
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of spiro-3H-indazoles via 1,3-dipolar cycloaddition of arynes with 6-diazocyclohex-2-en-1-one derivatives and fused-2H-indazoles by subsequent rearrangement†
Bin Cheng *a,
Bian Baoa,
Bing Zua,
Xiaoguang Duana,
Shengguo Duana,
Yun Lia and
Hongbin Zhai*abc
*a,
Bian Baoa,
Bing Zua,
Xiaoguang Duana,
Shengguo Duana,
Yun Lia and
Hongbin Zhai*abc
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: chengb@lzu.edu.cn
bKey Laboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen 518055, China. E-mail: zhaihb@pkusz.edu.cn
cCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
First published on 24th November 2017
Abstract
A route to rare spiro-3H-indazoles bearing a carbonyl group adjacent to the spirocyclic quarternary carbon via 1,3-dipolar cycloaddition reaction of arynes with 6-diazocyclohex-2-en-1-one derivatives under mild conditions has been developed. Further transformation of these unique spiro-3H-indazoles via an acid- or heat-mediated rearrangement to fused-2H-indazoles and an interesting reduction/ring-opening/reduction sequence are also described.
Introduction
Since o-(trimethylsilyl)aryl triflate was introduced as a new aryne precursor,1 aryne chemistry has enjoyed impressive advances over the last decades.2 Among them, the 1,3-dipolar cycloaddition reaction of arynes with dipoles as an efficient synthetic method to construct heterocycles has been extensively studied. However, to use diazo compounds as the dipoles, these accomplishments mainly dealt with open-chain diazo substrates,3,4 and there were seldom reports involving cyclic diazo compounds.5 It is well-known that ordinary small molecule diazo compounds, like diazomethane, are highly unstable, dangerous and potentially explosive. Thus, an electron-withdrawing group (e.g., carbonyl group) conjugated to the diazo functional group is usually introduced to make them relatively stable. However, using this type of diazo substrate to construct spiroindazoles would lead to a carbonyl group adjacent to the spirocyclic quarternary carbon, a skeleton liable to undergo rearrangement via acyl migration.4,6,7 Thus, this is sometimes a dilemma. To address this issue, Shi8 and Moses9 adopted safe surrogates of diazo compounds, e.g., N-tosylhydrazone and hydrazonyl chloride, to synthesize indazoles via dipolar cycloaddition, but their methods were only applied to the synthesis of non-spiroindazoles. Recently, we disclosed the first isolated spiro[indazole-3,3′-indolin]-2′-ones via 1,3-dipolar cycloaddition reaction from arynes and 3-diazoindolin-2-ones (Scheme 1, eqn (a)).10 We wondered whether ordinary cyclic diazo compounds could also realize the similar transformation to afford spiroindazole through slightly tuning substrate structure. In this regard, there was only one successful spiroindazole example documented in literature6 besides our previous report, although great efforts had been devoted to tackle this issue. For example, Shechter explored the reaction of diazo cyclohexone with diazotized anthranilic acid, and a dimer was obtained as the major product in low yield (eqn (b)).6 Larock's group tried in vain to prepare a spiroindazole from 2-diazocyclohexane-1,3-dione, as there was no reaction detected in their system (eqn (c)).4 Given impressive bioactivities and medicinal applications of indazoles, such as anti-inflammatory, anti-tumor, and anti-HIV activity, among others,11 we embarked on extensive studies of the synthesis of spiroindazoles through 1,3-dipolar cycloaddition of ordinary cyclic diazo compounds and arynes. | ||
| Scheme 1 Synthetic route to spiro-3H-indazoles and fused-2H-indazoles. | ||
Results and discussion
We first investigated the reaction of literature substrates 1a and 1b with o-(trimethylsilyl)phenyl triflate (2a) under our previous optimal conditions (Table 1, entries 1 and 2).10 There was no reaction for substrate 1a, probably due to the low reactivity of the diazo group between the two carbonyl groups, while a complex reaction mixture was formed for substrate 1b. Then we turned our attention to substrates 1c–e. Using 1c as the diazo substrate, again only a complex reaction system was obtained (entry 3). Delightedly, we isolated the desired product 3d in 91% yield, when 1d was employed (entry 4). Of note, this result was different from that of structural analogue 1c. After these initial trials we intended to modify the reaction conditions with diazo substrate 1d and aryne precursor 2a (entries 5–9). Increase of the loading of 2a from 1.2 equiv. to 1.5 equiv. slightly enhanced the yield of 3a to 93% (entry 5). Changing the fluoride source and solvent did not give any better results (entries 6–9). Oddly there was no reaction at all with the combination of CsF/THF (entry 8). In view of the substrates (1a–d) and respective results, the substrate 1e was also examined under the reaction conditions. To our delight, it worked smoothly as well to afford the spiroindazole 3e in 79% yield.
|
||||
|---|---|---|---|---|
| Entry | Diazo compound | Fluoride source | Solvent | Yieldb (%) |
| a Conditions: 1 (0.3 mmol), 2a (1.2 equiv.), fluoride source (1.5 equiv.), solvent (3 mL), 0 to 20 °C, air.b Isolated yields.c 2a (1.5 equiv.) was employed instead.d 18-crown-6 was added as the additive.e Complex system was got. TBAT = tetrabutylammonium triphenyldifluorosilicate. | ||||
| 1 | 1a | TBAT | THF | NR |
| 2 | 1b | TBAT | THF | e |
| 3 | 1c | TBAT | THF | e |
| 4 | 1d | TBAT | THF | 91 |
| 5c | 1d | TBAT | THF | 93 |
| 6c,d | 1d | KF | THF | 78 |
| 7c | 1d | TBAF | THF | 79 |
| 8c | 1d | CsF | THF | NR |
| 9c | 1d | CsF | CH3CN | 87 |
| 10c | 1e | TBAT | THF | 79 |

With the reaction conditions optimized, the scope of the 1,3-dipolar cycloaddition reaction was explored. As illustrated in Table 2, we first tested the derivatives of 1d. For F, Br and MeO substituted substrates (1f–h), all of them reacted well to afford the corresponding spiroindazoles (3f–h) in 78–95% yields. 3-Diazochroman-4-one was also a suitable substrate and the desired product 3i was obtained in 68% yield. Then we investigated a series of substituted 6-diazocyclohex-2-en-1-one derivatives (1j–m). The reaction of both mono-substituted and di-substituted diazocyclohex-2-en-1-ones proceeded smoothly to deliver the desired spiroindazoles in moderate to good yields. It's interesting to note that for 1m, an i-propenyl group adjacent to the diazo moiety did not have adverse effect on the yield. The performance of substituted aryne precursors was also examined. Symmetrical aryne precursors (2b–d) gave the desired spiro products (3n–p) in satisfactory yields and unsymmetrical 3-methoxybenzyne furnished a single isomer 3q in 92% yield.3b,4,12 The spiroindazoles 3r and 3s were determined just by their crude 1H and 13C-NMR spectra due to their lability resulting from high propensity towards isomerization to afford fused-indazoles (for the same transformation see Scheme 2). Unexpectedly, 2-diazo-2,3-dihydro-1H-inden-1-one gave a complex mixture instead of the desired product 3t. We speculated that it may be due to the presence of acidic proton at the benzyl position and also the adjacent position of diazo group.
|
|||
|---|---|---|---|
| Entry | Productb (yield) | Entry | Productb (yield) |
| a Conditions: 1 (0.3 mmol), 2 (0.45 mmol, 1.5 equiv.), TBAT (0.45 mmol, 1.5 equiv.), solvent (3 mL), 0 to 20 °C, air.b Isolated yields.c Pure sample could not be obtained.d No desired product was formed.e The ratio was determined by the quantity of isolated product. | |||
| 1 |  |
2 |  |
| 3 |  |
4 |  |
| 5 |  |
6 |  |
| 7 |  |
8 |  |
| 9 |  |
10 |  |
| 11 |  |
12 |  |
| 13 |  |
14 |  |
| 15 | 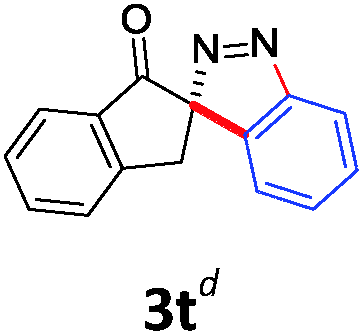 |
||

 | ||
| Scheme 2 Synthesis of fused-2H-indazoles. | ||
Migration of acyl, alkyl or aryl group of 3H-pyrazoles leading to 1H-pyrazoles has been well documented,13 and a similar rearrangement of spiro[indazole-3,3′-indolin]-2′-ones was also observed in our study.10 As a synthetic application of these spiro-3H-indazoles, we allowed 3e to be heated in PhMe at 120 °C, and the migration product 4e was isolated in 56% yield (Scheme 2, eqn (a)). We also found that TFA and even weakly acidic CHCl3 or SiO2 could promote isomerization of these spiro-3H-indazoles (Scheme 2, eqn (b) and (c)). For example, 4d and 4s could be obtained in satisfying yields with TFA/CHCl3 and SiO2 to promote isomerization respectively. With these satisfying results, we intended to synthesize a range of fused indazoles from spiroindazoles (3f–r) generated from 1,3-dipolar cycloaddition reaction. However, we observed that this kind of isomerization promoted by heat or acids was only applicable to a limited amount of spiroindazoles, because side-reactions such as ring-opening or polymerization sometimes took place, resulting in the difficulty in isolating pure samples.6 To further demonstrate the advantages of this method to introduce fused indazole motif under mild conditions, we applied it to the derivation reaction of biomolecule testosterone propionate (Scheme 2, eqn (d)). Fused indazole 4u could be readily prepared in 88% yield via a two-step manipulation.
On the other hand, we envisaged that if ketone 3d was reduced to alcohol followed by being treated with acid and heating, it might undergo carbon cationoic rearrangement to afford other fused heterocycles, for example cinnoline derivative. Unexpectedly, 5d was isolated as the major product in 68% yield under the reduction conditions and its exact structure was further confirmed by X-ray diffraction studies (Scheme 3).14 The formation of 5d could be rationalized by a sequence of reduction, ring-opening and a second reduction of the resulting aldehyde 7d.
 | ||
| Scheme 3 Unexpected transformation of spiro-3H-indazole 3d. | ||
Conclusions
In summary, we have demonstrated an efficient synthesis of spiro-3H-indazoles via 1,3-dipolar cycloaddition of arynes with 6-diazocyclohex-2-en-1-one derivatives under mild conditions in good to excellent yields. These unique spiro-3H-indazoles could be further transformed to fused-2H-indazoles via acid- or heat-mediated rearrangement. For spiroindazole 3d, an interesting sequential transformation involving reduction/ring-opening/reduction has been reported.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NSFC (21472072, 21302077, and 21290183), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT: IRT_15R28), FRFCU (lzujbky-2016-ct02, lzujbky-2017-90), and “111” Program of MOE for financial support. We thank Dr Hanwei Hu for his assistance in polishing the manuscript.Notes and references
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211 CrossRef CAS.
- For reviews, see: (a) S. A. Worlikar and R. C. Larock, Curr. Org. Chem., 2011, 15, 3214 CrossRef CAS; (b) S. S. Bhojgude and A. T. Biju, Angew. Chem., Int. Ed., 2012, 51, 1520 CrossRef CAS PubMed; (c) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140 RSC; (d) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191 RSC; (e) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766 CrossRef CAS PubMed; (f) R. Sanz, Org. Prep. Proced. Int., 2008, 40, 215 CrossRef CAS; (g) H. Pellissier and M. Santelli, Tetrahedron, 2003, 59, 701 CrossRef CAS; (h) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658 CrossRef CAS PubMed.
- (a) For seminal work using diazo compounds and benzynes to construct indazoles, see: R. Huisgen and R. Knorr, Naturwissenschaften, 1961, 48, 716 CrossRef; (b) T. Jin and Y. Yamamoto, Angew. Chem., Int. Ed., 2007, 46, 3323 CrossRef CAS PubMed; (c) Y. Hari, R. Sone and T. Aoyama, Org. Biomol. Chem., 2009, 7, 2804 RSC.
- Z. Liu, F. Shi, P. D. G. Martinez, C. Raminelli and R. C. Larock, J. Org. Chem., 2008, 73, 219 CrossRef CAS PubMed.
- (a) K. Hirakawa, T. Toki, K. Yamazaki and S. Nakazawa, J. Chem. Soc., Perkin Trans. 1, 1980, 1944 RSC; (b) K. Hirakawa, Y. Minami and S. Hayashi, J. Chem. Soc., Perkin Trans. 1, 1982, 577 RSC; (c) W. Burgert, M. Groβe and D. Rewicki, Chem. Ber., 1982, 115, 309 CrossRef CAS; (d) G. Baum, R. Bernard and H. Shechter, J. Am. Chem. Soc., 1967, 89, 5307 CrossRef CAS.
- T. Yamazaki and H. Shechter, Tetrahedron Lett., 1973, 14, 1417 CrossRef.
- T. Yamazaki and H. Shechter, Tetrahedron Lett., 1972, 13, 4533 CrossRef.
- P. Li, J. Zhao, C. Wu, R. C. Larock and F. Shi, Org. Lett., 2011, 13, 3340 CrossRef CAS PubMed.
- C. Spiteri, S. Keeling and J. E. Moses, Org. Lett., 2010, 12, 3368 CrossRef CAS PubMed.
- B. Cheng, B. Zu, B. Bao, Y. Li, R. Wang and H. Zhai, J. Org. Chem., 2017, 82, 8228 CrossRef CAS PubMed.
- For reviews, see: (a) S. Bräse, C. Gil and K. Knepper, Bioorg. Med. Chem., 2002, 10, 2415 CrossRef; (b) H. Cerecetto, A. Gerpe, M. González, V. J. Arán and C. O. de Ocáriz, Mini-Rev. Med. Chem., 2005, 5, 869 CrossRef CAS PubMed; (c) A. Schmidt, A. Beutler and B. Snovydovych, Eur. J. Org. Chem., 2008, 4073 CrossRef CAS.
- The regioselectivity of 3q was assigned according to the analogous reaction of 3-methoxybenzyne with diazo compounds. For theoretical studies of the regioselectivities of aryne reactions, see: (a) P. M. Tadross, C. D. Gilmore, P. Bugga, S. C. Virgil and B. M. Stoltz, Org. Lett., 2010, 12, 1224 CrossRef CAS PubMed; (b) J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 15798 CrossRef CAS PubMed; (c) P. H.-Y. Cheong, R. S. Paton, S. M. Bronner, G.-Y. J. Im, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 1267 CrossRef CAS PubMed.
- For reviews, see: (a) M. P. Sammes and A. R. Katritzky, Adv. Heterocycl. Chem., 1983, 34, 1 CrossRef CAS; (b) T. Nagai and M. Hamaguchi, Org. Prep. Proced. Int., 1993, 25, 403 CrossRef CAS; for two recent examples, see: (c) M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2013, 52, 7219 CrossRef PubMed; (d) D. Gladow, S. Doniz-Kettenmann and H. Reissig, Helv. Chim. Acta, 2014, 97, 808 CrossRef CAS.
- CCDC 1577009 (5d) contains the ESI crystallographic data.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and HRMS data. CCDC 1577009. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra12117b |
| This journal is © The Royal Society of Chemistry 2017 |