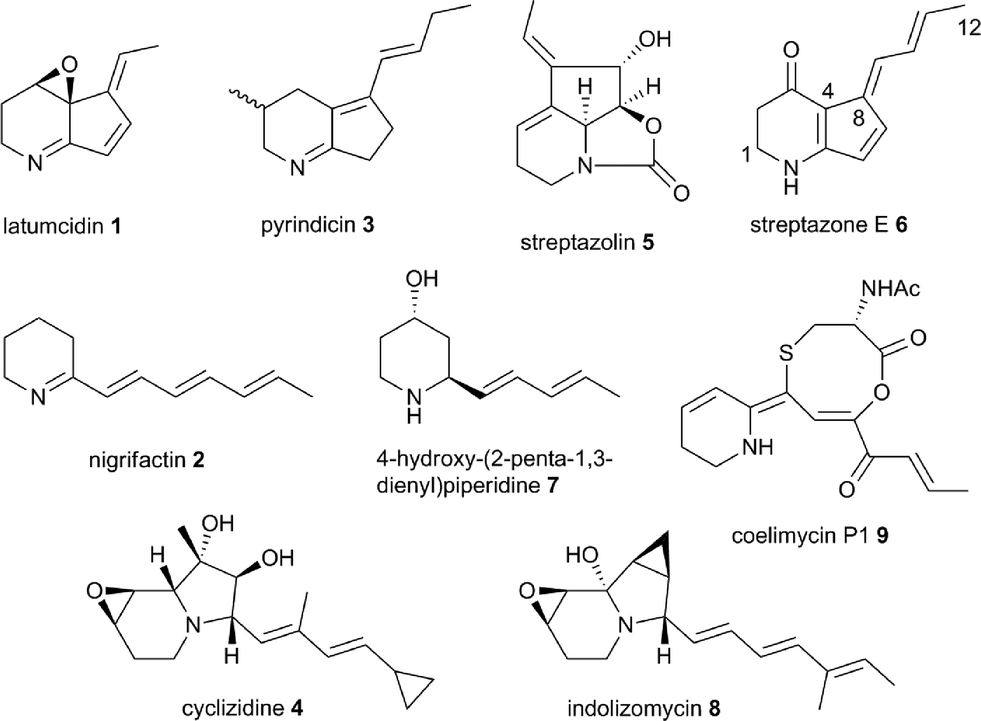 | ||
| Fig. 1 Structures of polyketide alkaloids from actinobacteria. | ||
The cpkA, cpkB and cpkC genes within the coelimycin biosynthetic gene cluster encode a hexamodular PKS, which is proposed on the basis of the predicted specificities of its catalytic domains to assemble thioester 10 (Fig. 2).15 This intermediate is thought to be released from the PKS by NAD(P)H-mediated reduction to the corresponding aldehyde 11, catalyzed by the thioester reductase (TR) domain at the C-terminus of CpkC.15 Reductive amination of aldehyde 11, catalyzed by the putative ω-transaminase encoded by cpkG, is proposed to give amine 12 (or its (Z)-Δ-3,4 isomer, depending on the timing of Δ-2,3 isomerization).15 Elaboration of 12 to 9 is believed to proceed via a series of oxidations, a cyclodehydration and bis-epoxide trapping by N-acetyl-L-cysteine, as illuminated by incorporation of labelled precursors.15
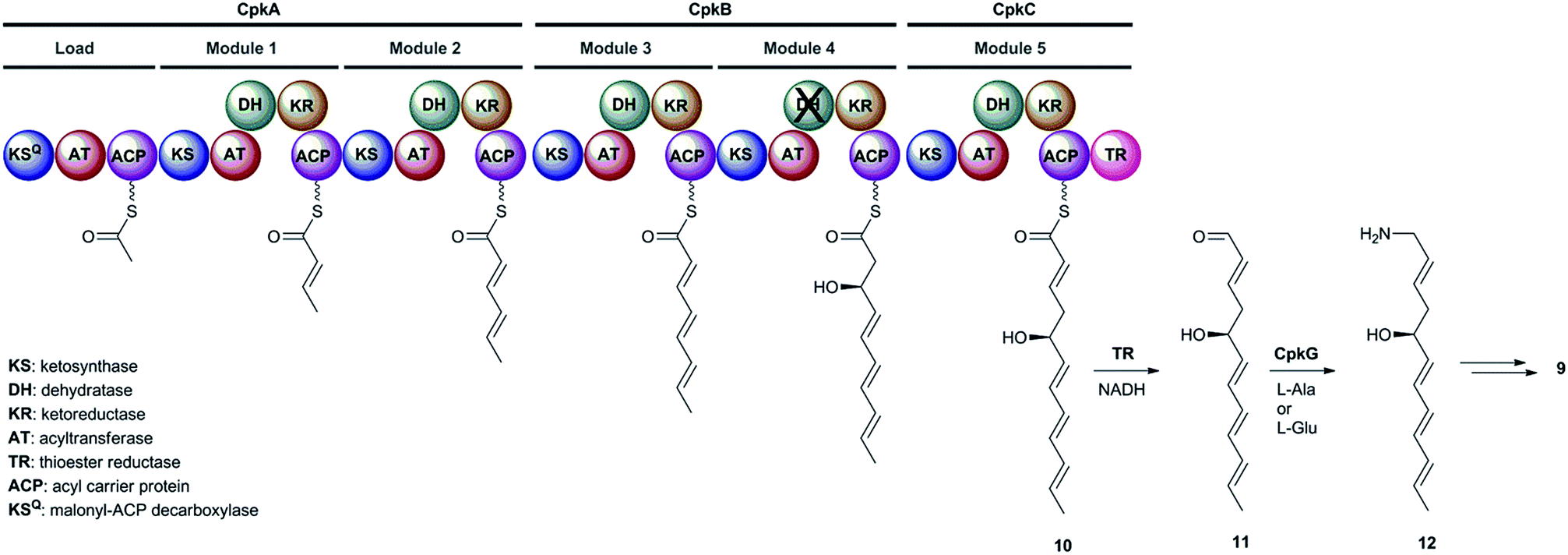 | ||
| Fig. 2 The proposed roles played by the CpkABC modular PKS and the CpkG ω-transaminase in coelimycin P1 9 biosynthesis. | ||
Results and discussion
To experimentally investigate the role played by the CpkC TR domain in coelimycin 9 biosynthesis, it was overproduced in E. coli as an N-terminal His6 fusion protein and purified to homogeneity using nickel affinity chromatography. UHPLC-ESI-Q-TOF-MS analysis confirmed that the purified protein had the expected molecular weight (see ESI†). The catalytic activity of the CpkC TR domain was investigated using octanoyl-CoA 13 as a mimic of the proposed natural substrate 10. NADH and NADPH consumption was examined by monitoring the decrease in absorbance at 340 nm. A substantial decrease in absorbance with time was observed when NADH was used, whereas no decrease in absorbance was observed for NADPH (Fig. 3), showing that octanoyl-CoA is reduced by the former but not the latter. No NADH consumption was observed in control reactions from which the enzyme was omitted (Fig. 3).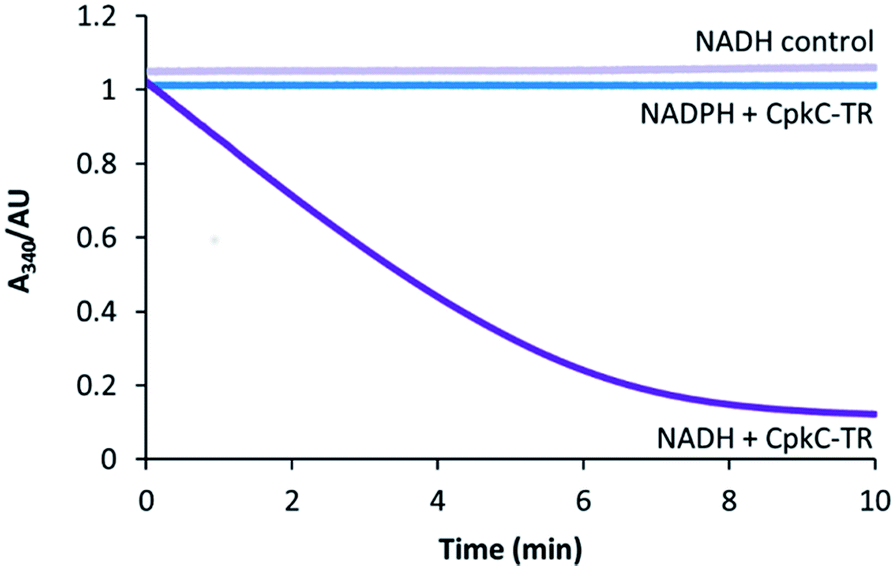 | ||
| Fig. 3 Analysis of NAD(P)H consumption in the CpkC-TR-catalyzed reduction of octanoyl-CoA using a continuous spectrophotometric assay. | ||
In the absence of CpkG, which is proposed to intercept the octanal 14 formed by reduction of octanoyl-CoA 13via transamination, CpkC-TR could catalyze further reduction of 14 to 1-octanol 15 (Scheme 1). Thus, the reaction mixture containing CpkC-TR, octanoyl-CoA 13 and NADH was extracted with chloroform and derivatized with N,O-bis-(trimethylsilyl)acetamide (BSA) to convert any alcohols present to the corresponding trimethylsilyl (TMS) ether derivatives. GC-MS analysis of the derivatized extract identified an analyte, absent from a control reaction containing the boiled enzyme, with m/z = 187.2. This corresponds to the fragment ion of trimethylsilyl-1-octanol 16 resulting from neutral loss of a methyl group and was found to have the same retention time as the m/z = 187.2 ion resulting from GC-MS analysis of BSA-derivatised 1-octanol (Fig. 4). An analogous experiment in which octanal 14 was used in place of octanoyl-CoA 13 gave a similar result (see ESI†). Taken together, the data show that CpkC-TR catalyzes the NADH-mediated reduction of octanoyl-CoA 13 to octanal 14, which is further reduced to 1-octanol 15 in the absence of a suitable transaminase to trap the transient aldehyde intermediate.
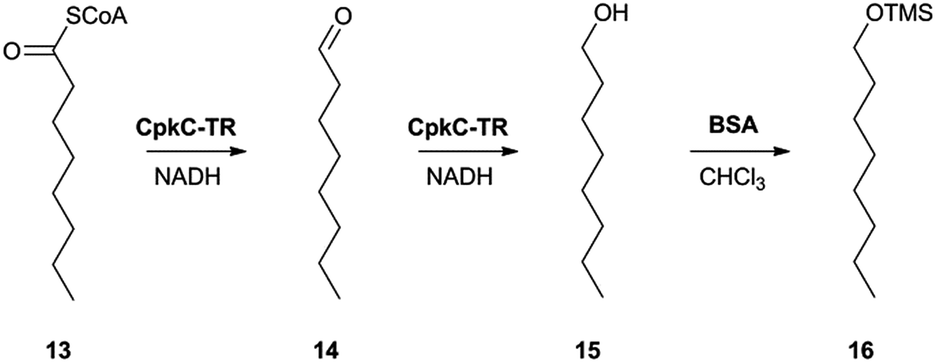 | ||
| Scheme 1 Reactions catalyzed by CpkC-TR using octanoyl-CoA 13 and octanal 14 as substrate analogues and conversion of 1-octanol 15 to its TMS ether 16 to facilitate GC-MS analysis. | ||
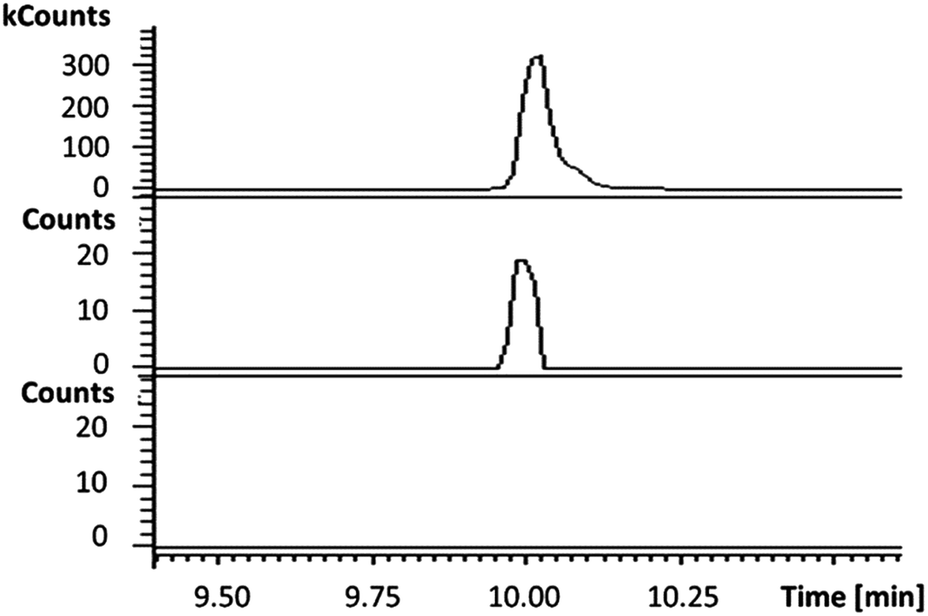 | ||
| Fig. 4 Extracted ion chromatograms at m/z = 187.2 (corresponding to the fragment ion of 16 resulting from loss of a methyl group) from GC-MS analyses of an authentic standard of 16 (top), the BSA-derivatized product of the CpkC-TR-catalyzed reduction of 13 with NADH (middle) and the BSA-derivatized product of a control reaction containing boiled enzyme (bottom). | ||
We next turned our attention to CpkG which shows sequence similarity to pyridoxal-5′-phosphate (PLP)-dependent ω-transaminases. CpkG was overproduced in E. coli and purified as described for CpkC-TR. UHPLC-ESI-Q-TOF-MS analysis confirmed that the purified protein had the molecular weight expected for apo-CpkG and gel filtration chromatography showed that it is a homodimer (see ESI†). To investigate the ability of PLP to bind to CpkG, the UV-vis absorbance spectrum of the enzyme purified in the presence of the cofactor was compared to that of PLP alone. At pH 8, a characteristic λmax shift from 410 nm to 418 nm was observed, indicating that PLP forms an aldimine with CpkG (see ESI†).16 We initially investigated whether holo-CpkG is able to catalyze the conversion of amines 17–21 to the corresponding aldehydes/ketones, along with concomitant conversion of either pyruvate 22 or α-ketoglutarate 23 to alanine 24 or glutamic acid 25, respectively (Scheme 2). A spectrophotometric assay that detects the production of amino acids via formation of a copper(II) complex was used.17 All of the amines tested were found to stimulate conversion of the two α-keto acids to the corresponding amino acids, demonstrating that CpkG has broad substrate tolerance (see ESI†). The highest level of activity was observed for octylamine 21 using pyruvate 22 as the co-substrate, indicating that CpkG prefers medium chain alkylamines, consistent with its proposed role in coelimycin biosynthesis. The production of octanal 15 in this reaction was confirmed by derivatization with D-cysteine and LC-MS comparison with an authentic standard of the resulting thiazolidine (see ESI†).18
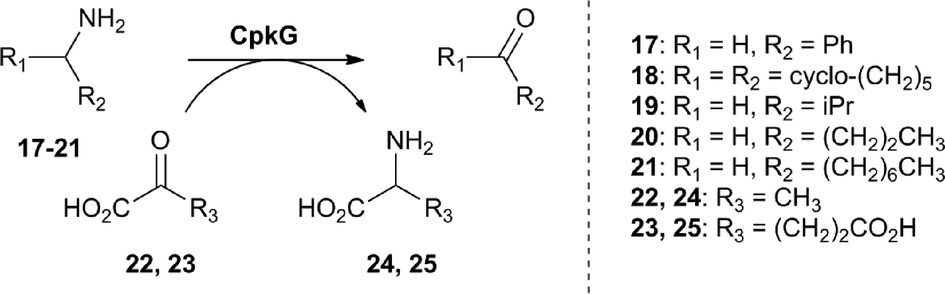 | ||
| Scheme 2 Amines tested as substrates for CpkG, alongside pyruvate and α-ketoglutarate as co-substrates. | ||
The stereochemical outcome of the CpkG-catalyzed reaction of octylamine 21 with pyruvate 22 was investigated by derivatizing the alanine 24 produced with Marfey's reagent.19 LC-MS comparisons with Marfey's derivatives of L- and D-alanine revealed that only L-alanine is formed (see ESI†), suggesting that CpkG has preference for L-amino acid substrates.
Having established that octylamine 21 and pyruvate 22 are good substrates for CpkG in the reverse of the proposed biosynthetic reaction, we investigated reductive amination of octanal 14 using alanine 24 as an amino donor. The pyruvate 22 produced was detected using a coupled assay with lactate dehydrogenase and NADH (see ESI†)20 and octylamine 21 formation was confirmed by LC-MS comparisons with an authentic standard (Fig. 5). Despite the exclusive production of L-alanine in the reverse reaction (see above), CpkG was found to accept both L- and D-alanine as amino donors in the forward reaction.
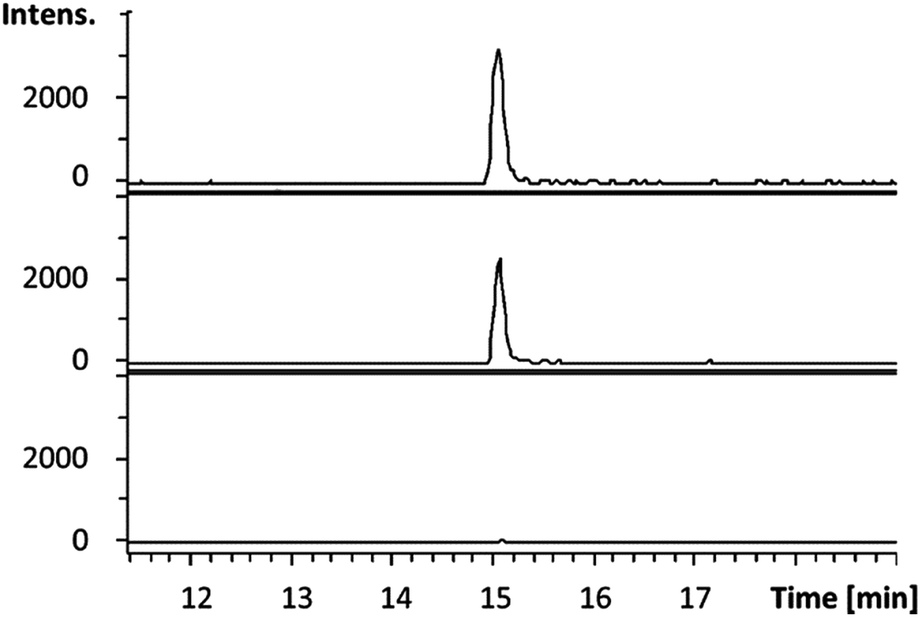 | ||
| Fig. 5 Extracted ion chromatograms at m/z = 130.1590 ± 0.005 (corresponding to the [M + H]+ ion for 21) from UHPLC-ESI-Q-TOF-MS analyses of an authentic standard of 21 (top), the CpkG-catalyzed reaction of 14 with L-alanine (middle) and a control reaction from which the enzyme was omitted (bottom). | ||
Taken together, our data strongly support the key proposed roles played by CpkC-TR and CpkG in diverting the product of a modular PKS into alkaloid metabolism. We thus reasoned that homologous enzymes are likely to be ubiquitous in bacterial polyketide alkaloid biosynthesis. To investigate this hypothesis, we searched the GenBank database for gene clusters containing genes encoding a type I modular PKS with a TR domain and a CpkG homolog within 35 kb of each other (see ESI† for details). Functional annotation of the sequences obtained using antiSMASH resulted in the identification of 22 actinobacterial gene clusters (in addition to the coelimycin cluster) that are hypothesized to direct the biosynthesis of polyketide alkaloids (see ESI†).21 The module and domain organization of the PKSs encoded by these gene clusters were used to predict the structures of the polyketide chains they assemble (assuming a co-linear relationship between the number of modules and the number of precursors utilized for chain assembly). Two of the gene clusters are highly similar to the cpk cluster in S. coelicolor and are predicted to assemble identical polyketide chains to CpkABC.15 It thus seems likely that these clusters direct the biosynthesis of coelimycin P1 9 or a closely related metabolite. Four other clusters produce a polyketide chain that corresponds to the carbon skeletons of nigrifactin 2 and streptazone E 6. These clusters contain a very similar set of genes to those in a gene cluster recently reported to direct the biosynthesis of streptazone E 6 in Streptomyces sp. MSC090213JE08.22 Similarly, one of the clusters assembles a polyketide chain corresponding to the carbon skeleton of cyclizidine 4 and this cluster is essentially identical to a cluster that was recently shown to direct cyclizidine biosynthesis in Streptomyces sp. NCIB11649.23 The polyketide chains assembled by two of the other clusters correspond to the carbon skeleton of pyrindicin 3 and two further clusters are proposed to direct the production of latumcidin 1, streptazolin 5, or a closely related derivative, based on the structure of the carbon chains assembled by the PKSs and the presence of genes encoding homologs of the cyclases recently reported to be responsible for formation of the C4–C8 bond in streptazone E biosynthesis (Fig. 6).22 The polyketide chains produced by the PKSs encoded by the remaining nine clusters do not correspond to the carbon skeletons of any known metabolites. We hypothesize that these clusters direct the biosynthesis of novel polyketide alkaloids.
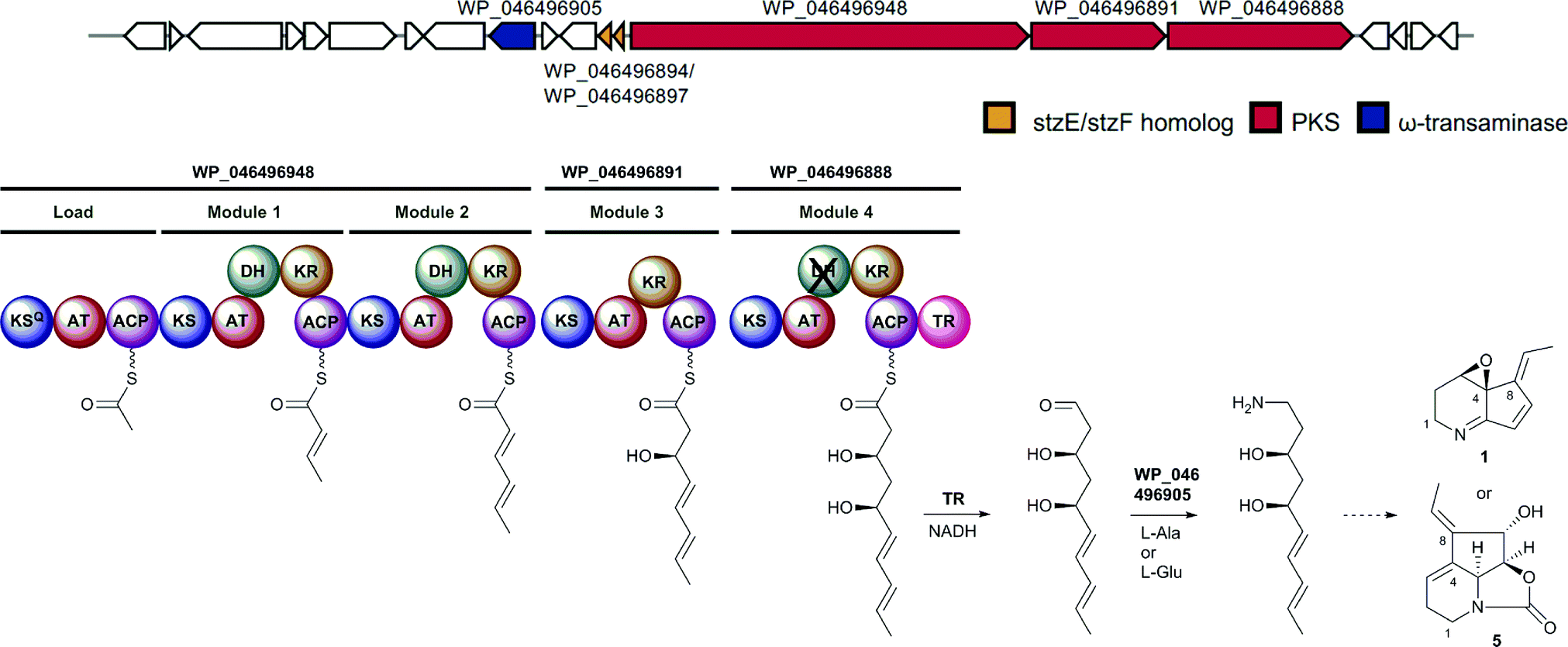 | ||
| Fig. 6 Example of a putative polyketide alkaloid biosynthetic gene cluster identified in Streptomyces sp. NRRL-B24891. Based on the predicted structure of the chain assembled by the PKS and the presence of two genes encoding homologs of the StzE and StzF enzymes, the cluster is hypothesized to direct the biosynthesis of latumcidin 1, streptazolin 5 or a structurally-related metabolite. | ||
Numerous nonribosomal peptide synthetase (NRPS) assembly lines have been reported to employ a TR domain for reductive chain release and, in one case, the initially formed aldehyde has been shown to be intercepted via transamination to form the corresponding amine.4,24–26 TR-catalysed reductive chain release has also been proposed to occur in several fungal PKS systems, although direct biochemical evidence for this is currently lacking.27–31 In contrast, relatively few bacterial modular PKSs have been reported, prior to this work, to contain a TR domain and to the best of our knowledge CpkC-TR is the first of these to be biochemically characterized. In common with a TR (Zmn14) that we recently showed to be responsible for chain release from a polyunsaturated fatty acid synthase-like assembly line in the biosynthesis of the zeamine antibiotics,32 CpkC-TR has a strict preference for NADH. This contrasts with most other TR domains, which are able to utilize NADPH. The three residues that interact with the phosphate group of NADPH in the TR domain from the myxalamid NRPS are completely conserved in CpkC-TR and Zmn14.26 Thus the structural basis for the cofactor preference of CpkC-TR and Zmn14 is unclear.
Conclusions
In conclusion, we have shown that reductive chain release and transamination are the key steps in the conversion of a modular PKS product to the unusual alkaloid coelimycin P1 9. Moreover, a further 22 gene clusters in actinobacteria that are known or predicted to direct the biosynthesis of diverse polyketide alkaloids have been identified. All of these encode a TR-terminated modular PKS and a CpkG homolog, suggesting a common enzymatic logic for actinobacterial polyketide alkaloid biosynthesis.Acknowledgements
This research was supported by a Chancellor's International Scholarship (to U. R. A.), a Chancellor's Scholarship (to J. L. R.), a Marie Sklodowska-Curie Fellowship (to J. M., contract no. 656067), and grants from the BBSRC (BB/K002341/1, BB/L502017/1 and BB/M017982/1).Notes and references
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155 CrossRef CAS PubMed.
- K. J. Weissman, Nat. Prod. Rep., 2015, 32, 436 RSC.
- L. Ray and B. S. Moore, Nat. Prod. Rep., 2016, 33, 150 RSC.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255 RSC.
- L. Vieweg, S. Reichau, R. Schobert, P. F. Leadlay and R. D. Süssmuth, Nat. Prod. Rep., 2014, 31, 1554 RSC.
- D. X. Hu, D. M. Withall, G. L. Challis and R. J. Thomson, Chem. Rev., 2016, 116, 7818–7853 CrossRef CAS PubMed.
- H. Seto, T. Sato, H. Yonehara and W. C. Jankowski, J. Antibiot., 1973, 26, 609 CrossRef CAS PubMed.
- T. Terashima, E. Idaka, Y. Kishi and T. Goto, J. Chem. Soc., Chem. Commun., 1973, 75 RSC.
- Y. Iwai, K. Kumano and S. Omura, Chem. Pharm. Bull., 1978, 26, 736 CrossRef CAS.
- F. J. Leeper, P. Padmanabhan, G. W. Kirby and G. N. Sheldrake, J. Chem. Soc., Chem. Commun., 1987, 505 RSC.
- M. Mayer and R. Thiericke, J. Org. Chem., 1993, 58, 3486 CrossRef CAS.
- Q. Liu, J. Wang, X. Wang, C. Liu, J. Zhang, Y. Pang, C. Yu and W. Xiang, J. Asian Nat. Prod. Res., 2013, 15, 221 CrossRef CAS PubMed.
- S. Grabley, P. Hammann, H. Kluge, J. Wink, P. Kricke and A. Zeeck, J. Antibiot., 1991, 44, 797 CrossRef CAS PubMed.
- S. Gomi, D. Ikeda, H. Nakamura, H. Naganawa, F. Yamashita, K. Hotta, S. Kondo, Y. Okami, H. Umezawa and Y. Iitaka, J. Antibiot., 1984, 37, 1491 CrossRef CAS PubMed.
- J. P. Gomez-Escribano, L. Song, D. J. Fox, V. Yeo, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 2716 RSC.
- R. A. Vacca, S. Giannattasio, G. Capitani, E. Marra and P. Christen, BMC Biochem., 2008, 9, 17 CrossRef PubMed.
- B. Hwang and B. Kim, Enzyme Microb. Technol., 2004, 34, 429 CrossRef CAS.
- H. J. Kim and H. S. Shin, Anal. Chim. Acta, 2011, 702, 225 CrossRef CAS PubMed.
- R. Bhushan and H. Brückner, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3148 CrossRef CAS PubMed.
- M. Sova, G. Čadez, S. Turk, V. Majce, S. Polanc, S. Batson, A. J. Lloyd, D. I. Roper, C. W. G. Fishwick and S. Gobec, Bioorg. Med. Chem. Lett., 2009, 19, 1376 CrossRef CAS PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Müller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237 CrossRef PubMed.
- S. Ohno, Y. Katsuyama, Y. Tajima, M. Izumikawa, M. Takagi, M. Fujie, N. Satoh, K. Shin-Ya and Y. Ohnishi, ChemBioChem, 2015, 16, 2385 CrossRef CAS PubMed.
- W. Huang, S. J. Kim, J. Liu and W. Zhang, Org. Lett., 2015, 17, 5344 CrossRef CAS PubMed.
- Y. Li, K. J. Weissman and R. Müller, J. Am. Chem. Soc., 2008, 130, 7554 CrossRef CAS PubMed.
- A. Chhabra, A. S. Haque, R. K. Pal, A. Goyal, R. Rai, S. Joshi, S. Panjikar, S. Pasha, R. Sankaranarayanan and R. S. Gokhale, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5681 CrossRef CAS PubMed.
- J. F. Barajas, R. M. Phelan, A. J. Schaub, J. T. Kliewer, P. J. Kelly, D. R. Jackson, R. Luo, J. D. Keasling and S. Tsai, Chem. Biol., 2015, 22, 1018 CrossRef CAS PubMed.
- O. Salo, F. Guzman-Chavez, M. I. Ries, P. P. Lankhorst, R. A. L. Bovenberg, R. J. Vreeken and A. J. M. Driessen, Appl. Environ. Microbiol., 2016, 82, 3971 CrossRef CAS PubMed.
- J. Davison, A. al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642 CrossRef CAS PubMed.
- T. Ugai, A. Minami, R. Fuiji, M. Tanaka, H. Oguri, K. Gomi and H. Oikawa, Chem. Commun., 2015, 51, 1878 RSC.
- K. M. Fisch, E. Skellam, D. Ivison, R. J. Cox, A. M. Bailey, C. M. Lazarus and T. J. Simpson, Chem. Commun., 2010, 46, 5331 RSC.
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053 RSC.
- J. Masschelein, C. Clauwers, U. R. Awodi, K. Stalmans, W. Vermaelen, E. Lescrinier, A. Aertsen, C. Michiels, G. L. Challis and R. Lavigne, Chem. Sci., 2015, 6, 923 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional tables, figures, chromatograms and results from biological assays. See DOI: 10.1039/c6sc02803a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |