
Impact of catalyst injection conditions on the gas phase polymerization of propylene
Aarón J.
Cancelas
ab,
Vincent
Monteil
a and
Timothy F. L.
McKenna
 *a
*a
aC2P2 - LCPP Group, UMR5265 CNRS, Université de Lyon, ESCPE Lyon, Bat 308F, 43 Bd du 11 novembre 1918, F-69616, Villeurbanne, France. E-mail: timothy.mckenna@univ-lyon1.fr
bDutch Polymer Institute DPI, POBox 902, 5600 AX Eindhoven, The Netherlands
First published on 16th January 2017
Abstract
In the current work, gas phase propylene polymerizations were performed on ZN catalysts in a stopped flow reactor to understand the effect that the injection conditions (dry, as received, or wetted with a commercially available paraffinic mineral oil) have on initial temperature profiles, nascent polymer properties, and polymerization kinetics. Temperature spikes followed by initially high activity and rapid deactivation were always observed earlier in the dry case. The initially high activities are attributed, at least in part, to lower resistance to monomer transport to the active sites, due to the absence of mineral oil in the dry case. This causes overheating of the catalyst, leading to faster reaction, but then to rapid thermal deactivation of the catalysts. Furthermore, it was found that oil can decrease the iPP crystallization temperature which makes the polymer more deformable, and is possibly one of the reasons for which one obtains better morphology using wet injection.
1. Introduction
By virtue of its broad range of applications, isotactic polypropylene (iPP) is the second most widely produced polymer in the world after polyethylene, with world production currently exceeding 60 MT per year and projected to grow to 90 million by the year 2022.1 This production is almost entirely based on Ziegler–Natta (ZN) catalysts and is carried out in both gas phase and monomer slurry (bulk) processes (there is also a process using supercritical propane). Bulk processes use condensed propylene as the process fluid, and gas phase processes use gaseous monomer (and other components) as the continuous phase. One important family of polypropylenes is high impact polypropylene (hiPP) polymers. During the production of hiPP, an initial part of the production process is used to make iPP homopolymer, and a second copolymerization step is used to form an ethylene–propylene rubber (EPR) elastomeric phase inside the still-reactive homopolymer particles. This second step must be carried out in the gas phase, since the amorphous EPR component is soluble in any organic solvent, including liquefied monomer. One of the process-related challenges in hiPP production is to obtain the best possible morphology (i.e., regular particle shape, narrow particle size distribution) during the homopolymerization step. This helps to avoid associated problems such as stickiness, lump formation, reactor fouling and a number of other problems.iPP kinetics and particle morphology are greatly influenced during the initial instants when fragmentation takes place.2 It is widely accepted that one of the surest ways to preserve catalyst properties and obtain reasonable particle morphology is to use a prepolymerization step before it undergoes full reaction conditions.2–9 Prepolymerization is conducted under mild conditions that promote slower growth and better heat transfer conditions, for instance, in liquid propylene, at temperatures of from 15 to 40 °C.10 The mild conditions allow particle expansion in a controlled manner and increase in the surface area available for heat and mass transfer. These conditions are known to help obtain good iPP morphology. Gas phase processes are particularly prone to heat transfer-related problems far more so than slurry processes, especially during the initial moments of the reaction following injection of the catalyst and, in particular, if prepolymerization steps are not used.11 However they are less expensive to install and run than slurry processes since liquefication steps and the pumping of liquids around the process add extra costs to the slurry process operation. On the downside, using a prepolymerization step in addition to the main polymerization(s) increases the capital and operating costs of production. Gas phase polymerization would therefore be even more attractive if certain problems could be solved, such as carrying out the polymerization in a thermally controlled manner to avoid hot spots and obtaining a polymer morphology which replicates that of the catalyst.
Among the gas phase technologies available is the INNOVENE™ G process, based on the horizontal stirred bed reactor developed by Standard Oil.12–14 Here the precatalyst is not prepolymerized, but rather the catalyst is injected in a manner carefully designed to steadily activate it directly in the reactor. It is claimed that it is important to inject the catalyst and cocatalyst at a well-defined separation distance since this facilitates the gradual activation of the catalyst by thoroughly mixing it with the polymer material within the reactor vessel. Some related patents claim that adding a small amount of hydrocarbon to the catalyst prior to its injection in the gas phase reactor improves polymer morphology and reduces the level of fines.15–17 An additional advantage is revealed in another patent: the reduction of the amount of nitrogen fed to the polymerization reactor, which otherwise would be used to feed the catalyst.18 In a patent to BASF on what is now the Novolen process,19 the authors show that it is necessary to inject the different components of the catalyst formulation into different parts of a stirred powder bed to avoid overheating. The authors of patent US3965083 to Standard Oil say that they try and inject the different catalyst components into the bed at different spots, and often one tries to put the dry catalyst at a spot where quench liquid (for cooling) is also injected.20 Clearly how the catalyst is treated during its activation is a key point from a commercial point of view in gas phase olefin polymerizations.
Despite this, investigations of what happens to the catalyst particles during the activation of Ziegler–Natta catalysts for gas phase propylene polymerization are scarce in the literature. In a previous publication from our group,21 it was revealed that injecting the catalyst in mineral oil22 in a gas phase iPP process had a beneficial impact on catalyst activity (see Fig. 1) and polymer particle morphology. Even when an in situ prepolymerization was applied on dry injected catalyst particles (and therefore ensuring avoidance of particle overheating), poor morphology (see Fig. 2) and lower activities were always obtained as compared to when they were injected wet.23 It was hypothesized that morphology improvement was attributed to the influence that the hydrocarbon exerts over the initial polymer chains, acting as a solvent, plasticizing them and making the resulting polymer more ductile, leading to a more controlled particle fragmentation. Since it is well known that typical gas phase catalysts for PP homo- and copolymers are relatively sensitive to thermal deactivation,24,25 the activity improvement was attributed to avoidance of deactivation owing to the influence of the hydrocarbon. Most likely it would slow the diffusion of monomer to the active sites and cause the polymerization to occur more slowly, akin to an “in situ” prepolymerization step. These hypotheses need to be investigated further.
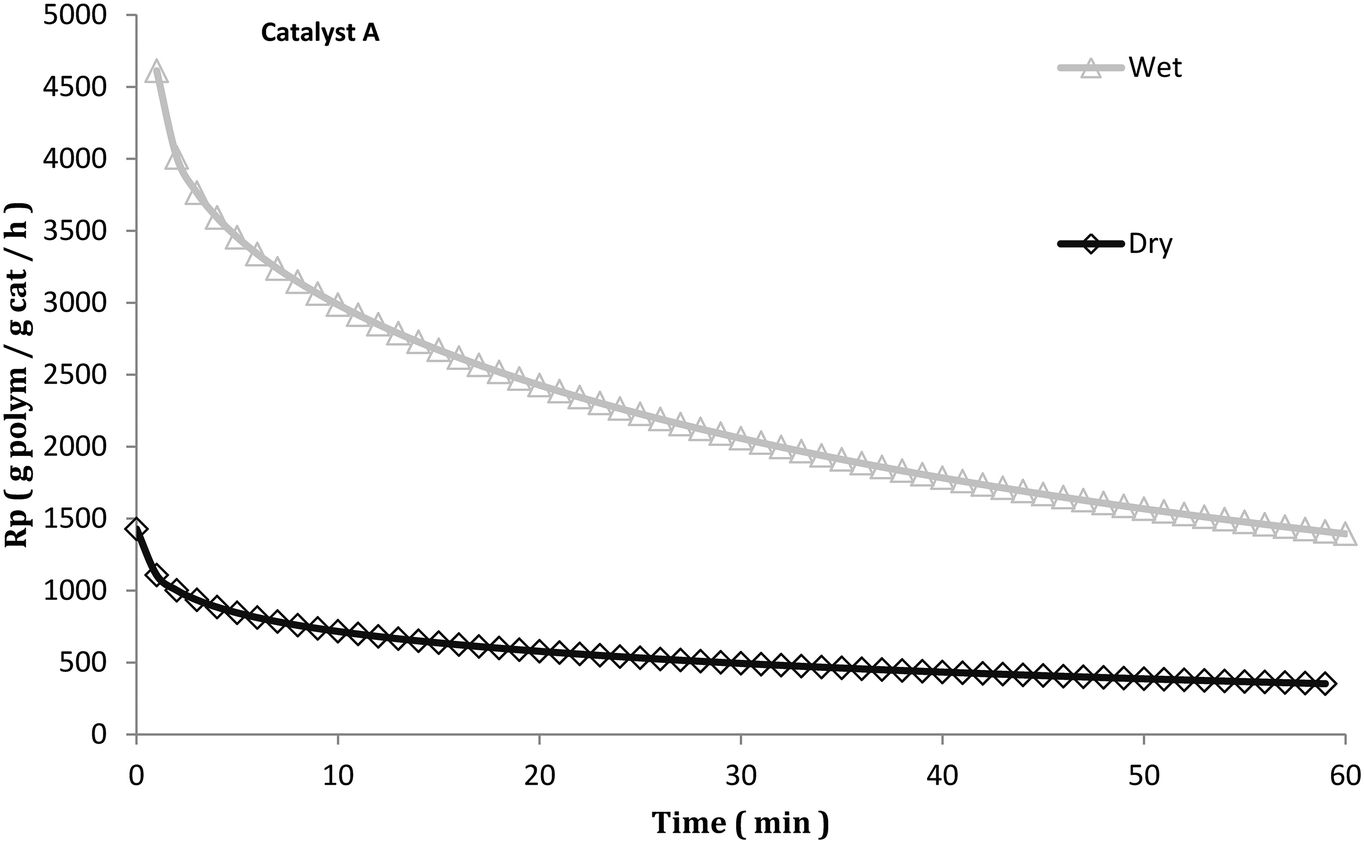 | ||
| Fig. 1 Instantaneous rate of gas propylene polymerization at 70 °C with 5 bar propylene with the injection of A catalyst particles (same as used in this publication) suspended in mineral oil (wet) and coarse salt (dry). Reproduced with permission.21 Copyright 2016, John Wiley and Sons. | ||
 | ||
| Fig. 2 SEM micrographs of polymer particles produced using prepolymerized ZN catalyst A (same as used in this publication) and injected pre-wetted with oil (left) and dry (right). Reproduced with permission.23 Copyright 2016, John Wiley and Sons. | ||
Since it is well known that the initial instants of a polymerization on supported ZN catalyst are extremely important in the development of the polymer particles2,3,6 and that standard reactors such as the semi-batch stirred bed reactors used to polymerize olefins at fixed pressures are ill-adapted to studying very short reaction times, a gas phase stopped flow reactor was developed by our group to study the initial instants of gas phase ethylene polymerization.26–30 Here, this same tool is used for the first time to investigate systematically the polymerization of propylene. It has also been found that the use of an approximate energy balance around the stopped flow reactor can give a better estimate of the relationship between the polymerization rate and the particle temperature than is possible in a standard stirred powder bed reactor.29,30 This approach is used to investigate the temperature rise of pre-wetted and dry ZN catalysts during the initial moments of propylene polymerization. This will allow us to determine whether temperature excursions at short times can be correlated with lower long time yields. Furthermore, analyses of the resultant polymers explain the morphological and activity differences observed.
2. Experimental section
2.1. Chemicals
Commercial fourth generation MgCl2 supported TiCl4 Ziegler–Natta catalyst with a titanium content of 2.02% and a narrow particle size distribution (shown in Fig. 3) was used for polymerization. Triethylaluminium (Witco, Germany) and dicyclopentyldimethoxysilane (DCPDMS) were used as cocatalyst and external electron donor, respectively. | ||
| Fig. 3 Differential (solid line) and integral (dashed line) particle size distributions of the precatalyst used in this work. | ||
All heptane used was pre-treated on 3 Å molecular sieves. Mineral oil Primol 352,22 a medicinal grade white mineral oil, was purchased from Fisher Scientifics. It is an inert and protective carrier commonly used industrially for polypropylene processes, and its main properties are summarized in Table 1. Before use, it was degassed cold under vacuum to eliminate any trace of oxygen and then kept under argon atmosphere at room temperature. Sodium chloride (purity >99.5%) was obtained from Acros Organics, France, and then dried under vacuum (10−3 bar) at 200 °C for 5 hours and kept under argon atmosphere. Propylene and propane with a minimum purity of 99.5% were purchased from Air Liquide (France). Propylene was purified using a three stage system of columns before use: the first column filled with BASF R3-16 catalyst (CuO on alumina), the second one filled with molecular sieves (13X, 3A, Sigma-Aldrich), and the last one filled with Selexsorb COS (Alcoa). Argon provided by Air Liquide, France, with a minimum purity of 99.5%, was used in order to keep the reaction free of oxygen.
| Oil Primol 352 | |
|---|---|
| Appearance | Clear and bright |
| Density @ 20 °C, kg m−3 | 865 |
| Flash point, °C | 240 |
| Average molecular weight | 480 |
| Paraffinic/naphthenic/aromatic | 66/34/0 |
2.2. Gas-phase stopped-flow polymerization
A schematic view of the 3.14 mL fixed packed bed stopped flow reactor system is shown in Fig. 4. The reactor and feed lines are plunged into a water bath to set the bed and feed temperatures. The inlet and outlet gas phase temperatures are measured with thermocouples. The polymerization occurs when a pulse of gas from the second ballast in Fig. 4 is fed to the reactor for a fixed time (possible range 0.01 to 100 seconds). At the end of the pre-set polymerization time, the reactor is simultaneously vented and fed with CO2. For a more detailed description of the system and reactor technology, the reader is referred to previous publications.29,30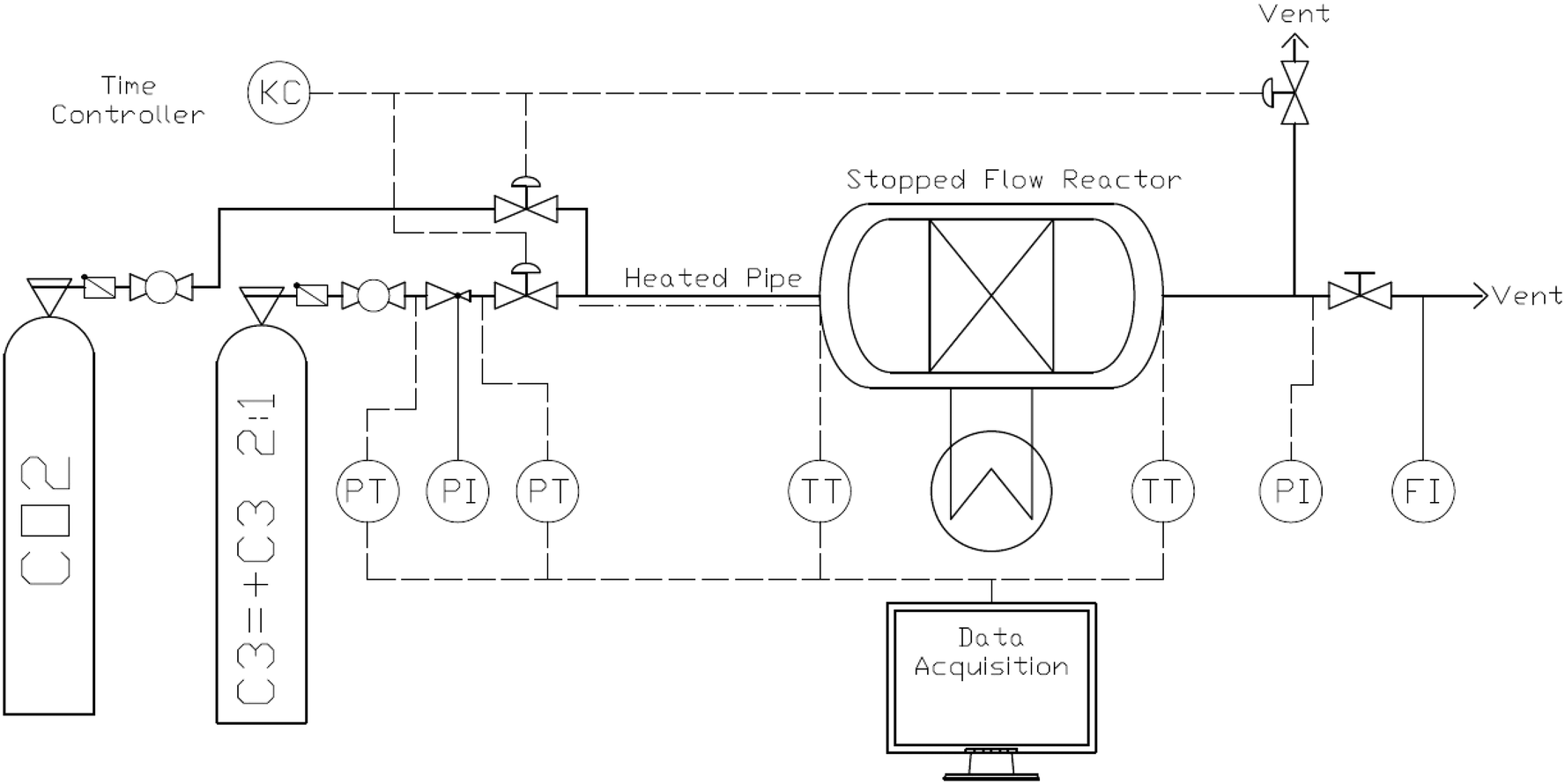 | ||
| Fig. 4 Stopped-flow reactor experimental setup and control system. | ||
Reactors were prepared in a glovebox (Jacomex, France) under argon atmosphere. Every reactor carried 15 mg of ZN precatalyst, either as a powder (from now on this is referred to as “dry method”) or 15 mg of ZN precatalyst suspended in 250 μl of mineral oil (from now on this is referred to as “wet method”). In both cases they were mixed and packed with 2 g of finely divided NaCl salt, consisting of single cubes of 5–10 μm that are slightly agglomerated to give a final single object of around 30 μm.30 TEA (1 M, heptane solution) was added to the catalyst/salt bed in an Al/Ti molar ratio of 26. As compared to previous papers where reactions were done in a typical semibatch polymerization reactor of 2.5 L,21,23 the Al/Ti ratio was diminished to avoid over-reduction, since a smaller volume will boost the local TEA concentration. The Si/Ti molar ratio was kept at 10 (DCPDMS, 0.42 M, heptane solution). Heptane from donor and cocatalyst solutions was removed by applying a vacuum to the solid mixture containing the activated catalyst before the reaction was run. Otherwise, heptane and its vaporisation would affect the measurements. All reactions were run at 4.5 bar (gauge pressure) of a propylene–propane mixture (33% propane) at 70 °C. The propylene was diluted with propane to improve heat transfer conditions inside the packed bed. Polymerizations were run at 1, 5, 10, 20, 40 and 75 seconds, always followed by degassing for 5 seconds and then 30 seconds of a CO2 stream to halt the reaction.
Reaction yield is measured by weighing the reactor before and after polymerization. It was considered that there were no losses of mineral oil during the process because of its properties. Afterwards, salt and most of the mineral oil not mixed within the polymer was washed away with distilled water and the polymer was recovered for further analysis.
2.3. Slurry phase semibatch polymerization
Slurry phase polymerizations were conducted in a semi-batch mode in a 2.5 L spherical stirred-bed reactor (turbosphere) equipped with injection valves for the catalyst and monomer feeds. During reaction the system was kept under isothermal conditions. A pressure reducer was used to maintain constant pressure. The experimental setup used to carry out the polymerization reactions was similar to the one described by Kittilsen and McKenna.31Before every experiment, the reactor was heated to 60 °C and kept under vacuum for 1 h. The reactor was purged with argon before each experiment. Once the reactor had reached the desired initial temperature, 500 mL of heptane were transferred under argon. TEA and DCPDMS were used as cocatalyst and external Lewis base, respectively, setting the Al/Ti and Si/Ti ratios to 260 and 20, respectively. Subsequently, the required amount of 1 M TEA/heptane mixture and the donor/heptane mixture (0.42 M) was injected under argon and agitation (∼400 rpm) was started. The flask containing the precatalyst suspension in mineral oil (60 mg mL−1) was pre-heated to the desired temperature (25, 80, 100 and 120 °C) by plunging it in a thermostated silicon oil bath. To ensure that the entire precatalyst batch reached the desired temperature, it was heated for at least 10 min. Note that the catalyst suspension was heated to the next temperature only if all the experiments at a determined injection temperature were finished. About 10 minutes after TEA injection, 167 μL of the precatalyst suspension (10 mg of precatalyst) were injected under propylene. Therefore, the precatalyst was not pre-activated before propylene injection. This method was used to ensure that reaction starts at injection temperature. The reaction was conducted at 8 bar propylene and 2% hydrogen for 1 h at 60 °C. For activity measurements, every five minutes the reactor inlet was closed and the time for a determined pressure difference to drop was measured. Pressure drop was interpreted using the Soave–Redlich–Kwong equation of state (SRK-EoS) to convert it to grams of PP. Finally, the integral of the instantaneous reaction rate was fitted to the real obtained polymer yield.
Prepolymerization was not performed. Nevertheless, note that the reaction is started by reactor pressurization. This takes around 60 seconds, making a softer initiation compared to industrial conditions where monomer is already present.
Once the reaction is finished the monomer inlet is closed and the reactor is rapidly cooled and depressurized. The polymer is recovered by filtration, and the final powder is dried overnight to remove the remaining traces of heptane.
2.4. Polymer characterization
The crystallinity and melting temperatures of the polymer samples were measured by DSC (DSC 1 by Mettler Toledo). Two heating steps were performed from −80 to 200 °C at a heating rate of 10 °C min−1 separated by a cooling from 200 to −80 °C at a rate of 10 °C min−1. The crystallinity of the samples was calculated using a value of 207 J g−1 for a full crystalline polypropylene.32 We considered data obtained during the second heating step.The molecular weight distributions (MWDs) of polymer samples were characterized using HT-SEC (Viscotek-Malvern Instruments). The system was equipped with three detectors (a refractometer, a viscometer and a light scattering detector) and three columns (PSS POLEFIN analytical 1000–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–1000000 Å). Analyses were performed in 1,2,4-trichlorobenzene stabilized with butylhydroxytoluol at 0.2 g L−1 at 150 °C at a flow rate of 1 mL min−1. The molecular weight distributions were calculated by a triple detection method. Refractometer, viscometer, and light scattering signals were used in order to erase possible artifacts.
000–1000000 Å). Analyses were performed in 1,2,4-trichlorobenzene stabilized with butylhydroxytoluol at 0.2 g L−1 at 150 °C at a flow rate of 1 mL min−1. The molecular weight distributions were calculated by a triple detection method. Refractometer, viscometer, and light scattering signals were used in order to erase possible artifacts.
Nuclear magnetic resonance (NMR) characterization was performed on a Bruker Avance III 400 spectrometer operating at 100.6 MHz for 13C NMR. 13C NMR spectra were obtained with a 10 mm PA-SEX probe at 393 K. A volume mixture of o-C6H4Cl2 (90%) and p-C6D4Cl2 (10%) was used as solvent. Chemical shifts were measured in ppm using the resonance of polypropylene (Tββ, m-m-PPP) as internal reference at 28.65 ppm.33
3. Results and discussion
3.1. Average calculated solid temperature
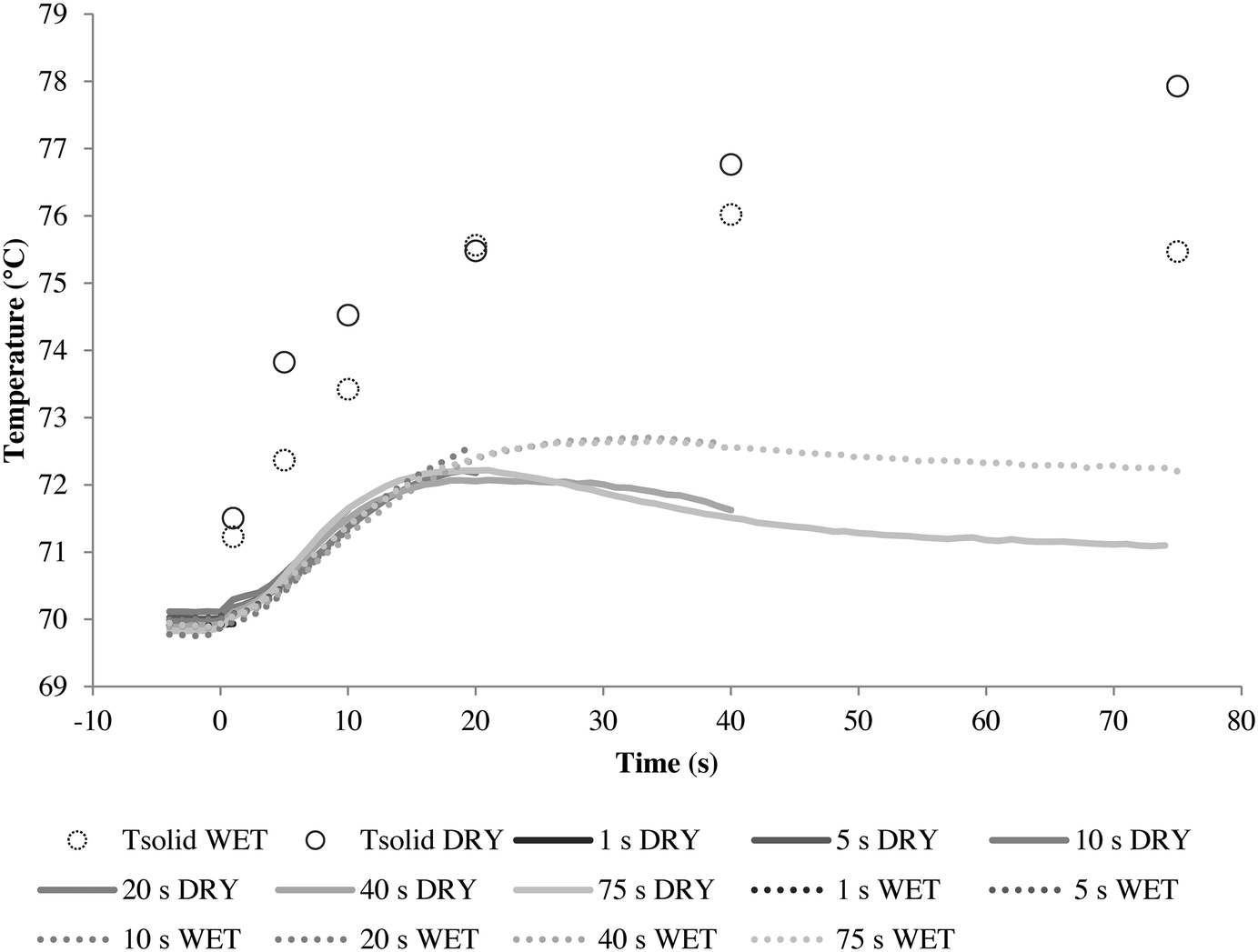
A series of runs using different injection conditions and a relative gas/particle velocity on the order of 8.75 cm s−1 were carried out to investigate the impact of wet vs. dry catalyst injection at short times. To ensure that the results were reproducible, all polymerizations were repeated at least three times under a given set of operating conditions. The results are shown in Fig. 5, where one can see the outlet gas temperature profiles as a function of time (the inlet temperature is constant at 70 °C) and estimates of the temperature of the solid particles in the bed at 5, 10, 20, 40 and 75 seconds of reaction versus time for both dry and wet methods. The average solid temperature of the bed (catalyst and salt, Tsolid) at the end of each reaction is calculated with eqn (1), derived by Tioni et al.29,30 Here, T0 is the initial solid temperature (70 °C) and msolid and Cp,solid are the mass and heat capacity of the bed, respectively.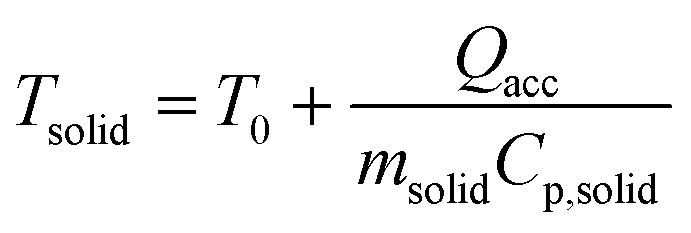 | (1) |
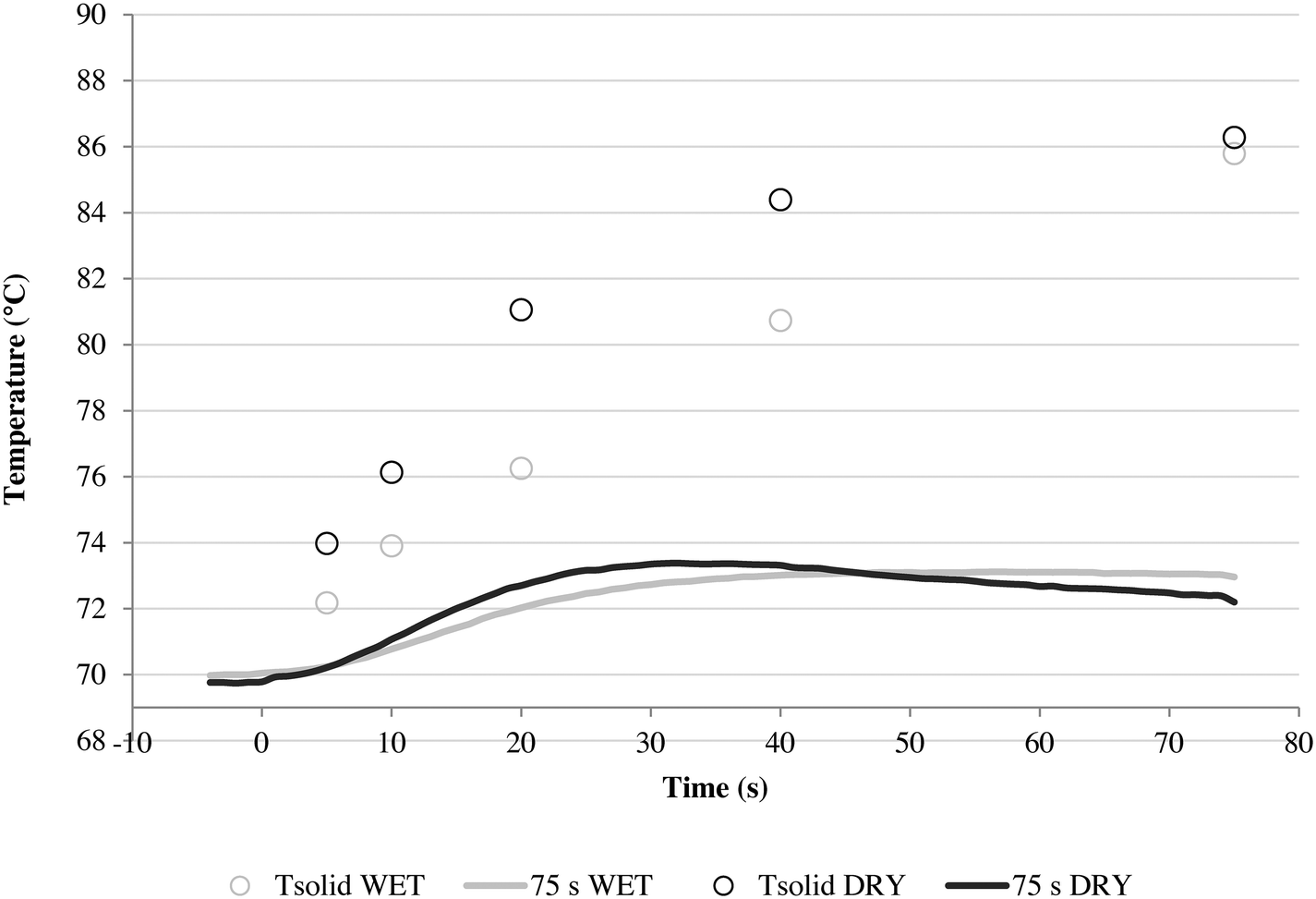 | ||
| Fig. 5 Outlet gas temperature profile and average calculated solid temperature (Tsolid) for 5, 10, 20, 40 and 75 s propylene homopolymerization using a linear gas velocity of 8.75 cm s−1 through the packed bed, at 70 °C and 4.5 bar propylene/propane 2:1 molar mixture using supported ZN catalyst in wet and dry status. All polymerizations were done with Al/Ti and Si/Ti ratios of 26 and 10, respectively. | ||
Note that eqn (1) represents a lower limit of the temperature of the catalyst particles, especially at very short times, since it assumes that the entire mass of the bed reaches the same average temperature almost instantaneously. It is entirely possible that the catalyst particles themselves are hotter than the salt particles and in particular for the first few seconds of the reaction. Nevertheless, this equation certainly gives one a better picture of the state of the catalyst particles than simply measuring the gas phase temperatures.
The wet and dry outlet temperature profiles shown in Fig. 5 are quite similar, and if one monitored only the gas phase temperature it might be concluded that the reactions are similar for both methods. For the wet case, the outlet gas temperature increased by approximately 3 °C within 50 s and remained constant, and in the dry case it increased by 3.5 °C in 25 s and afterwards started to decay. If we consider the solid temperatures, the story is somewhat different as Tsolid is always higher than the observed outlet temperature for both cases. However, the packed bed (and in consequence catalyst particles) from the series of dry polymerizations overheats more than the runs using the wet protocol, which shows a slower, steadier increase of temperature. This is logically correlated with higher initial activities (5, 10, 20 and 40 s) for the dry mode, as shown in Fig. 6.
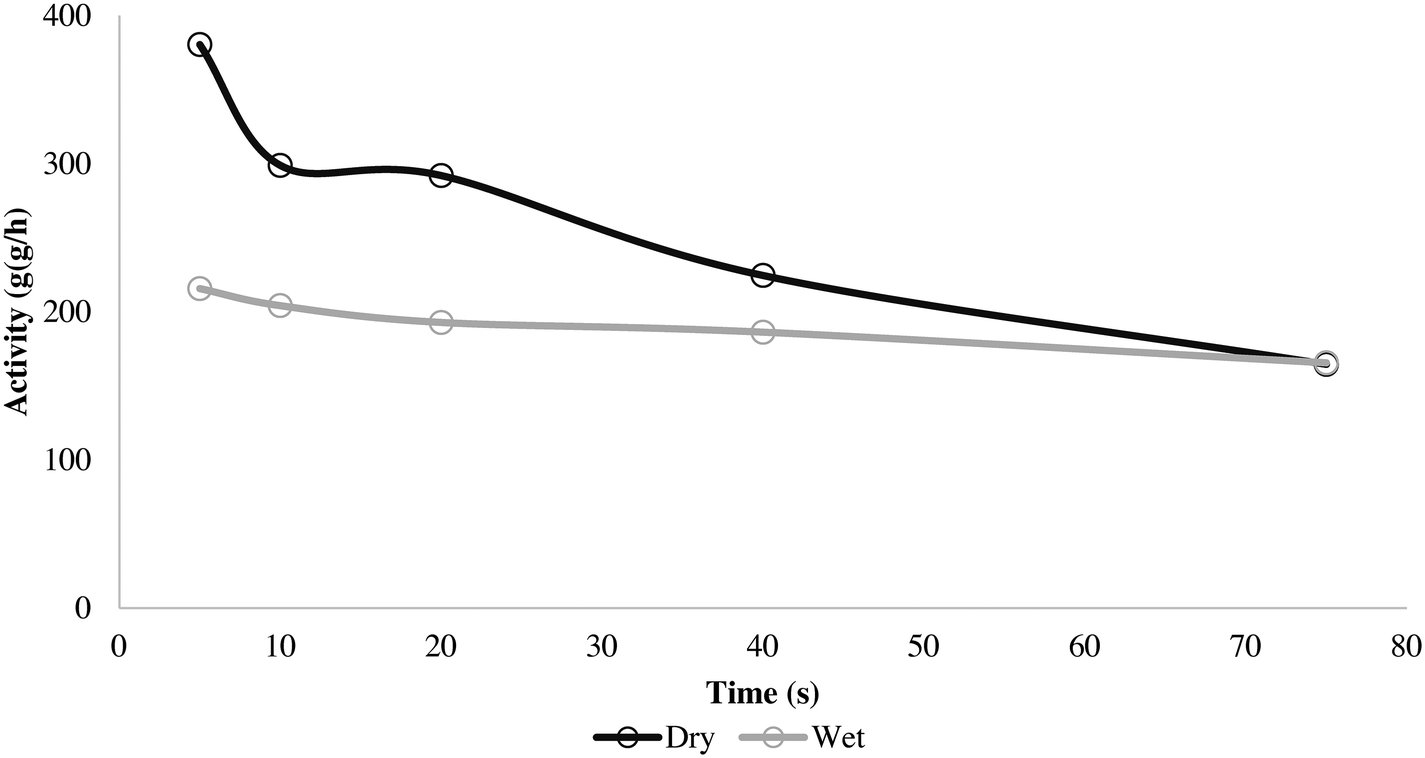 | ||
| Fig. 6 Activity for 5, 10, 20, 40 and 75 s propylene homopolymerization at 70 °C and 4.5 bar propylene/propane 2:1 molar mixture using supported ZN catalyst in wet and dry status. | ||
It is known that PP catalysts are somewhat more sensitive to thermal deactivation than those used in PE processes and so will lose some activity at higher temperatures, which might be the cause of the faster deactivation seen in the dry instantaneous rate profile from Fig. 6.
In order to investigate the possibility that the deactivation is due to overheating, a series of semibatch experiments was planned. Precatalyst was pre-heated to a determined temperature (in Table 2 referred to as catalyst injection temperature) and then slurry phase polymerization was conducted at 60 °C in the turbosphere reactor. It is not unreasonable to assume that slurry phase polymerizations provide much better heat transfer conditions as compared to gas phase ones. Therefore, overheating of the catalyst will be much less significant in slurry than in gas phase reactions, and any difference in the final polymer yield can be attributed to the applied precatalyst pre-heating.
| Injection temperature (°C) | Run | Productivity (g) |
|---|---|---|
| a All reactions were conducted at 60 °C, with 500 mL of heptane, and 8, 0.2, and 1 bar partial pressure of propylene, hydrogen and argon, respectively. | ||
| 25 | 1 | 31 |
| 2 | 27.2 | |
| 80 | 1 | 25.7 |
| 2 | 17 | |
| 100 | 1 | 21.5 |
| 2 | 15.5 | |
| 120 | 1 | 9.5 |
| 2 | 7.2 | |
To ensure that the results were reproducible, all polymerizations were repeated twice under a given set of operating conditions. Table 2 and Fig. 7 show the resulting yields for every reaction performed and the averaged activity profiles for each set of conditions, respectively.
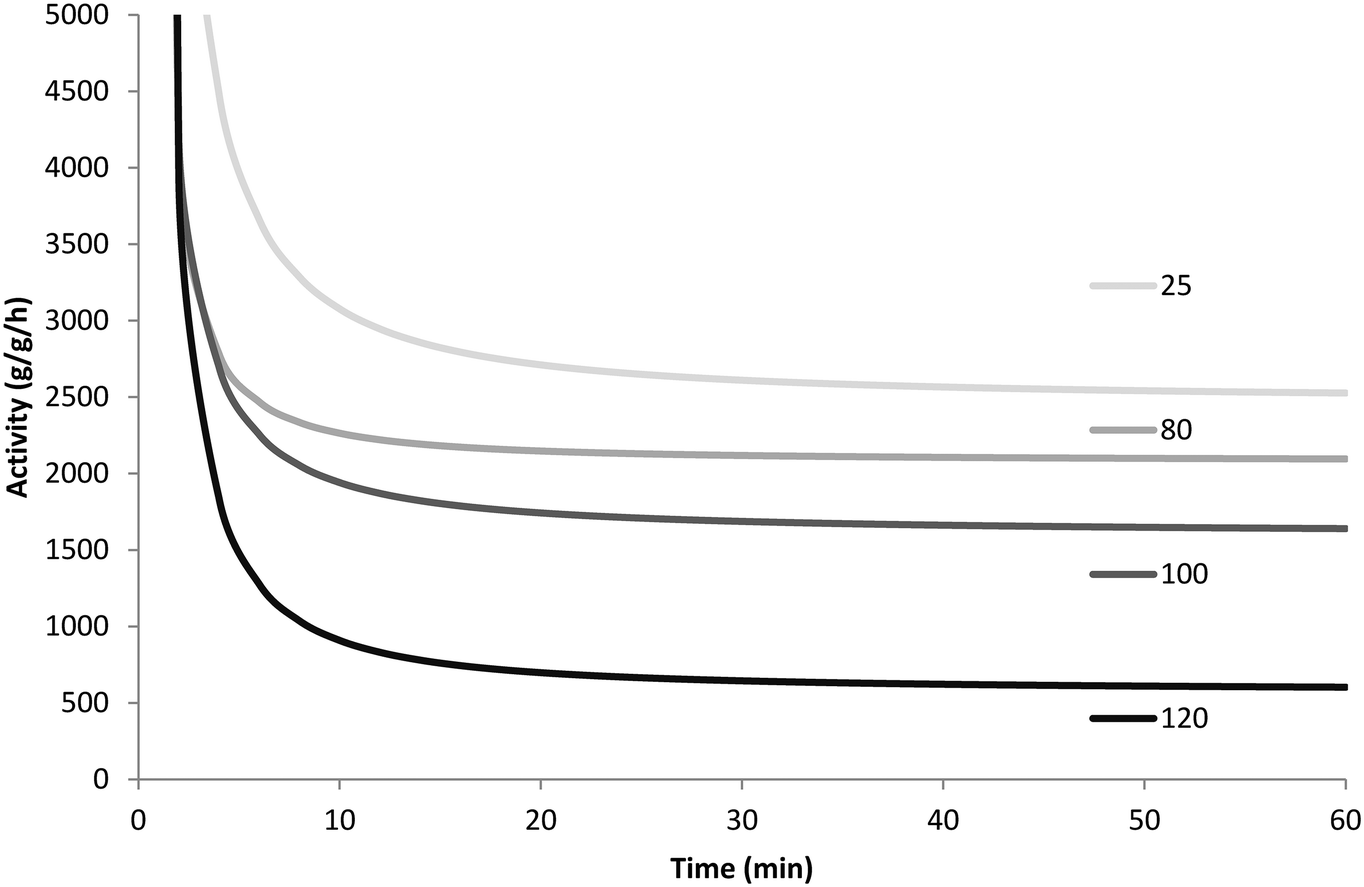 | ||
| Fig. 7 Instantaneous rate of propylene polymerization at 60 °C with 8 bar propylene in slurry phase injecting precatalyst at 25, 80, 100 and 120 °C. | ||
Since the catalyst is injected into the semi-batch reactor before the propylene, there is a short period of time during which propylene from the feed ballast is consumed by both the polymerization and the equilibration of the propylene–heptane mixture until heptane is saturated with propylene. Since the reaction rate is measured directly from the pressure drop in the ballast, this makes the initial activity appear higher than its true value, and this should be taken into consideration when interpreting the results shown in Fig. 7.
Based on the results from Fig. 7 and Table 2, it seems that the precatalyst can be thermally deactivated for propylene polymerization due to pre-heating. In general, the higher the temperature the precatalyst is treated at, the lower its productivity is. The precatalyst treated at 80 °C is less active and less productive than that prepared at room temperature, and a big drop in productivity is found with the increase of pre-heating temperature from 100 °C to 120 °C. If the thermal shock required to deactivate the catalyst does not need to be particularly long, these results could explain in large part the results obtained in Fig. 1 (and Fig. 5). The dry injection in Fig. 1 probably has a higher initial temperature than the wet injection curve. Care should be taken when trying to compare the rate curves in Fig. 1 and 5 since the experiments in Fig. 1 were run at 5 bar propylene rather than 3.3 (in Fig. 5), and while TEA was added to the seed bed in the stopped-flow experiments, none was added to the gas fed to the reactor. This means that TEA can be rapidly blown out of the bed and that the Al/Ti ratios are different in the 2 sets of experiments. Nevertheless, the results presented in Fig. 7 show that the catalyst is sensitive to overheating. Those in Fig. 5 show that the dry catalyst overheats more than the wet injected catalyst, and those in Fig. 1 show that the long-term activities of wet and dry catalyst are quite different.
As further proof, consider the results in Fig. 8 where the wet and dry series of runs were repeated but with a higher gas flow rate of 17.5 cm s−1, which gives better heat transfer conditions.
 | ||
| Fig. 8 Outlet gas temperature profile and average calculated solid temperature (Tsolid) for 1, 5, 10, 20, 40 and 75 s propylene homopolymerization using a linear gas velocity through the packed bed of 17.5 cm s−1 at 70 °C and 4.5 bar propylene/propane 2:1 molar mixture using supported ZN catalyst in wet and dry status. | ||
Here it can be seen that the wet and dry runs give very similar solid temperature profiles, especially during the initial 20 seconds. The dry method overheats slightly faster, but the difference is minimal. This is reflected in the activity curves seen in Fig. 9.
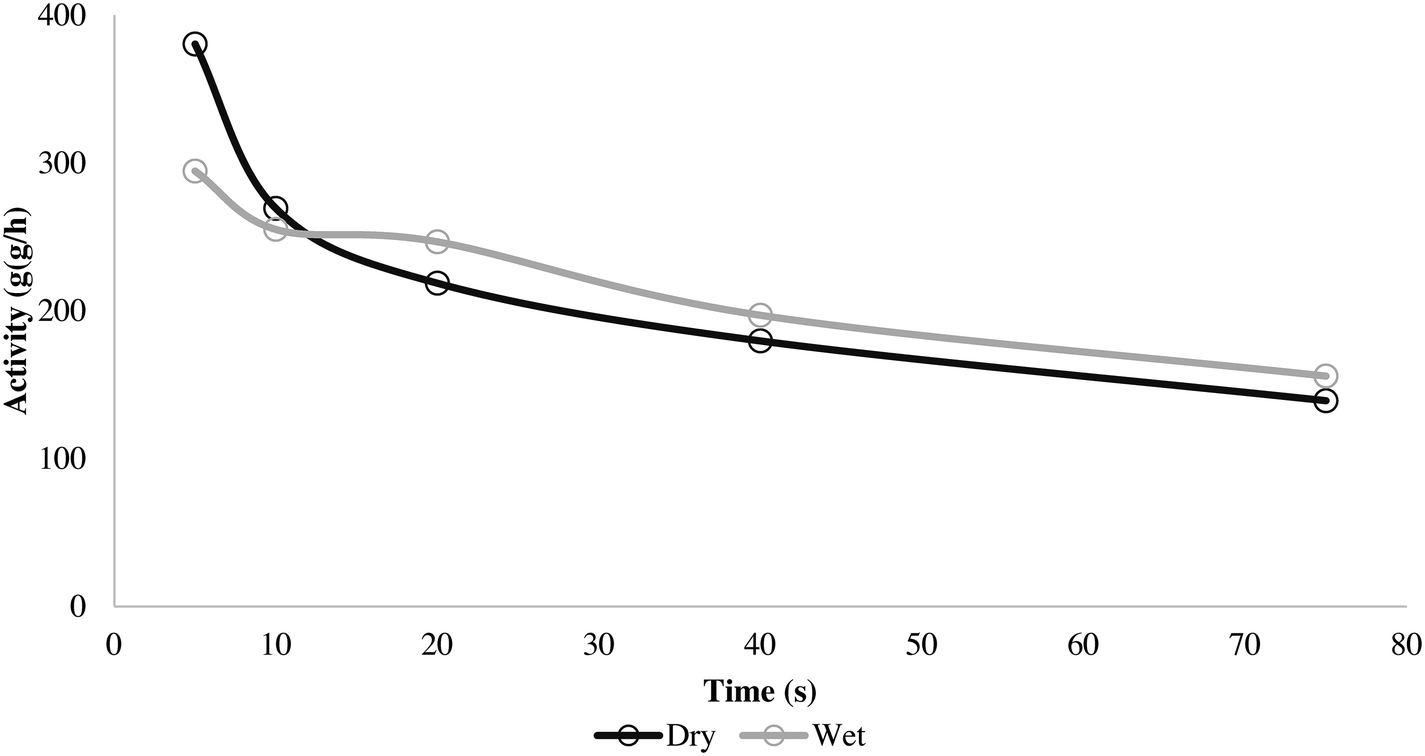 | ||
| Fig. 9 Activity for 5, 10, 20, 40 and 75 s propylene homopolymerization at 70 °C and 4.5 bar propylene/propane 2:1 molar mixture using supported ZN catalyst in wet and dry status. | ||
The PP microstructure does not appear to be affected by wet injection. NMR studies were done for the stopped flow samples, showing isotacticity values ranging from 96 to 98% mm in any case. Therefore, in this sense polymer properties are the same as those of common commercialized iPP, so withdrawn conclusions can be extrapolated.
Difference scanning calorimetry analyses were run on polymers obtained from Fig. 8 experiments. Fig. 10 and 11 show the obtained thermograms for crystallization and melting curves, respectively.
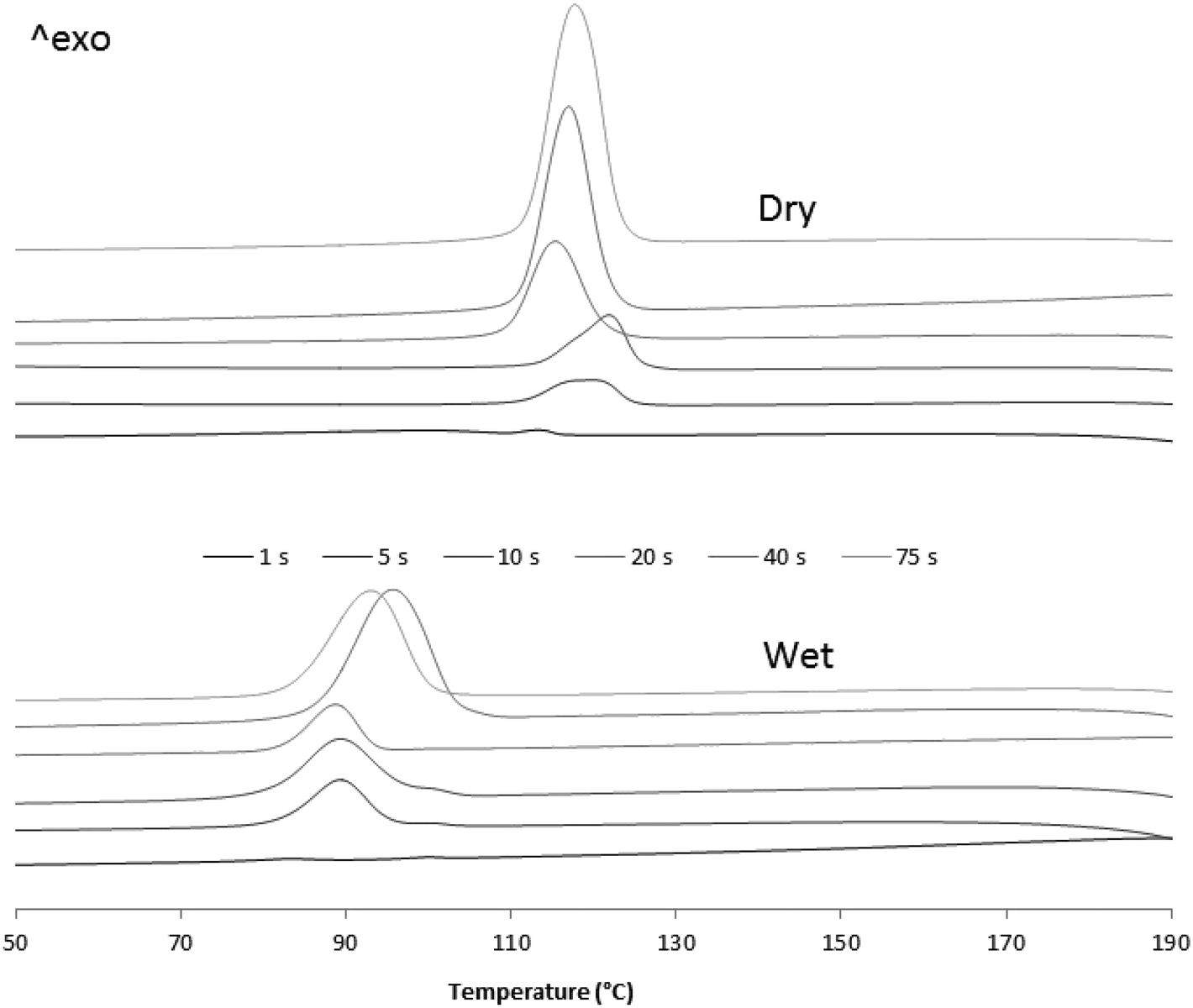 | ||
| Fig. 10 DSC crystallization curves of dry and wet samples from 1, 5, 10, 20, 40 and 75 seconds reaction time. | ||
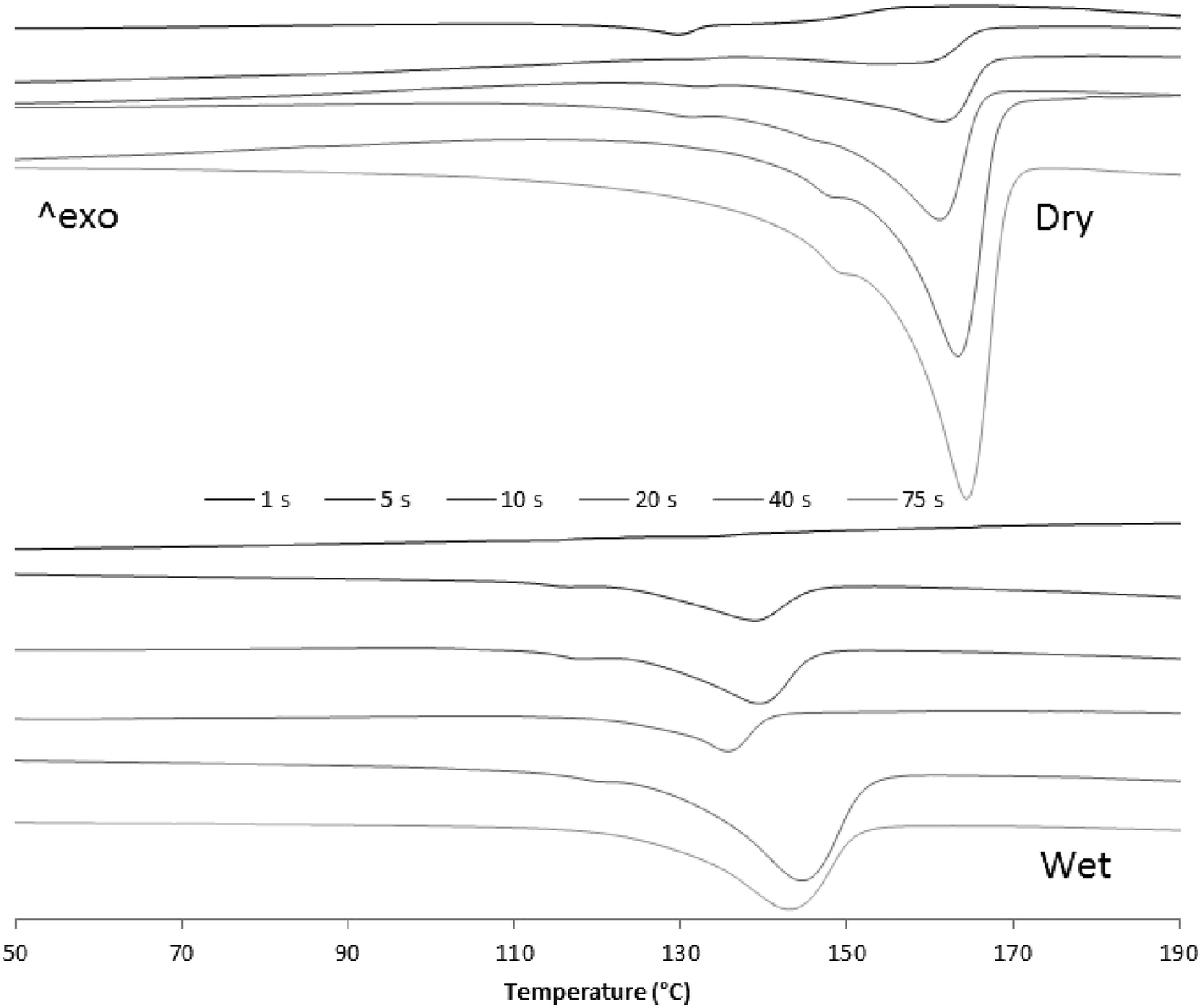 | ||
| Fig. 11 2nd DSC melting curves of dry and wet samples from 1, 5, 10, 20, 40 and 75 seconds reaction time. | ||
Fig. 10 and 11 show how, in both cases, crystallization and melting temperatures (Tc and Tm, respectively) gradually increase with reaction time, and Fig. 12 summarizes the evolution of the volume fraction of crystalline material for both runs. While polymer made with dry catalyst at 75 s nearly shows the same values as those typical for iPP (∼30% crystallinity, Tc of 117 °C and Tm of 165 °C), polymer resulting from wet injection is still far from there (∼13% crystallinity, Tc <100 °C and Tm <150 °C).
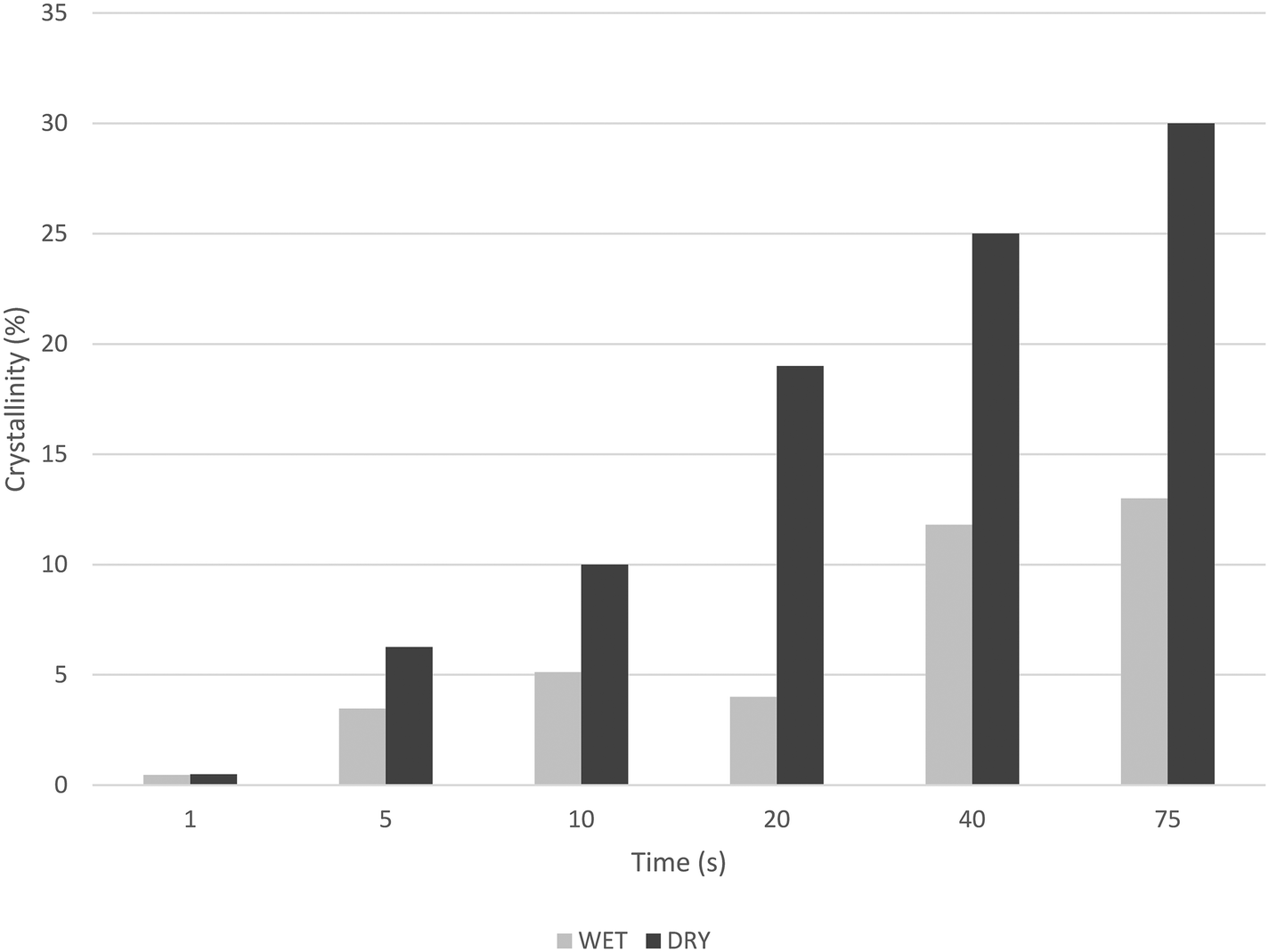 | ||
| Fig. 12 Evolution of crystallinity (2nd heating curve) with reaction time for PP powders produced by stopped flow reactor with dry or wet catalyst. | ||
In fact, it can be seen in Fig. 12 that the crystallinity of the nascent powders more than doubles in the dry samples than in the corresponding wet samples, essentially because the wet samples still contain some of the oil with which they were injected. (After washing them with water, PP particles contain about 60 wt% oil for 75 s reaction.) This means that not only does the oil impact the heat transfer conditions, but it also has a non-negligible impact on the physical properties of the polymer.
In previous articles from our group,21,23 it was observed that dry injection yields a broad PP particle size distribution mainly composed of flaky particles and fines, whereas the catalysts injected with the oil had a more regular structure (cf.Fig. 2). A more crystalline polymer structure is more brittle and less flexible and it would have more probabilities of breaking apart during catalyst fragmentation, leading to fine formation. On the contrary, the wet injection protocol leads to a more amorphous structure (composed of iPP and mineral oil) which would deform more easily during fragmentation and thus help to keep the spherical shape of the catalyst particle and avoid formation of fines during the polymerization reaction.
As noted before, looking at Fig. 10 and 11, the Tc and Tm of newly formed PP seem to be influenced by the oil surrounding catalyst particles. To better understand this phenomenon, the melting (2nd pass) and crystallization temperatures of a series of physical mixtures of an iPP homopolymer (semi-batch gas phase polymerization) with Primol 352 oil are shown in Fig. 13.
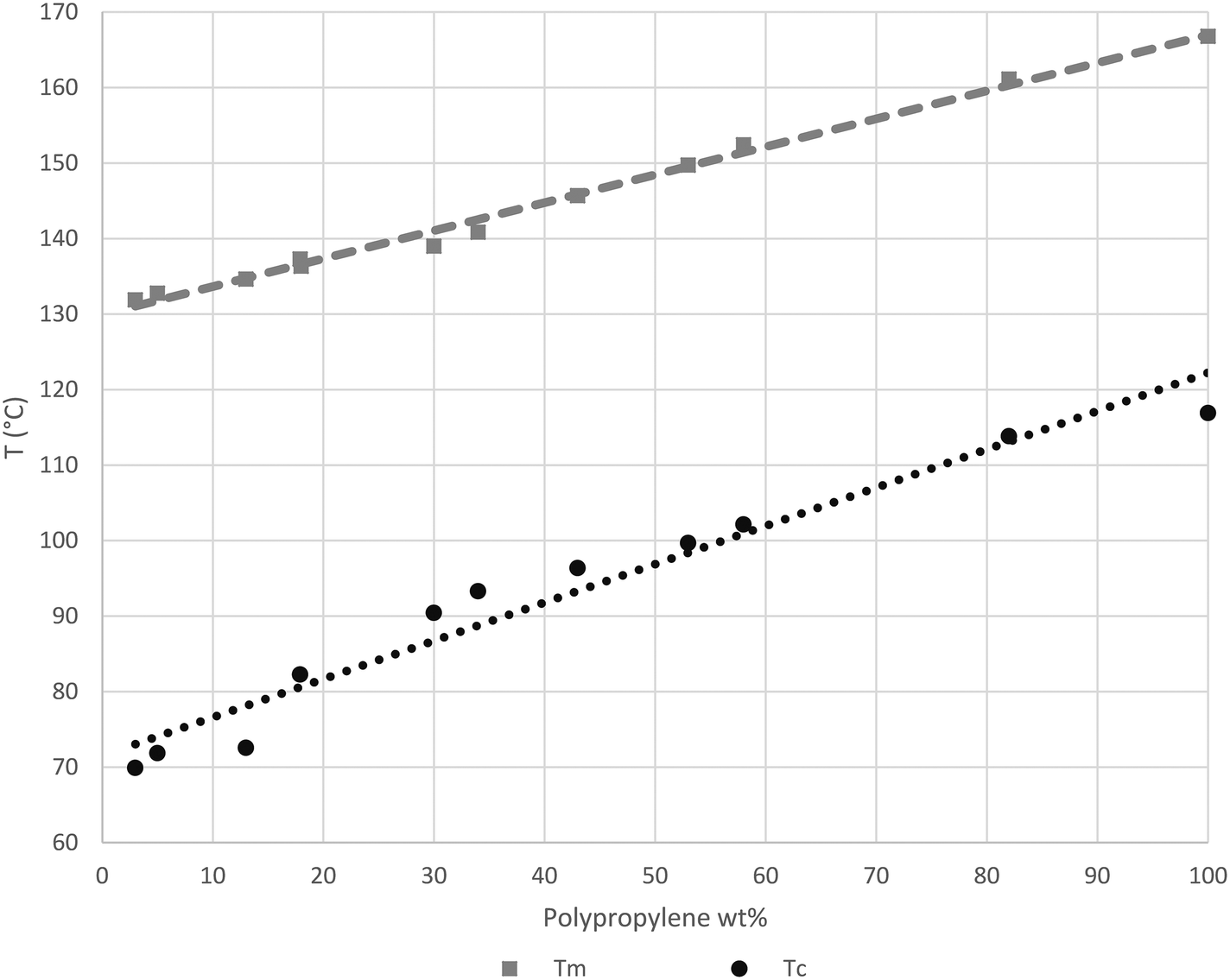 | ||
| Fig. 13 DSC study showing Tc and Tm of iPP depending on oil concentration. | ||
Fig. 13 shows how the Tc of iPP can decrease to as low as 70 °C when the oil/iPP ratio is high enough. During the first instants of the gas propylene polymerization runs in the current work we will most certainly be in the same situation at 70 °C and the initial PP chains will not able to crystallize if oil is present. In fact, in the current experiments, given that the temperature rises to 78–80 °C, the PP chains will not be able to crystallize until the polymer represents 20 weight percent of the mixture of polymer + oil. If one considers Fig. 14, it can be seen that after 75 seconds of polymerization, the PP particles formed in the presence of oil have a significantly more spherical form than those injected dry. It is quite probable that this can, at least in part, explain the observed improvement of polymer morphology and reduced level of fines during gas phase propylene polymerization claimed in industrial patents when the catalyst is injected with oil instead of dry.15,16
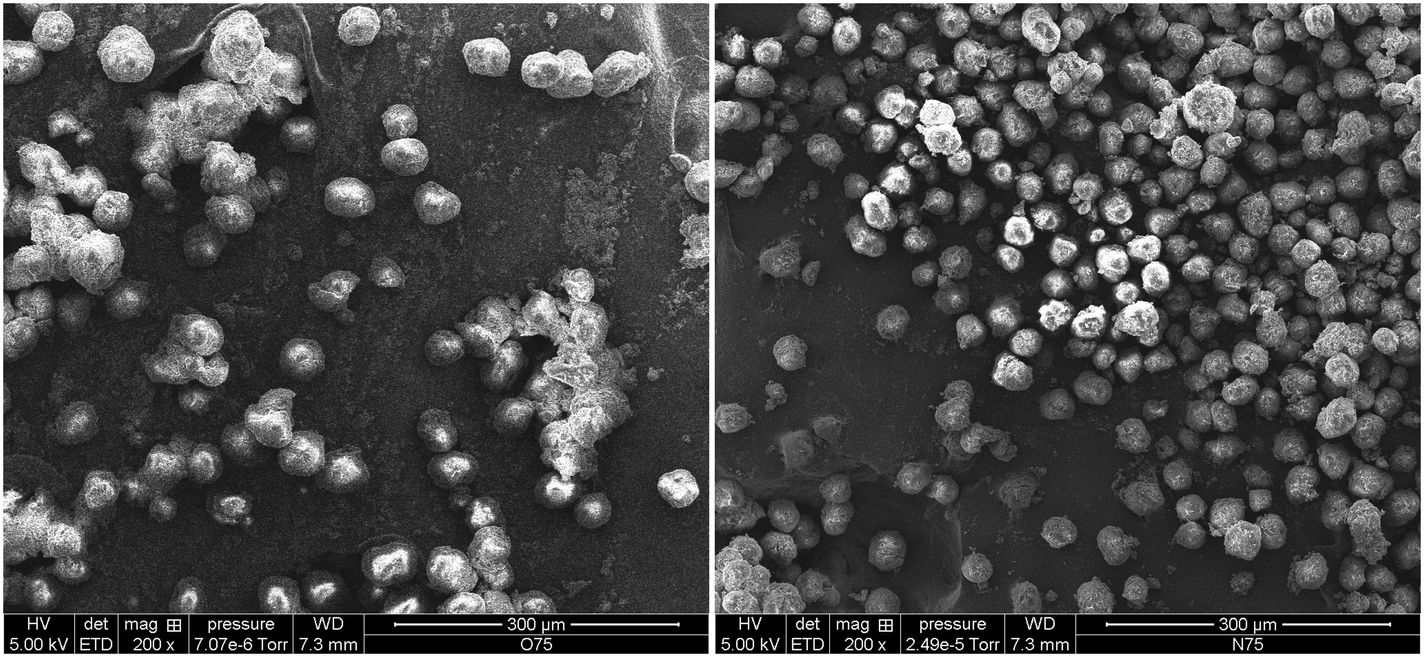 | ||
| Fig. 14 SEM micrographs of polymer particles produced using precatalyst injected wet (left) and dry (right) in a 75 s reaction at a linear gas velocity of 17.5 cm s−1. | ||
Analysis of the SEM micrographs (and additional similar images not shown here for the sake of brevity) allowed us to compute the average particle size and standard deviation of the wet and dry powders shown in Fig. 14. The values shown in Table 3 reveal that for a similar productivity (wet and dry runs were performed under favorable heat transfer conditions), the particles injected dry were smaller.
| Run | Diameter (max. dimension) | Productivity (g per gcatalyst) | |
|---|---|---|---|
| Average (μm) | Deviation (μm) | ||
| Wet 75 s | 52 | 7 | 3.2 |
| Dry 75 s | 37 | 6 | 2.9 |
Since productivities are similar for both cases, the observed trend of bigger particles seems to be attributable to the presence of the paraffinic oil, which would swell polymer particles (it is hard to say whether the bulk densities are different since there is so little polymer made during the experiments). It seems entirely possible that this difference will have also a great impact on other reaction aspects. Firstly, particles will overheat less since the larger particles have a higher surface area (by a factor of 2 after 75 seconds). The higher surface area for the same quantity of active sites per particle means that it is easier to evacuate energy during this critical stage of the polymerisation. In addition, activity will be enhanced due to a higher solubility of propylene, caused by two factors: (1) a higher amorphous fraction and (2) the co-solubility effect caused by the mineral oil (which has an average molecular weight of 480 g mol−1, as shown in Table 1).22 Hence, it is the combination of all these factors which makes the fact of pre-wetting catalyst particles with oil provoke an enhancement of polymerization rate and a better morphology control in the gas phase polymerization of propylene.
4. Conclusions
Gas phase propylene polymerizations were performed at times ranging from 1 to 75 s in a stopped flow reactor using a ZN precatalyst in two ways: dry or wetted with a commercially available paraffinic mineral oil. Online measurement of the inlet and outlet temperatures was used to get an approximation of the solid average temperature of the bed as a function of time.When heat transfer limitations exist, dry particles overheat more rapidly, whereas wet particles show a steadier, slower increase of temperature. This is explained as a result of a more direct exposure to the monomer of the dry particles causing a faster reaction start in the entire catalyst particle, whereas in the second case better particle temperature control is obtained because of the slower diffusion of the monomer in the oil blocked pores and the fact that the oil itself can also absorb some of the heat produced. In this way, thermal degradation is avoided, which explains the enhancement of propylene polymerization rate observed in ‘typical’ semibatch polymerizations. A series of runs where the precatalyst was heated at different temperatures clearly demonstrated that overheating the catalyst leads to thermal deactivation.
DSC analysis of the resulting polymers reveals that mineral oil provokes a decrease in crystallization temperature. A series of mixtures of an iPP homopolymer with the oil normally used for catalyst dosing were carried out, and it was found that the crystallization temperature of iPP can decrease to as low as 70 °C when the oil/iPP ratio is high enough. This absence of crystallinity during initial instants of life of the particles explains the better morphology observed by different patents and previous work from our group when mineral oil is present. Another consequence is that the solubility of propylene is enhanced due to polymer swelling, a higher amorphous fraction and the co-solubility effect due to mineral oil. This provokes a further enhancement of the polymerization rate in the gas phase polymerization of propylene.
Acknowledgements
The authors are grateful for financial support from the Dutch Polymer Institute (DPI), project #785, HIPSTER.References
- Expertise Beyond Borders - Business and Technology Management Advisory for the global Chemical and Materials Industries, http://www.jansz-ebb.com, (accessed August 2016).
- J. T. M. Pater, G. Weickert and W. P. M. V. Swaaij, Polymerization of liquid propylene with a fourth-generation Ziegler–Natta catalyst: Influence of temperature, hydrogen, monomer concentration, and prepolymerization method on powder morphology, J. Appl. Polym. Sci., 2003, 87(9), 1421–1435 CrossRef CAS.
- J. T. M. Pater, G. Weickert and W. P. M. van Swaaij, Propene bulk polymerization kinetics: Role of prepolymerization and hydrogen, AIChE J., 2003, 49(1), 180–193 CrossRef CAS.
- M. Monji, S. Abedi, S. Pourmahdian and F. A. Taromi, Effect of prepolymerization on propylene polymerization, J. Appl. Polym. Sci., 2009, 112(4), 1863–1867 CrossRef CAS.
- J. J. C. Samson, B. van Middelkoop, G. Weickert and K. R. Westerterp, Gas-phase polymerization of propylene with a highly active ziegler-natta catalyst, AIChE J., 1999, 45(7), 1548–1558 CrossRef CAS.
- G. B. Meier, G. Weickert and W. P. M. Van Swaaij, Gas-phase polymerization of propylene: Reaction kinetics and molecular weight distribution, J. Polym. Sci., Part A: Polym. Chem., 2001, 39(4), 500–513 CrossRef CAS.
- M. Smit, X. Zheng, J. Loos, J. C. Chadwick and C. E. Koning, Effects of propylene prepolymerization on ethylene/1-hexene and ethylene/1-octene copolymerization with an immobilized metallocene catalyst, J. Polym. Sci., Part A: Polym. Chem., 2006, 44(22), 6652–6657 CrossRef CAS.
- M. Ruff and C. Paulik, Controlling Polyolefin Properties by In-Reactor Blending: 2 Particle Design, Macromol. React. Eng., 2013, 7(2), 71–83 CrossRef CAS.
- J. B. P. Soares and A. E. Hamielec, Effect of hydrogen and of catalyst prepolymerization with propylene on the polymerization kinetics of ethylene with a non-supported heterogeneous Ziegler-Natta catalyst, Polymer, 1996, 37(20), 4599–4605 CrossRef CAS.
- C. Grein, K. Bernreiter, M. Kylmala, B. R. Kona and E. Hebesberger, Process for the Production of Propylene Random Copolymers for Injection Moulding Applications, EP2403883A1, 2012 Search PubMed.
- A. J. Cancelas, G. Hongmanee, T. F. L. McKenna, F. N. Andrade, M. Terano and T. Taniike, A Comparison of the Influence of Temperature During Slurry and Gas Phase Propylene Polymerization on Ziegler-Natta Catalyst, Macromol. Symp., 2016, 370(1), 41–51 CrossRef CAS.
- J. W. Shepard, J. L. Jezl, E. F. Peters and R. D. Hall, Divided Horizontal Reactor for the Vapor Phase Polymerization of Monomers at Different Hydrogen Levels, US3957448 (A), 1976 Search PubMed.
- J. L. Jezl and E. F. Peters, Horizontal Reactor for the Vapor Phase Polymerization of Monomers, US4101289A, 1978 Search PubMed.
- C.-H. Lin, J. F. Stevens, B. S. Tovrog and P. M. Rose, Method and Apparatus for Minimizing Polymer Agglomerate or Lump Formation in a Gas-Phase Polypropylene Polymerization Reactor, US4921919 (A), 1990 Search PubMed.
- B. Kimberley, G. Lacane and S. Mastroianni, Polymerisation Process, WO/2005/058978, 2005 Search PubMed.
- E. F. Peters, Catalyst Feeding System, US4610574A, 1986 Search PubMed.
- G. O. Michaels and M. J. Spangler, Process for Controlling Polymer Particle Size in Vapor Phase Polymerization, US4287327 (A), 1981 Search PubMed.
- D. A. Wright, J. R. Parrish, I. J. L. Swecker and M. G. Goode, Methods and Systems for Catalyst Delivery, US9023958 (B2), 2015 Search PubMed.
- T. Hans-Georg, R. Wolfgang, J. Theodor and P. Helmut, Polymerization of Propylene with Ziegler Catalysts in a Stirred Gas Phase Reactor, US3652527 (A), 1972 Search PubMed.
- J. L. Jezl, E. F. Peters, R. D. Hall and J. W. Shepard, Process for the Vapor Phase Polymerization of Monomers in a Horizontal, Quench-Cooled, Stirred-Bed Reactor Using Essentially Total off-Gas Recycle and Melt Finishing, US3965083 (A), 1976 Search PubMed.
- A. J. Cancelas, V. Monteil and T. F. L. McKenna, Influence of Activation Conditions on the Gas Phase Polymerisation of Propylene, Macromol. Symp., 2016, 360(1), 133–141 CrossRef CAS.
- Primol 352, http://www.exxonmobil.com/Denmark-English/Specialties/PDS/GLXXENSPCEMPrimol_352.aspx, (accessed August 2016).
- A. R. Martins, A. J. Cancelas and T. F. L. McKenna, A Study of the Gas Phase Polymerization of Propylene: The Impact of Catalyst Treatment, Injection Conditions and the Presence of Alkanes on Polymerization and Polymer Properties, Macromol. React. Eng., 2016 DOI:10.1002/mren.201600011.
- R. Spitz, C. Bobichon and A. Guyot, Synthesis of polypropylene with improved MgCl2-supported Ziegler-Natta catalysts, including silane compounds as external bases, Makromol. Chem., 1989, 190(4), 707–716 CrossRef CAS.
- G. Guastalla and U. Giannini, The influence of hydrogen on the polymerization of propylene and ethylene with an MgCl2 supported catalyst, Makromol. Chem., Rapid Commun., 1983, 4(8), 519–527 CrossRef CAS.
- F. M. Silva, J. P. Broyer, C. Novat, E. L. Lima, J. C. Pinto and T. F. McKenna, Investigation of Catalyst Fragmentation in Gas-Phase Olefin Polymerisation: A Novel Short Stop Reactor, Macromol. Rapid Commun., 2005, 26(23), 1846–1853 CrossRef CAS.
- F. Machado, E. L. Lima, J. C. Pinto and T. F. McKenna, An experimental study on the early stages of gas-phase olefin polymerizations using supported Ziegler-Natta and metallocene catalysts, Polym. Eng. Sci., 2011, 51(2), 302–310 CAS.
- B. Olalla, J.-P. Broyer and T. F. L. McKenna, Heat Transfer and Nascent Polymerisation of Olefins on Supported Catalysts, Macromol. Symp., 2008, 271(1), 1–7 CrossRef CAS.
- E. Tioni, J. P. Broyer, V. Monteil and T. McKenna, Influence of Reaction Conditions on Catalyst Behavior during the Early Stages of Gas Phase Ethylene Homo- and Copolymerization, Ind. Eng. Chem. Res., 2012, 51(45), 14673–14684 CrossRef CAS.
- E. Tioni, R. Spitz, J. P. Broyer, V. Monteil and T. McKenna, Packed-bed reactor for short time gas phase olefin polymerization: Heat transfer study and reactor optimization, AIChE J., 2012, 58(1), 256–267 CrossRef CAS.
- P. Kittilsen and T. F. McKenna, Study of the kinetics, mass transfer, and particle morphology in the production of high-impact polypropylene, J. Appl. Polym. Sci., 2001, 82(5), 1047–1060 CrossRef CAS.
- H.-S. Bu, S. Z. D. Cheng and B. Wunderlich, Addendum to the thermal properties of polypropylene, Makromol. Chem., Rapid Commun., 1988, 9(2), 75–77 CrossRef CAS.
- P. Masson, M.-F. Llauro-darricades, R. Spitz and H. N. Cheng, A Study of the Dispersity of Catalytic Sites in Ethylene/Propylene Copolymerization Through Computer-Assisted 13C NMR Analysis, Int. J. Polym. Anal. Charact., 1996, 2(4), 379–393 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |