
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The dual role of thiourea in the thiotrifluoromethylation of alkenes†
Paolo
Ricci‡
a,
Tanatorn
Khotavivattana‡
a,
Lukas
Pfeifer
a,
Maurice
Médebielle
b,
John Richard
Morphy
c and
Véronique
Gouverneur
*a
aChemistry Research Laboratory, Department of Chemistry, Oxford University, OX1 3TA, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk; Fax: +44 (0)1865 285002
bUniversité de Lyon, Université Claude Bernard Lyon I, ICBMS UMR CNRS – UCBL – CPE – INSA 5246, Equipe Synthèse de Molécules d'Intérêt Thérapeutique (SMITh), 43 bd du 11 Novembre 1918, Villeurbanne 69622, France
cMedicinal Chemistry, Eli Lilly and Company Limited, Erl Wood Manor, Sunninghill Road, Windlesham, Surrey GU20 6PH, UK
First published on 30th September 2016
Abstract
Alkenes substituted with a thiourea undergo C–CF3 followed by intramolecular C–S bond formation with the Togni reagent and trifluoroacetic acid (TFA) at room temperature; thiols and thioamides are not suitable S-sources for this reaction. This anti-addition process involves a CF3 radical, and affords CF3-substituted thiazolines and thiazines for medicinal applications. A metal or photoredox catalyst is not required as the thiourea acts as a reductant, as well as serving as an S-source capable of adding to a C-centered radical. Mechanistic work comparing the reactivity of thiourea, urea, thioamide and thiol in the context of alkene trifluoromethylation demonstrates that in this series, the thiourea is unique for its ability to release CF3 radical from the Togni reagent, and to orchestrate trifluoromethylation followed by S-cyclization with both activated and unactivated alkenes.
Introduction
A large number of pharmaceuticals contain a trifluoromethyl group because this structural motif affects the properties of organic molecules.1 The installation of trifluoromethyl groups onto sp3 hybridized carbon has progressed significantly with numerous addition reactions of CF3 across alkenes. Alkene vicinal functionalizations featuring C–CF3 combined with C–H, C–C or C–heteroatom bond formation have been disclosed, most requiring a transition metal or photoredox catalyst to activate the CF3 reagent (Scheme 1a).2 Vicinal difunctionalizations involving sulfur heteroatom are notoriously rare; this process is much more challenging as, in contrast to amines and alcohols, thiols undergo facile S-trifluoromethylation with the Togni or Umemoto reagents in the absence of catalyst.3 A case of alkene thiotrifluoromethylation was reported by Langlois in 2000.4 In this process, photolysis of CF3SO2SPh generates a CF3 radical (CF3˙) that adds to the alkene; this step affords a weakly nucleophilic radical that reacts with CF3SO2SPh to provide the thioether product and the chain propagating trifluoromethylsulfonyl radical. The reagent in this reaction serves both as CF3 and S-source, thereby minimizing S–CF3 bond formation. In a related approach, Zard reported the net addition of S-trifluoromethyl xanthates reagents onto alkenes, a process initiated with lauroyl peroxide.5 The abundance of sulfur containing heterocycles in medicinal chemistry6 prompted us to study alkene difunctionalization via C–CF3 and C–S bond formation where the CF3 and SR groups would not stem from a single reagent. In 2015, Liu and co-workers reported a case of intermolecular difunctionalization with the copper-catalyzed trifluoromethylthiocyanation of alkenes; this process requires trimethylsilylisocyanate, a silicon-based S-source that acts as Lewis acid to activate the Togni reagent.7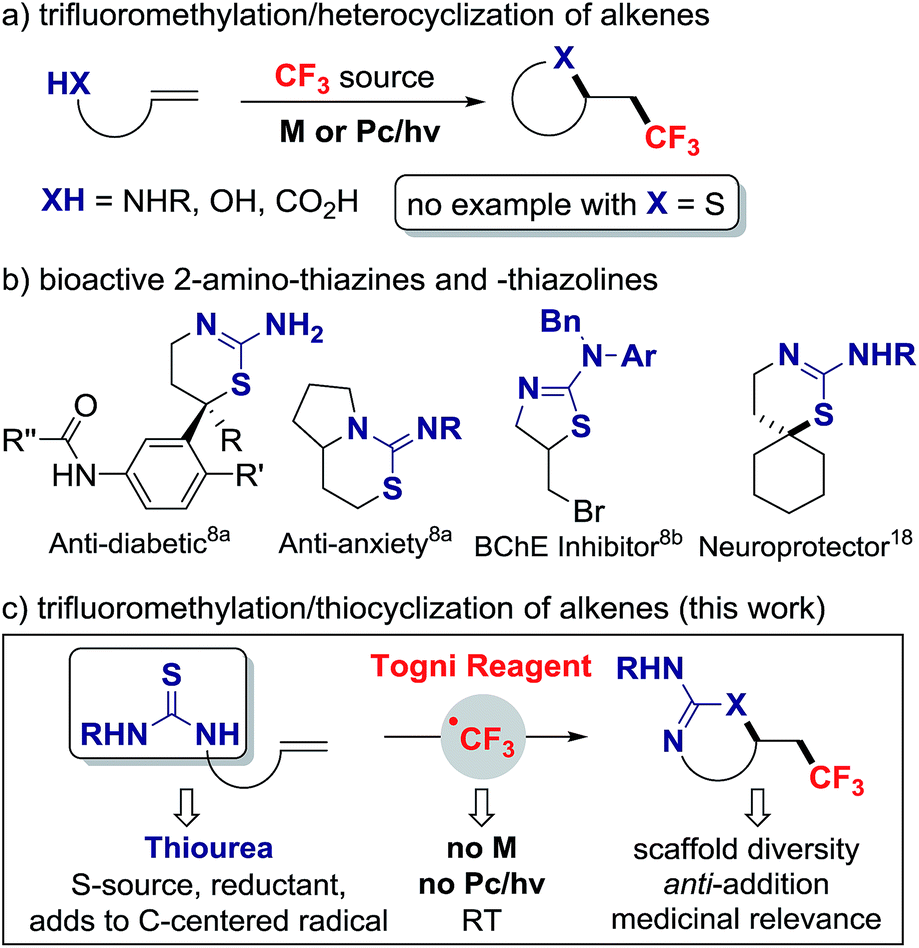 | ||
| Scheme 1 Trifluoromethylation/thiocyclization of alkenes (M = metal, Pc = photoredox catalyst). | ||
In our design plan, we opted to examine the reactivity of olefins with pending thioureas, a decision driven by synthetic and mechanistic considerations. Trifluoromethylation followed by C–S bond formation would afford novel trifluoromethylated 2-amino-thiazolines and 2-amino-thiazines for applications in medicinal chemistry.8 Selected 2-amino-thiazines and -thiazolines are important scaffolds in the development of aspartate beta-secretase enzyme (BACE-1) inhibitors, a therapeutic target for Alzheimer's disease,9 and are common motifs in several bioactive compounds (Scheme 1b). Mechanistically, the ability of thioureas to act as reducing agent10 and radical scavenger11 suggests that this group may induce the release of CF3˙ from the Togni reagent,12 and serve as an S-source capable of adding on a C-centered radical. Here we report that thiourea-substituted alkenes undergo C–CF3 followed by C–S bond formation with the Togni reagent and TFA. This operationally simple reaction does not require a metal catalyst, and affords diverse CF3-substituted 2-amino-thiazolines and thiazines resulting from overall anti-addition across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π bond (Scheme 1c).
C π bond (Scheme 1c).
Results and discussion
To identify suitable reaction conditions, we selected the unactivated alkene 1aa, and the Togni I, II13 and Umemoto III14 reagents as CF3 source (Table 1).15 The desired 2-amino-thiazoline 2aa resulting from trifluoromethylation followed by S-cyclization was formed in low yield when the reaction was carried out at room temperature in CHCl3 with I, II or III in the absence of catalyst or additive (Table 1, entries 1–3). No side-products resulting from oxidative dimerization or S–CF3 bond formation were detected. The conversion of 1aa into 2aa decreased at 60 °C (Table 1, entries 2 and 3).|
|
||||
|---|---|---|---|---|
| Entry | CF3 source | Catalyst | Additive | Yielda (%) |
| a Determined by 19F NMR integration relative to an internal standard (C6H5CF3). b Reaction at 60 °C. c Reaction time is 1 h. d Reaction in CH3CN. e 14 W bulb as light source (λmax = 452 nm). A = Cu(CH3CN)4PF6. B = 1,10-phenantroline. C = Ru(bpy)3(PF6)2. D = methylene blue. TFA = trifluoroacetic acid. | ||||
| 1 | I | — | — | 7 |
| 2 | II | — | — | 33, 21b |
| 3 | III | — | — | 12, 7b |
| 4 | II | — | TFA (2 equiv.) | 76, 62 |
| 5 | II | — | TFA (1 equiv.) | 69 |
| 6 | III | — | TFA (2 equiv.) | 13 |
| 7d | II | — | TFA (2 equiv.) | 59 |
| 8 | II | A (5 mol%) | — | 21 |
| 9 | II | A (100 mol%) | B (1 equiv.) | 33 |
| 10e | II | C (5 mol%) | — | 20 |
| 11e | II | D (2 mol%) | — | 31 |
| 12e | III | C (5 mol%) | — | 40 |
| 13e | III | D (2 mol%) | — | 38 |
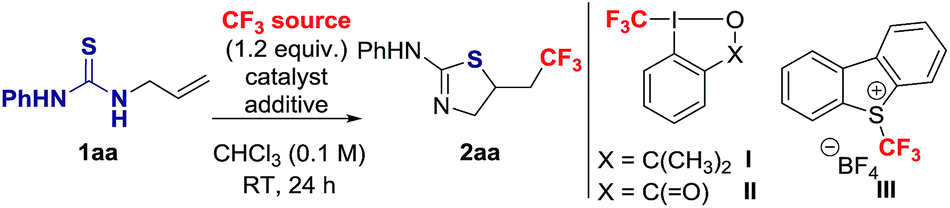
Activation of the Togni reagents by protonation with BrØnsted acid is well documented,16 but not typically considered for CF3 addition onto alkenes. We envisioned that upon protonation of II with trifluoroacetic acid (TFA), the resulting highly electrophilic iodine centre could undergo S–I(III) coordination with the thiourea functionality followed by single electron transfer (SET) with more effective release of CF3 radical. Gratifyingly, 62% of 2aa was observed after 1 h when the reaction was conducted in the presence of 2 equiv. of TFA, and the yield reached 76% after 24 h (Table 1, entry 4). The reaction was less effective using 1 equiv. of TFA (Table 1, entry 5). The presence of the acid did not induce protocyclization, and its benefit was not significant with Umemoto III (Table 1, entry 6).
With the conditions described in entry 4 of Table 1, the scope of the thiotrifluoromethylation was investigated (Scheme 2). Allyl and metallyl thioureas afforded 2-amino-thiazolines 2aa and 2ba in 80% and 96%, respectively. A range of para-substituted styrenes underwent thiotrifluoromethylation with yields up to 92%. The reaction was extended to 1,2-dihydro-naphthalenes, 2H-chromene, 2H-thiochromene and indene; in this series, all thiazolines were formed as a single stereoisomer resulting from anti-addition (d.r. > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).17 The 1,2-dihydro-naphthalene scaffold was selected to investigate the tolerance of the reaction to variation of the thiourea N-substituent. The resulting products anti-2ga–2gl were isolated in yields ranging from 53% to 83%. No reaction occurred with 1gm, a substrate possessing the free NH2 sub-motif. The corresponding 2-amino-thiazoline 2gm was obtained by a detour pathway involving in situ deprotection of the N-tBu group of 2gi under acidic conditions. The thiotrifluoromethylation of the chiral substrate 1-(1-(3,4-dihydronaphthalen-1-yl)propyl)-3-phenylthiourea provided adduct 2gn in moderate yield as a mixture of diastereomers (ratio = 3.5:1).15 Thiazines are also within reach applying this methodology. The spirocyclic product 2ka was obtained in 53% yield and an eroded d.r. = 6:1 favoring the anti-isomer. Styrenes, with different points of attachment for the thiourea, delivered additional trifluoromethylated scaffolds. The 2,2,2-trifluoroethyl-substituted 4H-benzo[d][1,3]-thiazin-2-amines 2la and 2ma were obtained in moderate yields. Products possessing the CF3 group on the thiazine ring itself were accessible from 3-substituted 1-cinnamyl-thioureas; for example, 2na was isolated in 40% yield with a d.r. > 20:1. In this series, substituents on the aryl rings are well tolerated. The reaction with the internal alkyl-substituted alkene, (E)-1-(hex-2-en-1-yl)-3-phenylthiourea delivered a mixture of 5-exo- and 6-endo-regioisomers in a ∼1:1 ratio (isolated yields were 22% and 20%, respectively).15 The spirocyclic thiazine anti-2ra, a CF3-substituted analogue of a neuroprotector,18 was prepared in 60% yield (d.r. > 20:1, after purification). A larger scale reaction on 2.3 mmol provided consistent yield of 2ra (61%), an indicator of the robustness of the process. Thiazine 2rc is a trifluoromethylated analogue of a scaffold found in BACE-1 inhibitors for treating Alzheimer's disease;19 this thiazine was obtained by deprotection with CAN of the N-PMB group of 2rb. Finally, the method also gave access to the CF3-containing thiazepine 2sa.
:1).17 The 1,2-dihydro-naphthalene scaffold was selected to investigate the tolerance of the reaction to variation of the thiourea N-substituent. The resulting products anti-2ga–2gl were isolated in yields ranging from 53% to 83%. No reaction occurred with 1gm, a substrate possessing the free NH2 sub-motif. The corresponding 2-amino-thiazoline 2gm was obtained by a detour pathway involving in situ deprotection of the N-tBu group of 2gi under acidic conditions. The thiotrifluoromethylation of the chiral substrate 1-(1-(3,4-dihydronaphthalen-1-yl)propyl)-3-phenylthiourea provided adduct 2gn in moderate yield as a mixture of diastereomers (ratio = 3.5:1).15 Thiazines are also within reach applying this methodology. The spirocyclic product 2ka was obtained in 53% yield and an eroded d.r. = 6:1 favoring the anti-isomer. Styrenes, with different points of attachment for the thiourea, delivered additional trifluoromethylated scaffolds. The 2,2,2-trifluoroethyl-substituted 4H-benzo[d][1,3]-thiazin-2-amines 2la and 2ma were obtained in moderate yields. Products possessing the CF3 group on the thiazine ring itself were accessible from 3-substituted 1-cinnamyl-thioureas; for example, 2na was isolated in 40% yield with a d.r. > 20:1. In this series, substituents on the aryl rings are well tolerated. The reaction with the internal alkyl-substituted alkene, (E)-1-(hex-2-en-1-yl)-3-phenylthiourea delivered a mixture of 5-exo- and 6-endo-regioisomers in a ∼1:1 ratio (isolated yields were 22% and 20%, respectively).15 The spirocyclic thiazine anti-2ra, a CF3-substituted analogue of a neuroprotector,18 was prepared in 60% yield (d.r. > 20:1, after purification). A larger scale reaction on 2.3 mmol provided consistent yield of 2ra (61%), an indicator of the robustness of the process. Thiazine 2rc is a trifluoromethylated analogue of a scaffold found in BACE-1 inhibitors for treating Alzheimer's disease;19 this thiazine was obtained by deprotection with CAN of the N-PMB group of 2rb. Finally, the method also gave access to the CF3-containing thiazepine 2sa.
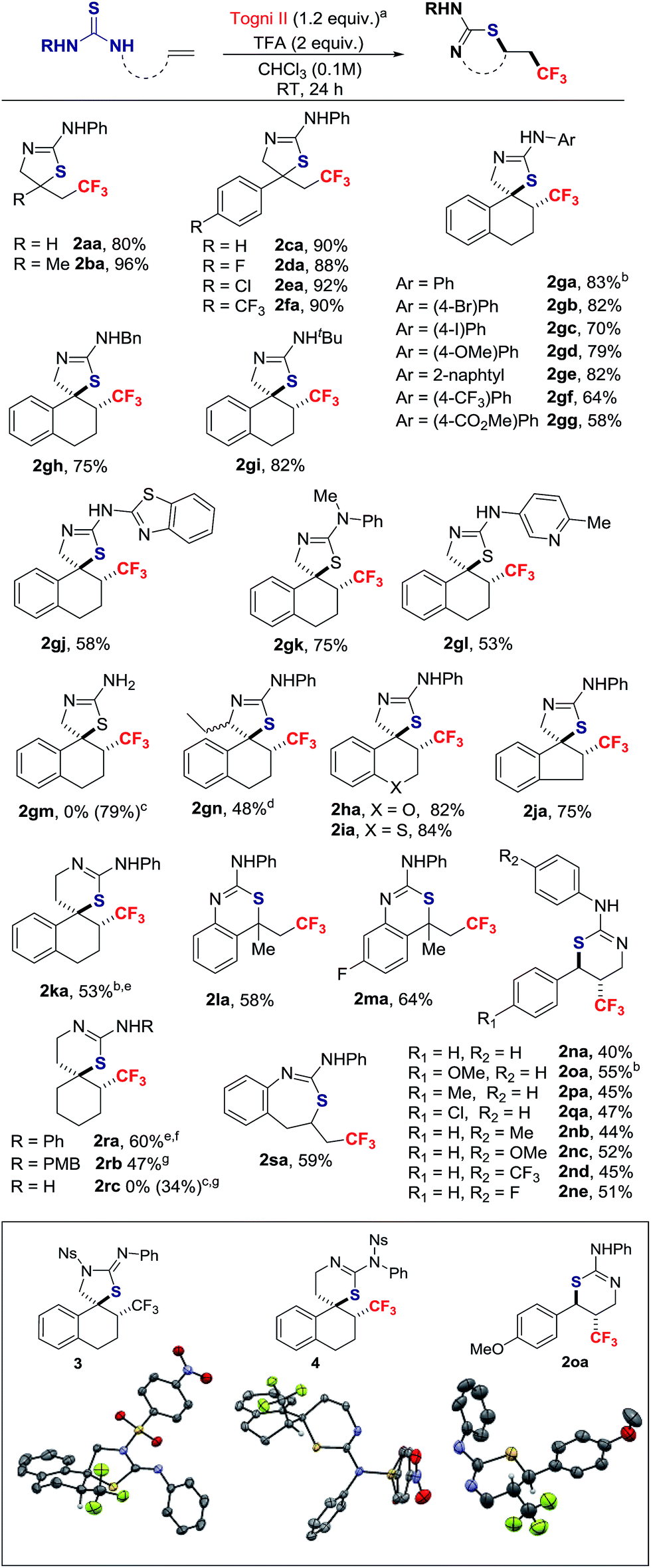 | ||
| Scheme 2 Substrate scope of the reaction. aThe reaction was performed on a 0.3 mmol scale; yield of isolated product; d.r. > 20:1 by 19F NMR of crude reaction. bRelative configuration established by single crystal X-ray diffraction analysis; for 2ga and 2ka, analysis was performed on the derivatives 3 and 4, respectively. c2gm and 2rc were obtained by in situ deprotection of 2gi and 2rb, respectively; yields from the alkene. dd.r. = 3.5:1. ed.r. = 6:1. f61% yield when the reaction was scaled up to 2.3 mmol. gd.r. = 5:1. PMB = para-methoxybenzyl. | ||
Mechanistic experiments
We probed the mechanism of this reaction with a series of experiments (Scheme 3). The presence of 1 equiv. of TEMPO significantly inhibited the thiotrifluoromethylation of 1aa, yielding 23% of TEMPO-CF3 and 6% of 2aa.15 Complete inhibition for the formation of 2aa was observed in the presence of benzoquinone. The cyclopentane 5 was isolated in 20% yield when diethyl 2,2-diallylmalonate was submitted to the reaction conditions in the presence of 1 equiv. of N,N-diphenylthiourea (DPTU);20 in the absence of thiourea, no reaction occurred (eqn (1)). Both E-1na and Z-1na gave anti-2na with d.r. > 20:1 (eqn (2)). Collectively, these data indicate that a CF3 radical is involved in the reaction.
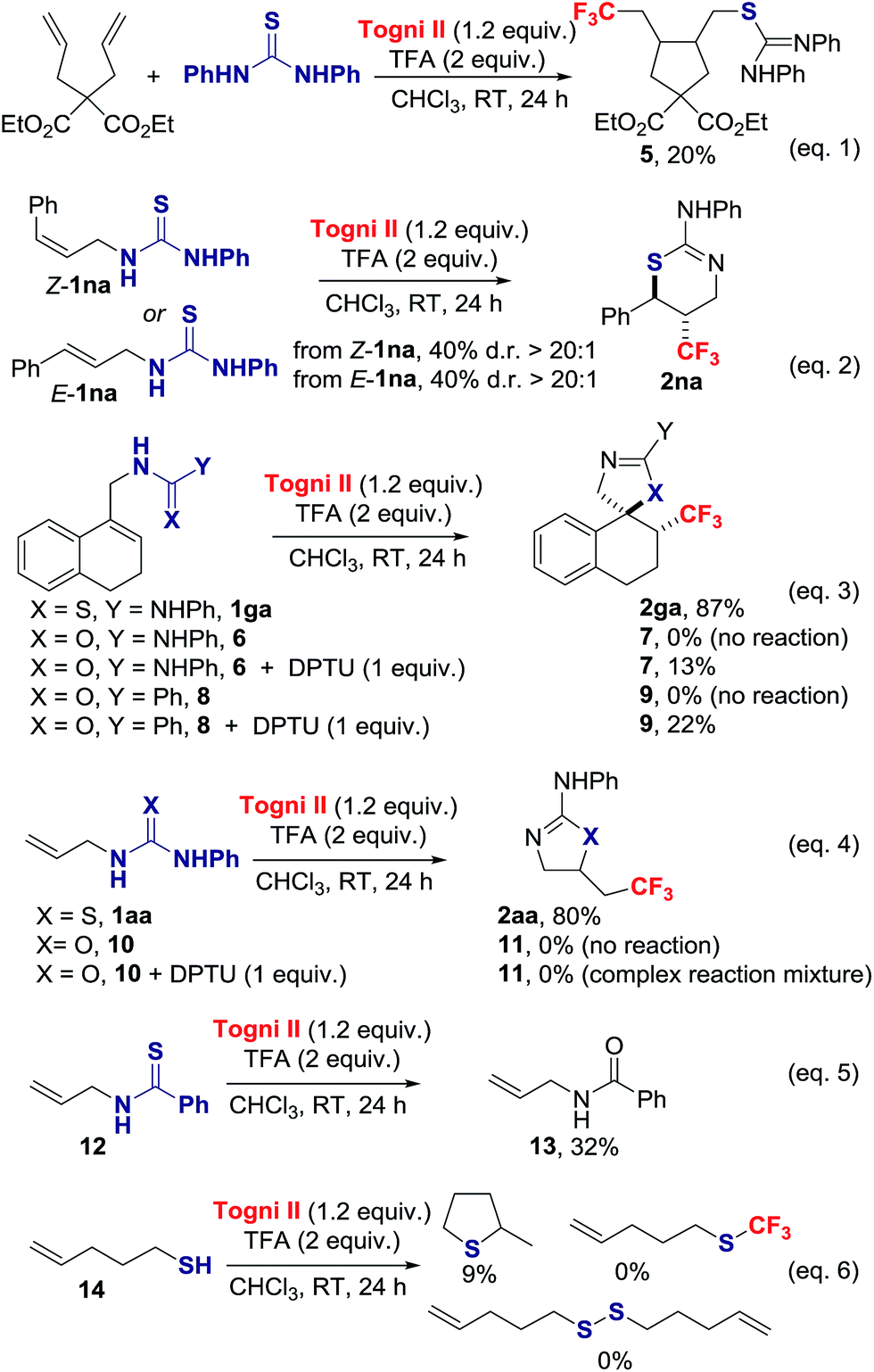 | ||
| Scheme 3 Mechanistic experiments. | ||
Next, we investigated the uniqueness of the thiourea functionality for its ability to induce CF3˙ formation. We compared the reactivity of the thiourea 1ga with the corresponding urea 6 and amide 8 (eqn (3)). We found that 6 and 8 did not react under the standard reaction conditions. Notably, the cyclized products 7 and 9 were isolated in 13% and 22% yield respectively, when the trifluoromethylation was performed in the presence of 1 equiv. of DPTU. In a similar vein, 1-allyl-3-phenylurea 10 did not react under the standard reaction conditions, but was consumed in the presence of DPTU with evidence that CF3 radical addition to the alkene took place, but cyclization to 11 did not occur (eqn (4)).15 The thiourea therefore acts as an activator leading to CF3˙ formation, and subsequent addition of this radical on the CC π bond. The contrasting reactivity of thiourea and urea is consistent with their oxidation potentials (+1.19 V vs. SCE in CH3CN for thiourea 1aa and +1.56 V vs. SCE in CH3CN for urea 10); similar values were found for cyclic voltammetry measurements performed in CH3CN in the presence of TFA.15 Moreover, thioureas are superior to ureas for their ability to react with radical acceptor, an additional factor that accounts for the observed difference of reactivity. We considered next thioamides and thiols as alternative S-sources. Under our standard reaction conditions, the thioamide 12 failed to provide the product of thiotrifluoromethylation, but led instead to the corresponding amide 13 (eqn (5)).15,21 Pent-4-ene-1-thiol 14 underwent intramolecular thiol–ene ring closure and side reactions other than S–CF3 bond formation or oxidative S–S dimerization (eqn (6)).15,22 The thiourea is therefore unique to enable orchestrated alkene trifluoromethylation followed by S-cyclization.
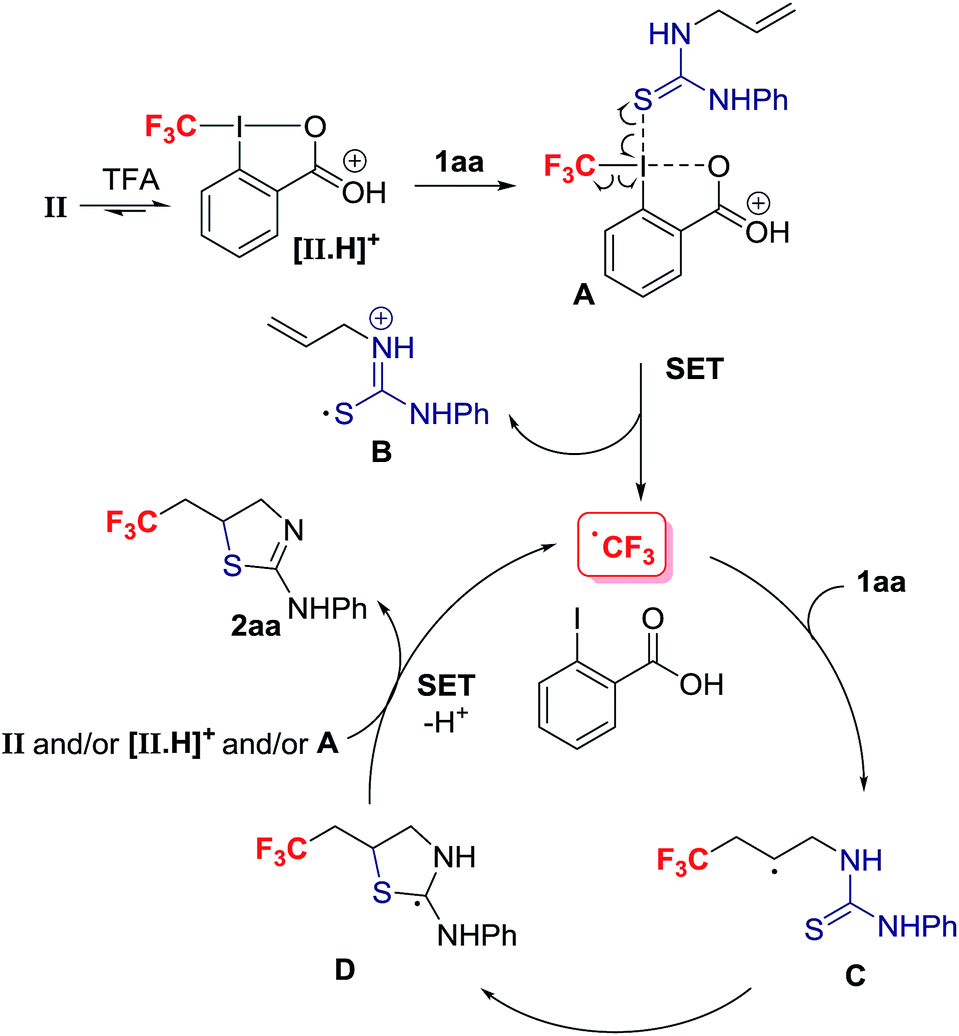
Mechanistically, we discarded the possibility of S-cyclization prior to trifluoromethylation because this sequence would convert alkenes such as 1na into a thiazoline via 5-exo-trig cyclization, and the thiazine anti-2na is the only product observed in the crude reaction mixture (eqn (2)).23 We propose that activation of the Togni reagent II with TFA affords the highly electrophilic iodine(III) species [II.H]+ that can associate with 1aavia iodine–sulphur coordination leading to A. Coordination of thiourea to the highly electrophilic I(III) in [II.H]+ is unprecedented, but S–I(III) coordination has been evoked in the S–CF3 bond formation for thiols reacting with the Togni reagent.24 Homolytic dissociation releases B, iodobenzoic acid and the electrophilic radical CF3˙, which is suited to add regioselectively to the alkene substrate 1aa. The alternative dissociative electron transfer pathway towards CF3 radical formation is also plausible. The resultant carbon radical C undergoes ring closure with C–S bond formation to provide adduct D, which should be easier to oxidize than C; SET to the Togni reagent II, [II.H]+ and/or A affords after proton transfer 2aa, and CF3˙ that starts a new reaction cycle.25 For radicals arising from CF3˙ addition to aryl-substituted alkenes, oxidation prior to S-cyclization is viable (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we developed the first trifluoromethylation followed by S-cyclization across CC π bonds using thiourea as the S-source. The substrate itself, through its thiourea functionality, acts as an initiator, thereby avoiding metal species or light/photoredox catalysts to induce facile formation of the CF3 radical that adds to the alkene. Thiourea can react with C-centered radical, so a range of alkenes including unactivated systems underwent facile thio-trifluoromethylation. This reaction is an attractive method for medicinal and other applications, because of its broad substrate scope, anti-selectivity and operational simplicity. The discovery that N,N-diphenylthiourea is an effective additive to induce the trifluoromethylation-cyclization of ureas and benzamides opens the possibility to investigate the value of this category of activators for the development of novel metal-free trifluoromethylation across double bonds.
Acknowledgements
The authors thank Eli Lilly (P. R.), the Royal Thai Government and SCI (T. K.), and the EU (FP7-PEOPLE-2012-ITN-RADIOMI-316882 to L. P.) for generous funding. V. G. thanks the Royal Society for a Wolfson Research Merit Award, and Prof. S. Zard (Ecole Polytechnique, France) and Prof. J. Burton (University of Oxford, UK) for very helpful discussions.Notes and references
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- For a recent review, see: E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598. For metal-catalysed and photochemical trifluoromethylation across alkenes, see: (a) A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120 ( Angew. Chem. , 2011 , 123 , 9286 ) CrossRef CAS PubMed; (b) J. Xu, Y. Fu, Y. D. Luo, Y. Jiang, B. Xiao, Z. Liu, T. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 15300 CrossRef CAS PubMed; (c) X. Wang, Y. Ye, S. Zhang, J. Feng, Y. Xu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 16410 CrossRef CAS PubMed; (d) R. Shimizu, H. Egami, Y. Hamashima and M. Sodeoka, Angew. Chem., Int. Ed., 2012, 51, 4577 ( Angew. Chem. , 2012 , 124 , 4655 ) CrossRef CAS PubMed; (e) S. Mizuta, O. Galicia-López, K. M. Engle, S. Verhoog, K. Wheelhouse, G. Rassias and V. Gouverneur, Chem.–Eur. J., 2012, 18, 8583 CrossRef CAS PubMed; (f) P. G. Janson, I. Ghoneim, N. O. Iichenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef CAS PubMed; (g) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 ( Angew. Chem. , 2012 , 124 , 9705 ) CrossRef CAS PubMed; (h) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462 CrossRef CAS PubMed; (i) R. Zhu and S. L. Buckwald, Angew. Chem., Int. Ed., 2013, 52, 12655 ( Angew. Chem. , 2013 , 125 , 12887 ) CrossRef CAS PubMed; (j) C. Feng and T.-P. Loh, Chem. Sci., 2012, 3, 3458 RSC; (k) X. Wu, L. Chu and F. Qing, Angew. Chem., Int. Ed., 2013, 52, 2198 ( Angew. Chem. , 2013 , 125 , 2254 ) CrossRef CAS PubMed; (l) H. Egami, S. Kawamura, A. Miyazaki and M. Sodeoka, Angew. Chem., Int. Ed., 2013, 52, 7841 ( Angew. Chem. , 2013 , 125 , 7995 ) CrossRef CAS PubMed; (m) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2013, 52, 12414 ( Angew. Chem. , 2013 , 125 , 12640 ) CrossRef CAS PubMed; (n) N. O. Ilchenko, P. G. Janson and K. J. Szabó, Chem. Commun., 2013, 49, 6614 RSC; (o) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheeelhouse, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505 CrossRef CAS PubMed; (p) X. Wu, L. Chu and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 2198 ( Angew. Chem. , 2013 , 125 , 2254 ) CrossRef CAS PubMed. For metal-free trifluoromethylation across alkenes, see: (q) Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221 ( Angew. Chem. , 2012 , 124 , 8345 ) CrossRef CAS PubMed; (r) B. Zhang, C. Muck-Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792 ( Angew. Chem. , 2013 , 125 , 10992 ) CrossRef CAS PubMed; (s) D. J. Wilger, N. J. Gesmundo and D. A. Nicewicz, Chem. Sci., 2013, 4, 3160 RSC; (t) W. Kong, M. Casimiro, N. Fuentes, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2013, 52, 13086 ( Angew. Chem. , 2013 , 125 , 13324 ) CrossRef CAS PubMed; (u) H. Egami, Y. Usui, S. Kawamura, S. Nagashima and M. Sodeoka, Chem.–Asian J., 2015, 10, 2190 CrossRef CAS PubMed; (v) N.-Y. Yang, Z.-L. Li, L. Ye, B. Tan and X.-Y. Liu, Chem. Commun., 2016, 52, 9052 RSC.
- X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed.
- T. Billard, R. Roques and B. R. Langlois, Tetrahedron Lett., 2000, 41, 3069 CrossRef CAS . For a trifluoromethylchlorosulfonylation, see: D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. N. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 ( Angew. Chem. , 2015 , 127 , 7105 ) CrossRef PubMed.
- F. Bertrand, V. Pevere, B. Quiclet-Sire and S. Zard, Org. Lett., 2001, 3, 1069 CrossRef CAS PubMed.
- For a review, see: M. Feng, B. Tang, S. H. Tang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed.
- Z. Liang, F. Wang, P. Cheng and G. Liu, Org. Lett., 2015, 17, 2438 CrossRef CAS PubMed.
- (a) M. Asif, Journal of Pharmaceutical and Applied Chemistry, 2015, 1, 49 Search PubMed; (b) G. Makhaeva, N. Boltneva, S. V. Lushchekina, O. G. Serebryakova, T. S. Stupina, A. A. Terentiev, I. V. Serkov, A. N. Proshin, O. G. Bachurin and R. J. Richardson, Bioorg. Med. Chem., 2016, 24, 1050 CrossRef CAS PubMed.
- (a) A. K. Ghosh and H. L. Osswald, Chem. Soc. Rev., 2014, 43, 6765 RSC; (b) R. Vassar, Alzheimer's Res. Ther., 2014, 6, 89 CrossRef PubMed; (c) L. L. Winneroski, M. A. Schiffler, J. A. Erickson, P. C. May, S. A. Monk, D. E. Timm, J. E. Audia, J. P. Beck, L. N. Boggs, A. R. Borders, R. D. Boyer, R. A. Brier, K. J. Hudziak, V. J. Klimkowski, G. P. Losada, B. M. Mathes, S. L. Stout, B. M. Watson and D. J. Mergott, Bioorg. Med. Chem., 2015, 23, 3260 CrossRef CAS PubMed.
- The ionization potential of urea and thiourea is 10.27 eV and 8.5 eV respectively. M. Baldwin, A. Kirkien-Konasiewicz, A. G. Loudon, A. Maccoll and D. Smith, Chem. Commun., 1966, 574 RSC.
- (a) M. Wasil, B. Halliwell, M. Grootveld, C. P. Moorhouse, D. C. Hutchison and H. Baum, Biochem. J., 1987, 243, 867 CrossRef CAS PubMed; (b) M. Whiteman and B. Halliwell, Free Radical Biol. Med., 1997, 22, 1309 CrossRef CAS; (c) M. C. Araujo, L. M. Antunes and C. S. Takahashi, Teratog., Carcinog. Mutagen., 2001, 21, 175 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed.
- P. Eisenberger, S. Gishig and A. Togni, Chem.–Eur. J., 2006, 12, 2579 CrossRef CAS PubMed.
- (a) T. Umemoto and S. Ishihara, Tetrahedron Lett., 1990, 31, 3579 CrossRef CAS; (b) T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156 CrossRef CAS; (c) T. Umemoto, Chem. Rev., 1996, 96, 1757 CrossRef CAS PubMed.
- For details, see the ESI.†.
- (a) R. Koller, Q. Huchet, P. Battaglia, J. M. Welch and A. Togni, Chem. Commun., 2009, 5993 RSC; (b) J. N. Brantley, A. V. Samant and F. D. Toste, ACS Cent. Sci., 2016, 2, 341 CrossRef CAS PubMed.
- N. Noto, K. Miyazawa, T. Koike and M. Akita, Org. Lett., 2015, 17, 3710 CrossRef CAS PubMed.
- S. V. Blokhina, T. V. Volkova, M. V. Ol'khovich, A. V. Sharapova, A. N. Proshin, S. O. Bachurin and G. L. Perlovich, Eur. J. Med. Chem., 2014, 77, 8 CrossRef CAS PubMed.
- A. E. Minatti, J. D. Low, J. R. Allen, J. Chen, Y. Cheng, T. Judd, Q. Liu, P. Lopez, W. Quia, R. Rumfelt, N. Rzasa, Q. X. Tamayo, B. Yang and W. Zhong, WO2013142613A1, 2014.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 ( Angew. Chem. , 2012 , 124 , 9082 ) CrossRef CAS PubMed.
- In the absence of TFA, thioamide 10 undergoes oxidative dimerization. I(III)-Mediated conversion of thioamide into amide is known N. K. Downer-Riley and Y. A. Jackson, Tetrahedron, 2008, 64, 7741 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 ( Angew. Chem. , 2010 , 122 , 1584 ) CrossRef CAS PubMed.
- P. D. Morse and D. A. Nicewicz, Chem. Sci., 2015, 6, 270 RSC.
- O. Sala, N. Santschi, S. Jungen, H. P. Lüthi, M. Iannuzzi, N. Hauser and A. Togni, Chem.–Eur. J., 2016, 22, 1704 CrossRef CAS PubMed.
- The oxidation of D with B regenerates 1aa.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1474239–1474241. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02790c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |