
Avoiding leaching of silica supported metallocenes in slurry phase ethylene homopolymerization
Muhammad Ahsan
Bashir
 ab,
Vincent
Monteil
a,
Christophe
Boisson
a and
Timothy F. L.
McKenna
*a
ab,
Vincent
Monteil
a,
Christophe
Boisson
a and
Timothy F. L.
McKenna
*a
aLaboratoire de Chimie Catalyse Polymères et Procédés (C2P2), LCPP team, Univ. Lyon 1, CPE Lyon, CNRS, UMR 5265, Université de Lyon, Bat 308F, 43 Bd du 11 novembre 1918, F-69616 Villeurbanne, France. E-mail: Timothy.mckenna@univ-lyon1.fr
bDutch Polymer Institute DPI, P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 18th May 2017
Abstract
Leaching of (n-BuCp)2ZrCl2/MAO catalyst from silica supports has been studied in slurry phase ethylene homopolymerisation by changing i) the polymerization protocol, ii) aluminum alkyl scavengers and iii) by adding butylated hydroxytoluene (BHT-H) along with aluminum alkyl scavengers. The impact of these three variations on the ethylene polymerization kinetics of the said metallocene/MAO/silica catalyst is also analyzed. The obtained results show that (n-BuCp)2ZrCl2/MAO does not leach from the silica support if pre-contact between the heterogeneous catalyst and aluminum alkyl scavenger is avoided. Furthermore, the addition of equimolar amounts of BHT-H and triisobutylaluminum (TIBA) into the reaction milieu generates Al(i-Bu)BHT2 which appears to be a non-interacting scavenger leading to no catalyst leaching (and therefore, no reactor fouling). This observation has been made by pre-contacting and avoiding the pre-contact between the silica supported (n-BuCp)2ZrCl2/MAO catalyst and the reaction mixture containing equimolar amounts of BHT-H and TIBA followed by the analysis of reactor conditions as well as the obtained polyethylene morphology. Molecular and physical properties of HDPE remain very similar by changing the polymerization protocol or the aluminum alkyl scavenger or by using BHT-H.
Introduction
Metallocene catalysts, which are generally composed of a transition metal sandwiched between two cyclopentadienyl or cyclopentadienyl-derivative rings, represent a versatile group of organometallic catalysts suitable for olefin(s) polymerization. In comparison to polyolefins produced by conventional Ziegler–Natta or Phillips catalysts, polyolefins from metallocene catalysts show a narrower molar mass distribution with a polydispersity index close to 2, more precisely defined microstructures due to controlled comonomer incorporation that is independent of molar mass, and comparable mechanical properties and clarity, all of which make metallocenes industrially attractive catalysts.1–5 This statement can be better understood if one considers that about 5 million tons of LLDPE produced worldwide in 2009 were based on single-site catalysts which are often metallocenes.6Metallocenes can be used to polymerize olefins in both homogenous or heterogeneous forms (i.e., supported on a carrier such as silica). The major benefits of using supported metallocenes are that they can be dropped into existing commercial reactors without significant process changes, they require lower amounts of co-catalysts than Ziegler–Natta catalysts, and, with respect to solution processes, they reduce the chance of reactor fouling and offer greater ease of handling.7 Most commonly used methods for supporting metallocene on a carrier are related to the absorption of the molecular catalyst during its activation via the formation of an ion pair.8 In conjunction with the type of the support and how it is conditioned, the method chosen for supporting the catalyst precursors can significantly affect the activity of heterogeneous metallocenes in olefin polymerization.8
Despite the various benefits obtained by supported metallocenes, reactor fouling (i.e., the formation of polyolefin film around the reactor walls and/or stirrer) can still be a major problem in slurry phase polymerizations. Desorption, or leaching, of the metallocene molecule, co-catalyst and/or metallocene/co-catalyst active species from the support can initiate homogeneous polymerization in the diluent, thereby causing reactor fouling.1,7–13 Different methods proposed in the open literature to overcome this problem of leaching include attempts to fix the metallocene covalently with the support via functional group of the ansa or cyclic ligand,14–16 and the consecutive-combined method where the ligands of desired metallocene are first chemically bonded to the silica support and then the transition metal is inserted to create the metallocene complex on the silica surface.17–19 However, these methods do not improve the yields of the supported metallocenes drastically, but do require additional steps during the synthesis procedure which makes them economically less attractive.17 Therefore, most academic and industrial research typically employs a technique such as the incipient wetness method (see below).6
Regardless of how the supporting procedure is carried out, the major cause of catalyst leaching from the support is the interaction of supported MAO (the main focus of the current discussion) and metallocenes with commonly used aluminum alkyls (e.g., triethyl aluminum (TEA), tri isobutylaluminum (TIBA) etc.), which are added to the reactor diluent either as co-catalysts or scavengers. The work of Sacchi et al.20 compares the impact of MAO and TIBA on the leaching of silica-supported metallocenes (i.e., Cp2ZrCl2, (Ind)2ZrCl2, rac-Et(Ind)2ZrCl2) which were prepared by either supporting the metallocene directly on the silica support, or by first impregnating the silica with MAO to prepare SMAO and then supporting the metallocene on SMAO. Their data suggests that in case of pre-contacting the SMAO-supported zirconocene with an MAO solution in toluene, about 18 wt% of the originally supported zirconocene was leached into the solution, whereas only 1.2 wt% of it was lost when the same catalyst was pre-contacted with a TIBA solution. Similarly, the work of Semikolenova et al.,21 also showed that interaction of MAO or TIBA with the silica-supported metallocenes (prepared by using different synthesis methods) led to catalyst leaching under polymerization conditions. About 50 to 60 wt% of their supported metallocene was found to leach when silica/MAO/Cp2ZrMe2 or silica/TEA/Cp2ZrMe2 catalysts were contacted with MAO solution at 70 °C for 30 minutes, whereas pre-contacting with TIBA at the same conditions caused 20 wt% loss in the zirconocene content of the same catalysts. Ernst et al.,22 reported 15 to 24 mole% desorption of the zirconocene from silica/MAO/Me2Si(2-Me-Ind)2ZrCl2 and silica/MAO/Me2Si(2-Me-4-Ph-Ind)2ZrCl2 catalysts when contacted with TIBA. In another study, Panchenko et al.,10 showed that contacting heptane solution of TIBA with SMAO-supported rac-Me2Si(Ind)2ZrCl2 can leach 15 to 40 wt% of Al and 50 to 60 wt% of Zr from the supported metallocene which leads to both homogeneous and heterogeneous polymerizations and bimodality in the molar mass distribution of the polyethylene produced via slurry phase reaction. Furthermore, it is also proposed that the interaction of TIBA with the silica surface bound MAO modifies the MAO in such a way that it becomes more soluble in aliphatic hydrocarbons and behaves like modified MAO (MMAO). It is important to mention here that these studies focused on showing that significant leaching occurs regardless of the method of catalyst synthesis employed; they do not explore the impact of polymerization protocol on the catalyst leaching or the ways to reduce it. In fact, there appears to be very few, if any such studies in the open literature.
Other commonly used scavengers in olefin polymerizations include compounds which are product of the reaction between aluminum alkyls and simple phenols or substituted phenols.23–32 These scavengers have been used mainly in homogeneous olefin polymerizations and termed as ‘non-interacting’ scrubbing agents. However, to the best of our knowledge these compounds have not been tested in slurry phase polymerizations employing silica-supported metallocene/MAO systems. Therefore, the question remains as to whether catalyst leaching and consequently reactor fouling, occurs with these scavengers (based on the reaction products of aluminum alkyls and simple phenols or substituted phenols) in aliphatic diluents commonly used in commercial processes.
In general, it is believed that either the use of diluents such as toluene in which co-catalysts and metallocenes have high solubility, or high Al/Zr molar ratios (greater than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of the supported catalysts are the principle reasons for catalyst leaching in aliphatic diluents. It appears that at lower Al/Zr ratios (on the order of 50–200:1), the metallocene/MAO catalysts are insoluble in common commercial aliphatic hydrocarbons.33 Therefore, we aim to determine whether or not leaching of silica-supported metallocene/MAO catalysts with Al/Zr molar ratios close to 150 occurs in slurry phase ethylene polymerizations conducted in heptane. We considered different polymerization protocols and types of aluminum alkyl used as scavenger. Furthermore, the impact of the scavengers based on the reaction between common aluminum alkyls and the substituted phenol 2,6-Di-tert-butyl-4-methylphenol (commonly known as butylated hydroxytoluene; BHT-H) on catalyst leaching and reactor fouling is also analyzed.
:1) of the supported catalysts are the principle reasons for catalyst leaching in aliphatic diluents. It appears that at lower Al/Zr ratios (on the order of 50–200:1), the metallocene/MAO catalysts are insoluble in common commercial aliphatic hydrocarbons.33 Therefore, we aim to determine whether or not leaching of silica-supported metallocene/MAO catalysts with Al/Zr molar ratios close to 150 occurs in slurry phase ethylene polymerizations conducted in heptane. We considered different polymerization protocols and types of aluminum alkyl used as scavenger. Furthermore, the impact of the scavengers based on the reaction between common aluminum alkyls and the substituted phenol 2,6-Di-tert-butyl-4-methylphenol (commonly known as butylated hydroxytoluene; BHT-H) on catalyst leaching and reactor fouling is also analyzed.
Experimental section
Materials
Two commercial silica have been used in this study as catalyst support. The first one is Grace 948 silica with a surface area of 290 m2 g−1, average particle diameter of 59 μm and a pore volume of 1.7 mL g−1. The second is PQMS 3040 silica with a surface area of 420 m2 g−1, average particle diameter of 45 μm and a pore volume of 3.0 mL g−1. 30 wt% methylaluminoxane (MAO) solution (5.24 wt% TMA) in toluene was obtained from Albermarle, and the (n-BuCp)2ZrCl2 precatalyst from Sigma-Aldrich, and both were used as received. Triethyl aluminum (TEA), triisobutyl aluminum (TIBA) and trioctyl aluminum were used as received from SGS and Witco Corporation, respectively. 2,6-Di-tert-butyl-4-methylphenol purchased from Sigma Aldrich was sublimed at room temperature and then dissolved in dry heptane to prepare 1 M stock solution. Dried heptane was used as diluent for slurry polymerizations. Ethylene (purity 99.95%) was purchased from Air Liquide (Paris, France) and passed through three different purification columns before use: a first one filled with reduced BASF R3-16 catalyst (CuO on alumina), a second one filled with molecular sieves (13X, 3A, Sigma-Aldrich), and a final one filled with Selexsorb COS (Alcoa).Supported catalyst synthesis
The incipient wetness8 method was used to prepare all the catalysts used in this work unless otherwise mentioned. Each silica was dehydroxylated at 600 °C under dynamic vacuum of 10−3 to 10−5 mbar prior to its impregnation with the metallocene + MAO mixture. In the first step, a weighed amount of (n-BuCp)2ZrCl2 was mixed with dry toluene and MAO (30 wt% toluene solution) and left for 1 hour at room temperature inside the glove box. The amounts of (n-BuCp)2ZrCl2 and MAO were selected by aiming Al/Zr molar of 150 in the final catalysts, whereas the volume of toluene used was in 150% excess of the pore volume of the used silica support. More precisely, the amount of MAO 30 wt% solution used was 1.77 mL g−1 for each catalyst. In the second step, the (n-BuCp)2ZrCl2 + toluene + MAO mixture was added to the weighed amount of silica dropwise followed by heating at 50 °C for 1 h without any stirring. In the third step, the supported catalyst was dried under vacuum at 75 °C, without applying any wash, for 2–3 h and then stored inside the glovebox. The same steps were used to make catalysts supported on Grace 948 silica and on PQMS 3040 silica.Polymerization protocols
Polymerization runs were carried out in a spherical laboratory scale 2.5 L semi-batch reactor which can be operated both in slurry and gas phases. The detailed reactor set-up has been described elsewhere.34 The reactor was conditioned for at least 2 h at 80 °C with a minimum of three argon-vacuum cycles, and then cooled down to the room temperature.Method one for slurry polymerization (MSP-1)
Two methods of catalyst injection into reactor were used during slurry phase homopolymerizations. In the first method, referred to hereafter as method one for slurry polymerization (MSP-1), 20 to 25 mg of catalyst and 1 mL of a 1 M TEA or TIBA solution in heptane were added to 500 mL of pure dried heptane under argon. This mixture (i.e., supported catalyst + scavenger + 500 mL heptane) was then injected into the reactor at room temperature under argon pressure followed by reactor heating to 80 °C under constant stirring at 450 revolutions per minute (rpm). Ethylene was then injected and maintained at 8 bar once the desired reaction temperature was achieved. Consumption of ethylene was recorded by the pressure drop in an ethylene cylinder equipped with a pressure gauge.Method two for slurry polymerization (MSP-2)
In the second method of catalyst injection in slurry polymerizations, referred to hereafter as method two for slurry polymerization (MSP-2), 25 to 30 mg of each catalyst were added to an injection cartridge with 5 mL of pure dried heptane inside the glove box. The injection cartridge was then attached to the reactor. 500 mL of pure dried heptane containing 1 mL of 1 M TEA or TIBA or ToA solution was injected separately into reactor under argon pressure, after which the reactor temperature was raised to 80 °C under constant stirring at 450 rpm. The mixture in the cartridge (i.e., the catalyst in 5 mL heptane) was then injected into the reactor under ethylene pressure at 80 °C and then the ethylene pressure was maintained at 8 bar throughout the reaction. Consumption of ethylene was recorded by the pressure drop in an ethylene cylinder equipped with a pressure gauge. It is important to highlight here that the major difference between these two methods is that MSP-2 does not entail any pre-contact between the scavenger and catalyst prior to polymerization, whereas MSP-1 does.All the reactions were of 1 hour and 15 minutes unless otherwise mentioned. For homogeneous polymerization, the desired amounts of metallocene and MAO were mixed in 3 mL toluene inside the glovebox in a shchlenk prior to their injection into the reactor.
Catalyst characterization
Al and Zr content of the final catalysts were obtained by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) at Mikroanalytisches Labor Pascher, Germany.Polymer characterization
High temperature size exclusion chromatography (HT-SEC) analyses were performed using a Viscotek system (from Malvern Instruments) equipped with three columns (PLgel Olexis 300 mm × 7 mm I. D. from Agilent Technologies). 200 μL of sample solutions with a concentration of 5 mg mL−1 were eluted in 1,2,4-trichlorobenzene using a flow rate of 1 mL min−1 at 150 °C. The mobile phase was stabilized with 2,6-di(tert-butyl)-4-methylphenol (200 mg L−1). The OmniSEC software was used for data acquisition and data analysis. Online detection was performed with a differential refractive index detector and a dual light scattering detector (LALS and RALS) for absolute molar mass measurement. The OmniSEC 5.02 software was used for calculations. Malvern Mastersizer 3000 was used for the measurement of particle size distribution (PSD) of silica supports in water with the specially designed Hydro unit whereas for polymer particles a dry module was used for the measurement. Eqn (1) was used to calculate the span of the PSD curves.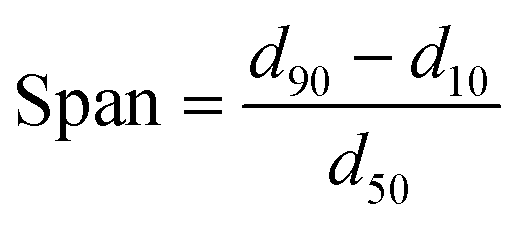 | (1) |
d 50 = diameter value (in μm) possessed by of 50 vol% of the particles present in the measured sample.
d 90 = diameter value (in μm) possessed by of 90 vol% of the particles present in the measured sample.
Results and discussion
Elemental analysis of the final catalysts is shown in Table 1. It can be seen here that both the catalysts obtained have very similar metal loadings, and the Al to Zr molar ratio is very close to the targeted value of 150 during the catalyst synthesis as mentioned in the experimental part. At this point it is important to mention that silica dehydroxylated at 600 °C contains only isolated silanols.35 When MAO solution containing a considerable amount of TMA (see experimental section for MAO description) is added to such silica, the isolated silanols of the silica are consumed by the TMA through protolysis, whereas the MAO adsorbs on the silica surface as proposed by Panchenko et al.36 In the present case since the metallocene was mixed with MAO prior to its impregnation on the silica supports, it is reasonable to assume that the metallocene remains coordinated with the MAO in the final supported catalyst and does not interact with the isolated silanols. Note also that the amount of MAO is significantly higher than that of the metallocene. Since complete spectroscopic characterization of these supported catalysts is beyond the scope of the present work, the interested reader is referred to our previous paper35 which describes the interaction of MAO and metallocenes with silica dehydroxylated at different temperatures.| Catalyst | Al (wt%) | Zr (wt%) | Al/Zr molar ratio |
|---|---|---|---|
| (n-BuCp)2ZrCl2/MAO/Grace 948 | 14.0 | 0.31 | 153 |
| (n-BuCp)2ZrCl2/MAO/PQMS3040 | 12.3 | 0.30 | 139 |
Effect of pre-contact between TEA and catalyst in slurry polymerization
As described above in the polymerization section, the two polymerization protocols used in this study differ from each other as to whether the catalyst and the scavenger are pre-contacted (MSP-1) or not (MSP-2) before the polymerization begins. Fig. 1 and 2 show the effect of pre-contacting the (n-BuCp)2ZrCl2/MAO/Grace948 and (n-BuCp)2ZrCl2/MAO/PQMS3040 supported catalysts with TEA, respectively. Pre-contacting each catalyst with TEA reduced the instantaneous catalytic activity with respect to the case when there was no pre-contact. In the case of Grace 948 based catalyst, the initial activation behavior is similar for the two polymerization protocols (i.e., compare first 5 minutes of reaction time in Fig. 1), whereas, for the catalyst supported on PQMS3040 silica, the polymerization rate starts off more slowly, and reaches a lower maximum when the catalyst is pre-contacted with TEA. The effect of pre-contacting is more pronounced in the case of catalyst supported on PMS3040 silica.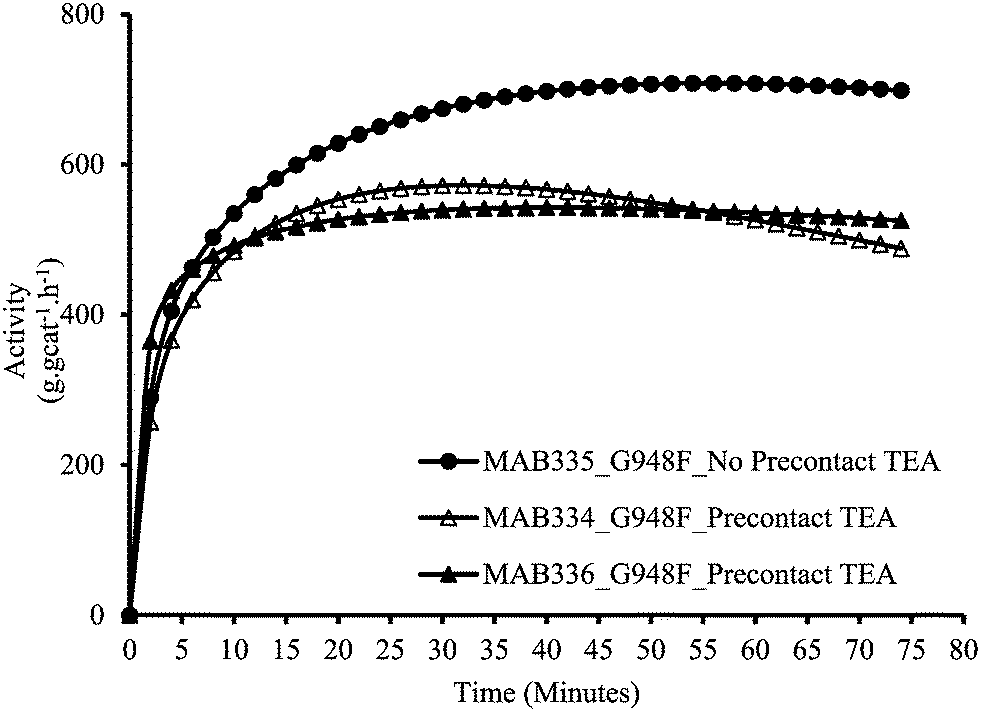 | ||
| Fig. 1 Effect of pre-contact between Grace 948 based catalyst and TEA in slurry phase polymerizations. | ||
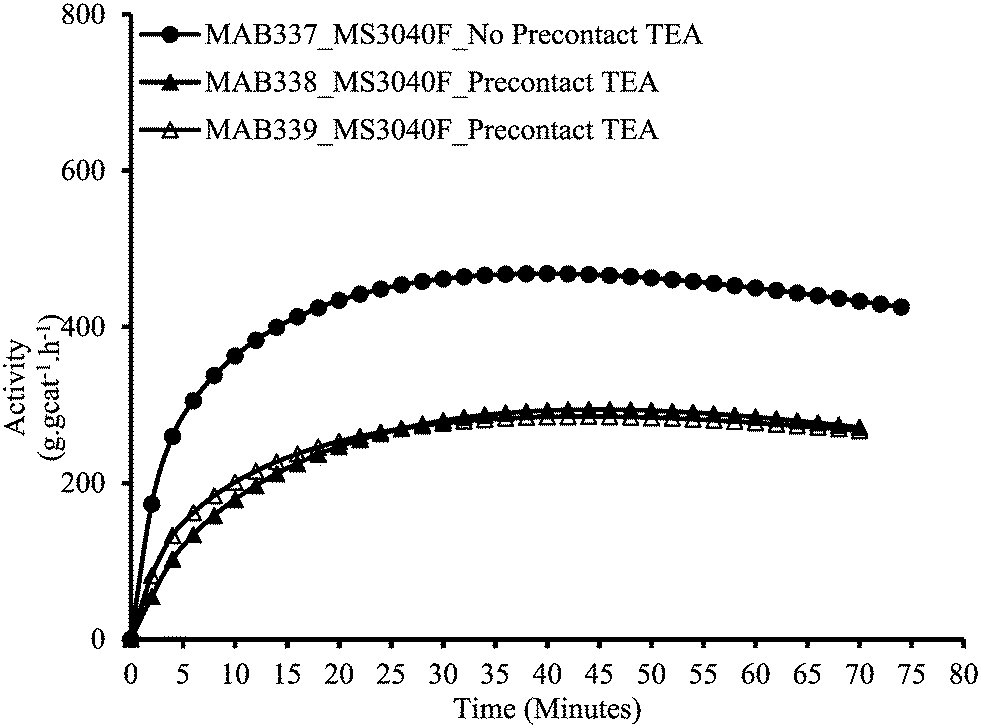 | ||
| Fig. 2 Effect of pre-contact between PQMS3040 based catalyst and TEA in slurry phase polymerizations. | ||
The effect of two polymerization protocols on the reactor fouling was also observed by verifying the condition of the reactor and its contents at the end the polymerization. Some representative results are shown in Fig. 3. A polymer film is visible in Fig. 3a on the reactor wall (precontact), whereas, no such film is present in Fig. 3b (no precontact). This supports the idea that pre-contacting the supported catalyst with TEA is linked with the leaching of supported (n-BuCp)2ZrCl2/MAO complex from the silica support, which remained active in the continuous phase and produced polyethylene that was deposited on the reactor walls. On the other hand, no pre-contact between the catalyst and TEA (i.e., MSP-2) led to heterogeneous catalysis without any leaching and no reactor fouling. It should be noted that the same behavior was observed for Grace 948 supported catalysts. These results clearly indicate that the polymerization protocol can significantly affect the reaction kinetics of silica supported metallocene/MAO complex as well as the fact that catalyst leaching can be avoided by selecting an appropriate polymerization protocol.
 | ||
| Fig. 3 Effect of polymerization protocols on reactor fouling for (a) MSP-1 with precontacting, and (b) MSP-2 with no precontacting. White colored PE film can be noticed on the reactor walls at the end of polymerization with MSP-1 protocol, whereas, no such film can be noticed on the reactor walls at the end of polymerization with MSP-2 protocol. | ||
As stated above, the major difference between the two protocols is the pre-contact, or absence thereof between the supported catalyst and the TEA. In both cases the TEA is well dispersed in the slurry phase, however in the case of MSP-1, the supported catalyst particles are exposed to the TEA in solution for a much longer period (pre-contacting lasted for approximately 10 minutes at room temperature before injection). This allows the active sites, especially those close to the exterior of the silica particles to eventually be dissolved in the heptane and diffuse out of the particles. The lower instantaneous polymerization rate obtained with the MSP-1 protocol (i.e., upon pre-contacting the supported catalyst with TEA) can be attributed to the fact that pre-contact of TEA with the supported (n-BuCp)2ZrCl2/MAO catalyst causes the generation of heterobimetallic species [L2M(μ-R)2AlR2]+, both in the reaction milieu where there is leached catalyst, and inside the supported catalyst. It is well-known that this species is very stable when it is generated via TEA, and decreases the activity of metallocenes in olefin polymerizations.37,38
In MSP-2, the TEA and catalyst come in contact at essentially the same time ethylene is injected and polymerization begins. Upon injection, the Al/Zr ratio inside the particles will be lower at the active sites with MSP-2 than with MSP-1, thus giving us a potentially different reaction rate. In addition, polymer forms immediately at the exterior of the particles, provoking mass transfer limitations for both C2 and TEA, but given that the TEA is bulkier, it will diffuse more slowly through the surface pores to the interior, thereby maintaining low Al/Zr ratios inside the particles, but still acting as a scavenger outside. Furthermore, polymer will rapidly cover accessible sites, thereby preventing leaching and consequently reducing the possibility of reactor fouling. The fraction of heterobimetallic species formed will therefore be lower with MSP-2 than MSP-1, leading to faster reactions.
The values of weight average molar mass (Mw) and dispersity of molar mass (Đ) are shown in Table 2. It is difficult to discern a clear influence of either polymerization protocol on the MWD or Đ values of the HDPE samples. It is tempting to say that the samples where the catalysts were pre-contacted (and thus leaching occurred) showed higher molecular weights than the non-pre-contacted case, but the differences could be simply experimental error; more on this below. In addition, the melting temperatures of the HDPE samples also remain unaffected by the polymerization protocol.
| Catalyst support | Sample name | Catalyst/TEA pre-contact | M w (g per mole) | Đ | T m (°C) |
|---|---|---|---|---|---|
| Grace 948 | MAB335 | No | 155000 |
2.4 | 135.7 |
| Grace 948 | MAB334 | Yes | 175000 |
2.4 | 134.5 |
| Grace 948 | MAB336 | Yes | 175000 |
2.4 | 135.2 |
| PQMS3040 | MAB337 | No | 165000 |
2.4 | 135.7 |
| PQMS3040 | MAB338 | Yes | 175000 |
2.3 | 135.3 |
| PQMS3040 | MAB339 | Yes | 180000 |
2.4 | 135.2 |
Since we have no leaching without pre-contact, we can also use this protocol to investigate the impact of TEA on the polymerization rate, and be sure that all the polymer is made inside the particles. Fig. 4 shows the effect of increasing TEA concentration inside reactor with MSP-2 on the rate profiles of ethylene homopolymerizations on a Grace 948 supported (n-BuCp)2ZrCl2/MAO catalyst. One observes a clear decrease in the instantaneous activity with increasing TEA concentration inside the reactor. It is important to mention here that no reactor fouling at all was observed in these reactions meaning that all the polymerization occurred on the supported catalyst particles. The reason for decrease in the catalytic activity by increasing TEA concentration inside reactor is probably due to the generation of a dormant heterobimetallic species [L2M(μ-R)2AlR2]+ formed by interaction between supported (n-BuCp)2ZrCl2/MAO catalyst and TEA. It is likely that the generation of this dormant, stable cationic species is more probable at higher TEA concentrations. A similar impact of increasing TEA on the kinetics of silica supported (n-BuCp)2ZrCl2 was observed by Tisse et al.38 In other words, an excess of TEA acts as a poison and helps to explain why the rates are higher in the absence of leaching as we saw above; when the catalyst is leached, it is exposed to higher levels of TEA in solution and thus polymerizes more slowly.
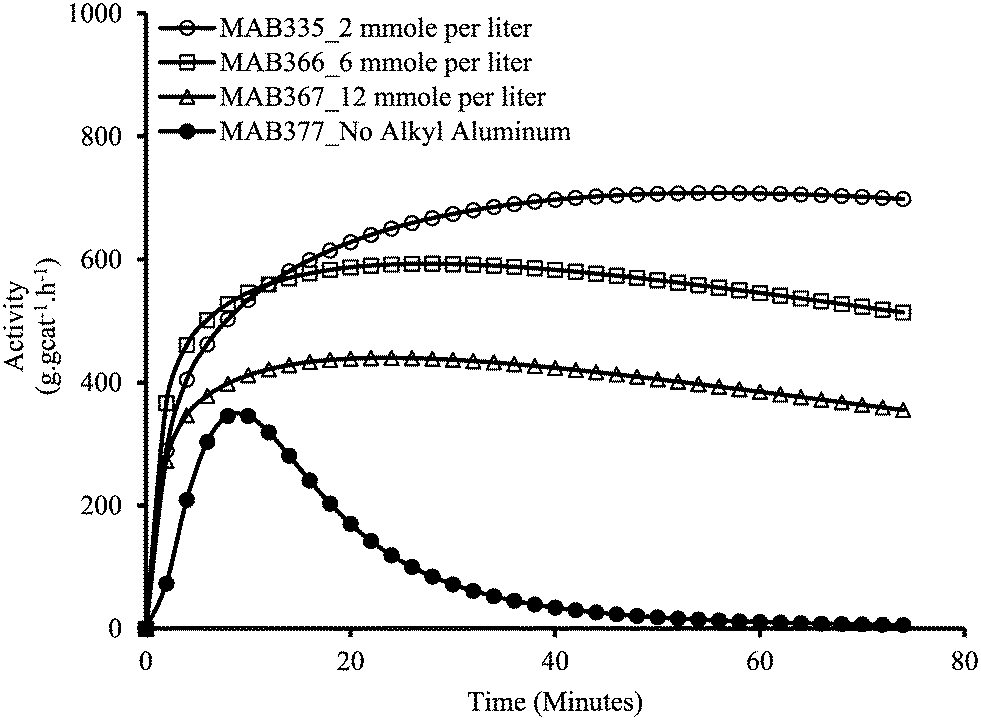 | ||
| Fig. 4 Effect of TEA concentration inside reactor on the instantaneous activity of silica supported (n-BuCp)2ZrCl2/MAO in ethylene homopolymerization with MSP-2. | ||
In addition to influencing the reaction rate, the TEA concentration seems to affect the particle size distribution and bulk density of the polyethylene as shown by Fig. 5. All the polymer samples replicate the support morphology indicating that there was no catalyst leaching with this polymerization protocol despite adding more TEA into the reactor. However, the relative concentration of fine particles (i.e., the particles with diameter ≤ 200 μm) seems to increase with increasing TEA concentration inside reactor. The decrease in the rate also corresponds to an increase in the bulk density of the polyethylene sample, shown in the inset of Fig. 8. The reason for this result is not immediately clear, although it is possible that the TEA penetrates a larger volume fraction of the small catalyst particles than it does with the large ones in the short time during which the catalyst and continuous reactor phase are in direct contact. This poisons (or at least slows down) polymerization in the small ones, causing that fraction of fine particles to appear more important.
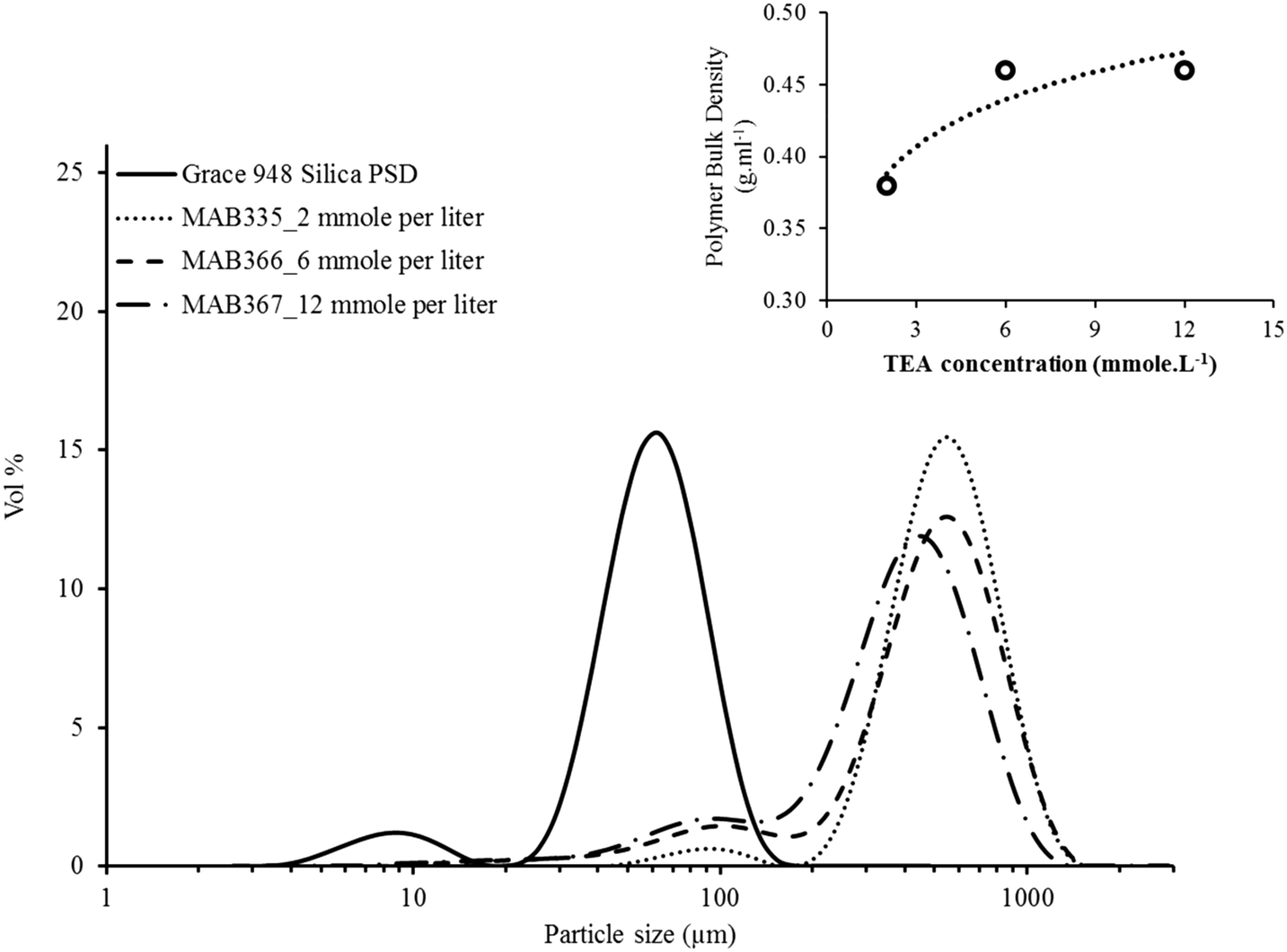 | ||
| Fig. 5 Effect of TEA concentration inside reactor on the PSD of HDPE produced in slurry phase polymerizations. Span of MAB335 PSD curve = 0.95, MAB366 PSD curve = 1.46 and MAB367 PSD curve = 1.48. Bulk density values are average values of minimum two measurements for one sample. | ||
Effect of pre-contact between TIBA and supported catalyst in slurry polymerization
Fig. 6 shows the rate profiles obtained by using the two different protocols for the catalyst supported on Grace 948 with TIBA as the scavenger. Similar runs were performed with the MS3040 supported catalyst and showed similar trends. In opposition to what is shown in the previous section, it can be observed that the instantaneous polymerization rate is higher for MSP-1 (i.e. pre-contacted with TIBA) than with MSP-2 (no pre-contact with TIBA).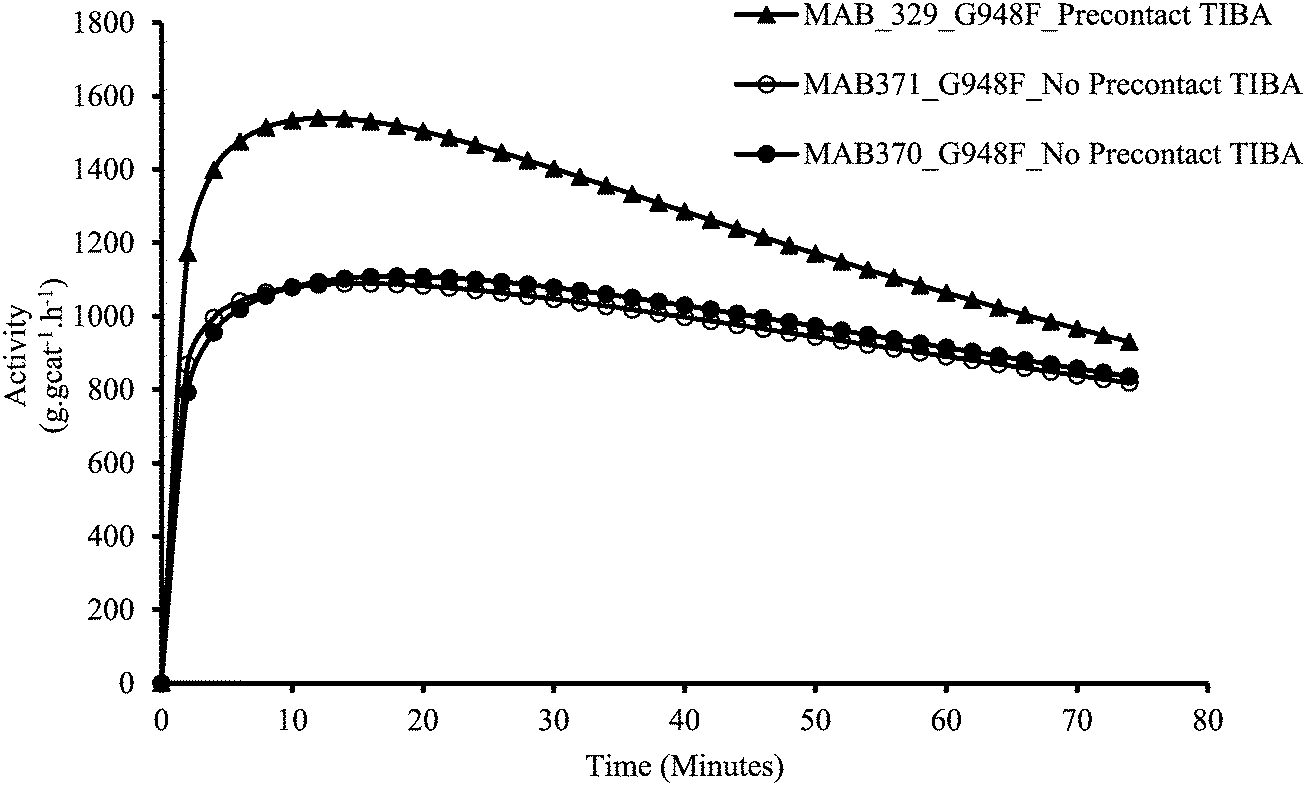 | ||
| Fig. 6 Kinetic profiles obtained with Grace 948 catalyst using TIBA as scavenger. | ||
Furthermore, the reactor condition at the end of reaction and morphology of the polyethylene samples produced with both the polymerization protocols was very different. With MSP-1, the Grace 948-supported catalyst led to extensive reactor fouling (Fig. 7a), the formation of film-like polymer and/or agglomerates of polyethylene, and virtually no well-defined granules that we would expect if the reaction proceeded ideally (Fig. 7e). Similarly, reactor fouling was also observed with MSP-1 when PQMS3040 based catalyst was used (Fig. 7c) but the polymer morphology was somewhat better than that obtained with Grace 948-supported catalyst, with some spherical particles being obtained (Fig. 7d). On the other hand, when polymerizations were conducted without pre-contacting TIBA and the supported catalyst (i.e., with MSP-2) no reactor fouling was observed in any of the polymerizations conducted by both the catalysts (Fig. 7e) and the obtained polymer particles possessed spherical shape indicating purely heterogeneous catalysis (Fig. 7f).
 | ||
| Fig. 7 Reactor fouling caused by the polymerization protocol MSP-1 (a–d) and the absence of reactor fouling in the case of MSP-2 protocol (e and f). | ||
The root cause of reactor fouling is actually what causes the higher activities observed in the case of MSP-1 with respect to MSP-2. It has been shown in the literature by various authors7,8,10,21 that pre-contacting supported metallocenes with an aluminum alkyl like TIBA causes the desorption or leaching of the metallocene/MAO activated complex from the support which leads to homogeneous polymerization in the diluent. As discussed in the introduction, the amount of zirconocene and aluminum desorbed into the diluent depends on the type of diluent, catalyst supporting method, properties of the support used, and the reaction conditions. Furthermore, Kaminsky et al.9,39 showed that the leached metallocene is active in polymerization if MAO is also present in the diluent. Therefore, the higher instantaneous activities showed by both supported (n-BuCp)2ZrCl2/MAO catalysts when pre-contacted with TIBA, as compared to the case when there is no pre-contact, is likely due to the leaching of (n-BuCp)2ZrCl2/MAO from the support leading to homogeneous polymerization in the diluent causing reactor fouling (as well as the heterogeneous polymerization which occurred due to the part of (n-BuCp)2ZrCl2/MAO remained on silica). In contrast to the previously discussed case of TEA pre-contacting with the same supported catalysts (where, although catalyst leaching occurred, the activity of the catalysts after pre-contacting was less than in the absence of any pre-contact), the increase of activity by pre-contacting TIBA with supported (n-BuCp)2ZrCl2/MAO catalyst can be explained by the fact that: i) the stability of heterobimetallic species [L2M(μ-R)2AlR2]+ formed by the presence of TIBA is less than that formed by the presence of TEA;37 and ii) TIBA interacts with MAO and partially replaces the Al–Me groups of MAO with Al–iBu groups which converts MAO into modified MAO (MMAO) which is better co-catalyst for metallocenes and has higher solubility in heptane then MAO.10,40,41 In case of no pre-contact between the supported catalysts and TIBA there was no leaching of the active species into the diluent. In this case we have purely heterogeneous catalysis, no fouling and kinetic profiles with lower catalyst deactivation than was seen with MSP-1.
In order to further investigate leaching, 28 mg of silica supported (n-BuCp)2ZrCl2/MAO/Grace 948 catalyst was pre-contacted with 1 mmole of TIBA in 500 mL of heptane for 10 minutes at 80 °C in a 500 mL Schlenk tube. Afterwards, the supernatant layer containing any eventual leached (n-BuCp)2ZrCl2/MAO + TIBA was transferred to the reactor under argon flow using a cannula whose dipped end was covered with a filter paper in order to avoid any solid catalyst from going into the reactor. The reactor was then heated and ethylene homopolymerisation was performed at 80 °C for 1 h 15 minutes at 8 bar monomer pressure. In a second reaction, the remaining solid catalyst was injected into the reactor after adding neat 500 mL heptane followed by reactor heating to 80 °C. At the reaction temperature, 1 mmole of TIBA was injected into the reactor under ethylene pressure so that there was no pre-contact between the freshly added TIBA and the solid catalyst part. Ethylene homopolymerisation was conducted for 1 h 15 minutes at 8 bar monomer pressure.
Fig. 8a and b confirm that upon pre-contacting the supported catalyst with TIBA, significant catalyst leaching occurred. In the case of polymerization of the supernatant, ∼10.5 g of polyethylene were produced (a small fraction of polymer was lost during cleaning), whereas 30 g of polyethylene were obtained with the supported catalyst. Fig. 9 shows the rate profiles of the polymerisation done without any pre-contact between the (n-BuCp)2ZrCl2/MAO/Grace948 and TIBA with the kinetic profile of the reaction done with the same catalyst after pre-contacting it with TIBA and removing the supernatant layer. While the curves eventually converge to similar activities after about 45 minutes, the initial reaction rates are quite different, with the pre-contacted catalyst particles activating more slowly.
 | ||
| Fig. 8 (a) Reactor stirrer showing polyethylene morphology formed by the leached catalyst in supernatant and (b) reactor stirrer showing polyethylene morphology formed by the supported catalyst. | ||
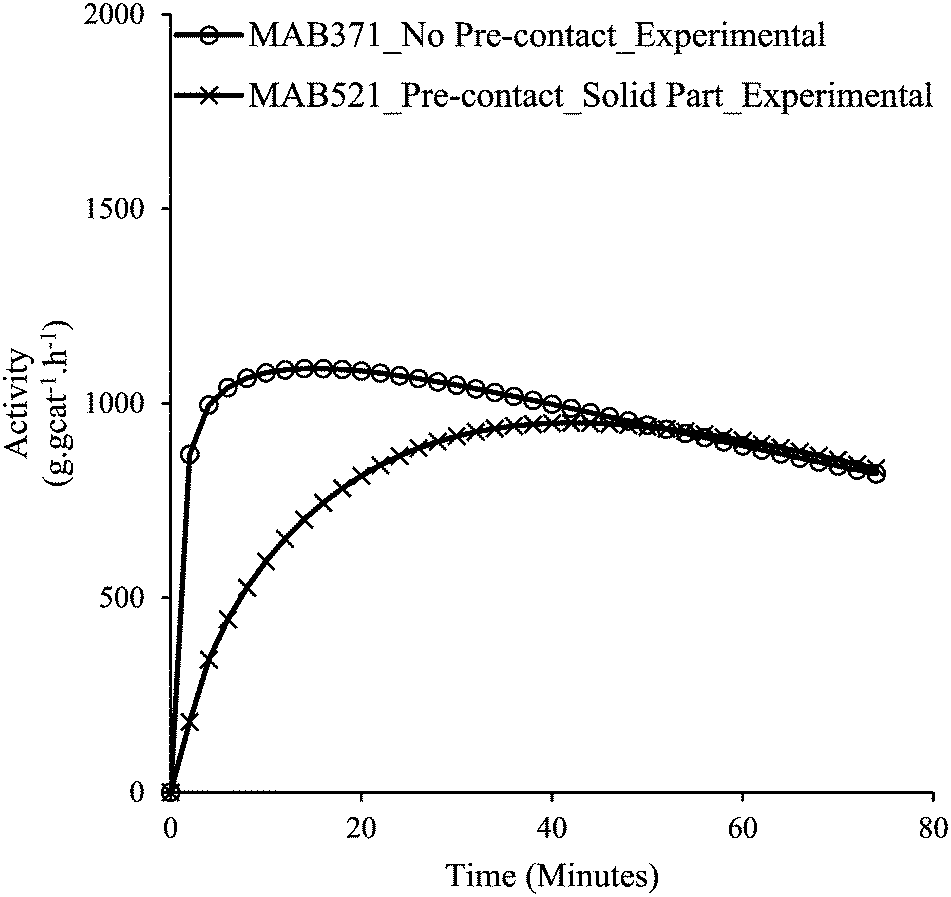 | ||
| Fig. 9 Activities of catalyst with no pre-contact (MAB371), and the residual solid catalyst particles resulting from the pre-contact and removal of the supernatant (MAB521). | ||
Impact of butylated hydroxytoluene (BHT-H) on the reaction kinetics of silica supported (n-BuCp)2ZrCl2/MAO catalyst
The effect of adding BHT-H into the reaction mixture was also studied with and without pre-contacting the aluminum alkyl (AlR3) + BHT-H + heptane mixture with the supported catalyst. The AlR3 + BHT-H + heptane mixture was prepared by first adding the desired amount of AlR3 to heptane under argon, and then adding the desired amount of BHT-H to that solution. A 1 M stock solution of BHT-H in heptane was used in all reactions. Note that the temperature and pressure of these polymerizations were kept the same to the conditions used in the previous polymerizations. All reactions are of 1 h 15 minutes reaction time. The rate curves are not shown for the sake of brevity, but it was found that adding BHT-H led to increases in the rate of polymerization of all polymerizations for all three alkyls tested with respect to the systems without the BHT-H. Similar rate profiles have also been observed by Reddy et al.,30 who used isolated AlMe(BHT)2 as a scavenger in zirconocene catalyzed ethylene slurry polymerization. Note that AlMe(BHT)2 is product of equimolar reaction between trimethyl aluminum (TMA) and BHT-H and we will discuss these reactions in the coming paragraphs. Furthermore, when TEA and TOA were used it was found that pre-contacting the silica supported catalyst with AlR3 + BHT-H + heptane mixture resulted in significant fouling. It can be seen in Fig. 10a and b (for TEA and TOA respectively) that reactor fouling, and poorly controlled morphologies were found for these systems. This suggests that both homogenous and heterogeneous modes of polymerization occurred in these reactions as well.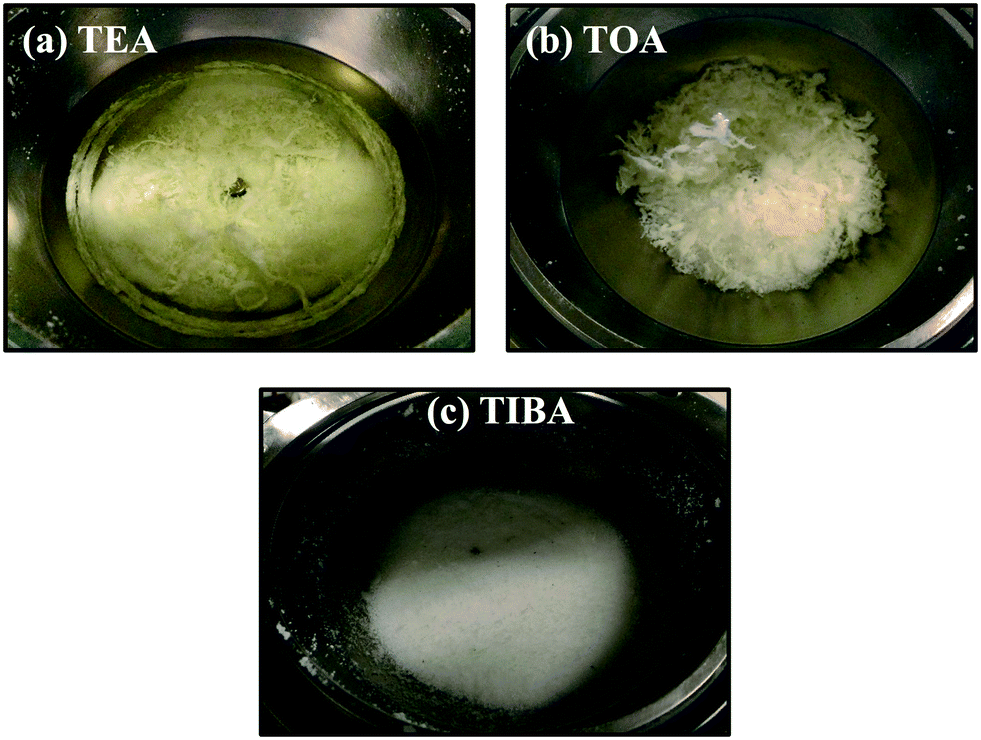 | ||
| Fig. 10 Effect of pre-contacting the aluminum alkyl + BHT-H + heptane mixture with the silica supported (n-BuCp)2ZrCl2/MAO on reactor fouling and polyethylene morphology for (a) TEA, (b) TOA and (c) TIBA. | ||
Surprisingly, absolutely no reactor fouling occurred when TIBA + BHT-H + heptane mixture was pre-contacted with the silica supported (n-BuCp)2ZrCl2/MAO catalyst as shown by the polymer morphology (see Fig. 10c) and the absence of any polymer film formation around the reactor walls or stirrer (not shown here). Replicate runs confirmed that these results were reproducible.
It is rather well-known that aluminum alkyls react readily with simple phenols to give phenoxide compounds which are bridged through the oxygen atom, whereas, the reaction of the same aluminum alkyls with substituted or sterically hindered phenols such as BHT-H under similar conditions yields compounds of the type AlR2(BHT) and AlR(BHT)2 (where R = ethyl or butyl group), which are monomeric in nature.23–27 According to Ittel et al.,27 the chemical reaction between 1 equivalent of TEA and 2 or more equivalents of BHT-H yields a mixture of TEA, AlEt2BHT and AlEtBHT2 which are in dynamic equilibrium, whereas, the same reaction between 1 equivalent of TIBA and 2 equivalents of BHT-H leads to complete consumption of TIBA and yields only the Al(i-Bu)BHT2 species; i.e., there is no dynamic equilibrium between the TIBA and Al(i-Bu)BHT2. AlEtBHT2 was shown to be reactive with t-BuOH whereas no discussion was made about the reactivity of Al-R bond in Al(i-Bu)BHT2. Later on it was shown by different authors26,28–32 that AlMeBHT2 (which is a product of the reaction of trimethyl aluminum (TMA) with BHT-H similar to the reaction of TEA with BHT-H) can be used as a ‘non-interacting’ scavenger or scrubbing agent for metallocene catalyzed olefin slurry polymerization due to its reactive nature (just like AlEtBHT2) as discussed by Ittel et al.27
The results observed in our work where pre-contact between the silica supported catalyst and AlR3 + BHT-H + heptane mixture lead to reactor fouling when TEA or TOA were used can be explained using the preceding concept. The presence of TEA + AlEt2BHT + AlEtBHT2 (due to the reaction between TEA and BHT-H) mixture in heptane causes catalyst leaching from the support due to the interactions between TEA, AlEt2BHT and the supported (n-BuCp)2ZrCl2/MAO catalyst which consequently leads to reactor fouling. The same explanation can be considered valid for the system involving TOA as reactor fouling was observed in that case also. On the other hand, the reaction of TIBA with BHT-H leads to complete transformation of TIBA into Al(i-Bu)BHT2, and since no fouling was observed in the reactions when the catalyst was pre-contacted with the TIBA + BHT-H + heptane mixture it is reasonable to assume that Al(i-Bu)BHT2 does not interact with the supported metallocene and therefore no catalyst leaching occurs. These results clearly indicate that only Al(i-Bu)BHT2 can be considered to be non-leaching scavenger for supported metallocenes whereas other similar species generated by the reaction of TEA or TOA with BHT-H generate species which interact with the supported metallocene and lead to catalyst leaching from the silica support.
Ethylene homopolymerizations were also conducted by avoiding any pre-contact between the silica supported (n-BuCp)2ZrCl2/MAO and AlR3 + BHT-H + heptane mixture using MSP-2. It was found that, as in the absence of this same compound, absolutely no reactor fouling occurred in these reactions, indicating that purely heterogeneous catalysis occurred. This confirms once again that avoiding any pre-contact between the supported catalyst and scavenger prohibits catalyst leaching from the support. MWD and melting temperature of the HDPE samples were found to be like those samples which were produced without using BHT-H.
Conclusion
In ethylene polymerization via silica supported metallocene/MAO complex the polymerization protocol can significantly affect the kinetic profile of the reaction, and, in particular, the morphology of the polyethylene produced and reactor fouling. The results obtained in this work clearly demonstrate that catalyst leaching will happen if an aluminum alkyl like TEA or TIBA is pre-contacted with the silica supported metallocene/MAO consequently leading to poor morphology of the polymer and reactor fouling due to homogeneous polymerization occurring parallel to heterogeneous reaction. On the other hand, one can get rid of these problems in slurry phase ethylene polymerizations by avoiding any pre-contact between the silica supported metallocene/MAO complex and the used aluminum alkyl. In addition to aluminum alkyls, the impact of adding BHT-H in the reactor diluent containing aluminum alkyls has also been discussed. It has been found that the reaction product of TIBA with BHT-H (i.e., Al(i-Bu)BHT2) is a non-interacting scavenger for silica supported metallocene/MAO catalysts and therefore, does not cause catalyst leaching and reactor fouling during slurry phase ethylene homopolymerization.Acknowledgements
Financial support by the Dutch Polymer Institute is gratefully acknowledged. This work is a part of the Research Programme of the Dutch Polymer Institute (DPI, Eindhoven, The Netherlands), project no. 753. The authors are also thankful to Dr. John Severn of DSM Ahead, Geleen, The Netherlands and Dr. Irfan Saeed from Borealis Polymers Oy, Porvoo, Finland, for their guidance in the synthesis of catalysts.Notes and references
- E. Casas, R. van Grieken and J. M. Escola, Appl. Catal., A, 2012, 437–438, 44–52 CrossRef CAS.
- S. Jungling, S. Koltzenburg and R. Mulhaupt, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1–8 CrossRef.
- W. Kaminsky, K. Külper and S. Niedoba, Makromol. Chem., Macromol. Symp., 1986, 3, 377–387 CrossRef CAS.
- O. Sperber and W. Kaminsky, Macromolecules, 2003, 36, 9014–9019 CrossRef CAS.
- K. A. Ostoja Starzewski, B. S. Xin, N. Steinhauser, J. Schweer and J. Benet-Buchholz, Angew. Chem., Int. Ed., 2006, 45, 1799–1803 CrossRef PubMed.
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722–9744 CrossRef CAS PubMed.
- J. P. J. Turunen and T. T. Pakkanen, J. Appl. Polym. Sci., 2006, 100, 4632–4635 CrossRef CAS.
- J. R. Severn, J. C. Chadwick, R. Duchateau and N. Friederichs, Chem. Rev., 2005, 105, 4073–4147 CrossRef CAS PubMed.
- W. Kaminsky and C. Strübel, J. Mol. Catal. A: Chem., 1998, 128, 191–200 CrossRef CAS.
- V. N. Panchenko, L. G. Echevkaya, V. A. Zakharov and M. A. Matsko, Appl. Catal., A, 2011, 404, 47–53 CAS.
- M. C. Sacchi, D. Zucchi, I. Tritto, P. Locatelli and T. Dall'Occo, Macromol. Rapid Commun., 1995, 16, 581–590 CrossRef CAS.
- H. Liu, C. W. M. Bastiaansen, J. G. P. Goossens, A. P. H. J. Schenning and J. R. Severn, Macromol. Chem. Phys., 2017, 218, 1600490–1600491 CrossRef.
- F. F. Karbach, J. R. Severn and R. Duchateau, ACS Catal., 2015, 5, 5068–5076 CrossRef CAS.
- M. Kristen, Top. Catal., 1999, 7, 89–95 CrossRef CAS.
- N. Suzuki, H. Asami and T. Nakamura, et al. , Chem. Lett., 1999, 28, 341–342 CrossRef.
- A. M. Uusitalo, T. T. Pakkanen and E. I. Iskola, J. Mol. Catal. A: Chem., 2002, 177, 179–194 CrossRef CAS.
- Y. Choi and J. B. P. Soares, Can. J. Chem. Eng., 2012, 90, 646–671 CrossRef CAS.
- K. Soga, H. J. Kim and T. Shiono, Macromol. Rapid Commun., 1994, 15, 139–143 CrossRef CAS.
- K. Soga, H. J. Kim and T. Shiono, Macromol. Chem. Phys., 1994, 195, 3347–3360 CrossRef CAS.
- M. C. Sacchi, D. Zucchi, I. Tritto, P. Locatelli and T. Dall'Occo, Macromol. Rapid Commun., 1995, 16, 581–590 CrossRef CAS.
- N. V. Semikolenova and V. A. Zakharov, Macromol. Chem. Phys., 1997, 198, 2889–2897 CrossRef CAS.
- E. Ernst, J. Reußner and P. Denifl, in Metalorganic Catalysts for Synthesis and Polymerization, ed. K. Walter, Springer, Berlin Heidelberg, 1999, p. 406 Search PubMed.
- T. Mole, Aust. J. Chem., 1966, 19, 373–379 CrossRef CAS.
- M. Skowronska-Ptasinska, K. B. Starowieyski, S. Pasynkiewicz and M. Carewska, J. Organomet. Chem., 1978, 160, 403–409 CrossRef CAS.
- K. B. Starowieyski, S. Pasynkiewicz and M. Skowroñska-Ptasiñska, J. Organomet. Chem., 1975, 90, C43–C44 CrossRef CAS.
- M. D. Healy, D. A. Wierda and A. R. Barron, Organometallics, 1988, 7, 2543–2548 CrossRef CAS.
- A. P. Shreve, R. Mulhaupt, W. Fultz, J. Calabrese, W. Robbins and S. D. Ittel, Organometallics, 1988, 7, 409–416 CrossRef CAS.
- R. Vollmerhaus, M. Rahim, R. Tomaszewski, S. Xin, N. J. Taylor and S. Collins, Organometallics, 2000, 19, 2161–2169 CrossRef CAS.
- V. C. Williams, C. Dai and Z. Li, et al. , Angew. Chem., Int. Ed., 1999, 38, 3695–3698 CrossRef CAS PubMed.
- S. S. Reddy, G. Shashidhar and S. Sivaram, Macromolecules, 1993, 26, 1180–1182 CrossRef CAS.
- V. C. Williams, G. J. Irvine and W. E. Piers, et al. , Organometallics, 2000, 19, 1619–1621 CrossRef CAS.
- V. Busico, R. Cipullo, R. Pellecchia, G. Talarico and A. Razavi, Macromolecules, 2009, 42, 1789–1791 CrossRef CAS.
- G. G. Hlatky, Chem. Rev., 2000, 100, 1347–1376 CrossRef CAS PubMed.
- M. Namkajorn, A. Alizadeh, E. Somsook and T. F. L. McKenna, Macromol. Chem. Phys., 2014, 215, 873–878 CrossRef CAS.
- M. A. Bashir, T. Vancompernolle and R. M. Gauvin, et al. , Catal. Sci. Technol., 2016, 6, 2962–2974 CAS.
- V. N. Panchenko, N. V. Semikolenova, I. G. Danilova, E. A. Paukshtis and V. A. Zakharov, J. Mol. Catal. A: Chem., 1999, 142, 27–37 CrossRef CAS.
- R. Kleinschmidt, Y. van der Leek, M. Reffke and G. Fink, J. Mol. Catal. A: Chem., 1999, 148, 29–41 CrossRef CAS.
- V. F. Tisse, C. Boisson and T. F. L. McKenna, Macromol. Chem. Phys., 2014, 215, 1358–1369 CrossRef CAS.
- W. Kaminsky and H. Winkelbach, Top. Catal., 1999, 7, 61–67 CrossRef CAS.
- K. P. Bryliakov, N. V. Semikolenova, V. N. Panchenko, V. A. Zakharov, H. H. Brintzinger and E. P. Talsi, Macromol. Chem. Phys., 2006, 207, 327–335 CrossRef CAS.
- D. E. Babushkin, V. N. Panchenko, M. N. Timofeeva, V. A. Zakharov and H. H. Brintzinger, Macromol. Chem. Phys., 2008, 209, 1210–1219 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |