
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnt-abietane diterpenoids and their probable biogenetic precursors from the roots of Euphorbia fischeriana†
Jia Zhang‡
 ab,
Jun He‡c,
Xiao-Xue Wangc,
Ying-Xue Shia,
Nuan Zhanga,
Bing-Zhi Mac,
Wei-Ku Zhang*b and
Jie-Kun Xu*a
ab,
Jun He‡c,
Xiao-Xue Wangc,
Ying-Xue Shia,
Nuan Zhanga,
Bing-Zhi Mac,
Wei-Ku Zhang*b and
Jie-Kun Xu*a
aSchool of Life Sciences & School of Chinese Medicine Sciences, Beijing University of Chinese Medicine, Beijing 100029, China. E-mail: xjkbucm@163.com
bInstitute of Clinical Medical Sciences, China-Japan Friendship Hospital, Beijing 100029, China. E-mail: cpuzwk@163.com
cDepartment of Pharmacy, China-Japan Friendship Hospital, Beijing 100029, China
First published on 8th December 2017
Abstract
Five new ent-abietane diterpenoids (1–5), along with nine known analogues (6–14), were isolated from the roots of Euphorbia fischeriana. Their structures and absolute configurations were determined by the analysis of extensive spectroscopic data (UV, IR, HRESIMS, NMR) and the comparison of the experimental and calculated electronic circular dichroism (ECD). Among the isolates, it's notable that the ent-abietane diterpenoids bearing an additional five-membered lactone ring might be derived from the proposed precursors of fischeriabietane B (2) and C (3), and their plausible biosynthetic relationship was detailed discussed. All the isolates were evaluated for cytotoxicity against three human cancer cell lines (Panc-28, Bel-7402, and HT-29), 2 and 3 exhibited moderate cytotoxicity.
Introduction
The genus Euphorbia comprises about 2000 species around the world, which is the largest in the spurge family. In particular, there are about 80 species growing in China.1 Phytochemical investigations into plants of the genus Euphorbia have led to a variety of bioactive secondary metabolites, consisting of diterpenoid, phloracetophenones, cerebrosides, and flavonoids.2The roots of Euphorbia fischeriana are well known as “Lang-du” in traditional Chinese medicine, and are mostly used to cure cancer, edema, and ascites.3 Previous chemical studies of the roots of E. fischeriana indicated the presence of various types of diterpenoids.4–7 In particular, jolkinolide B and 17-hydroxy-jolkinolide B, the ent-abietane diterpenoids bearing an additional five-membered lactone ring, exhibited significant pharmacological anti-tumor and anti-inflammatory activities. Moreover, these two ent-abietane diterpenoids were also thought to be the major active constituents of the roots of E. fischeriana.8–10 In a continuing search for the novel anti-tumor agents and fascinating structures, five new ent-abietane diterpenoids (1–5), along with nine known analogues (6–14) were isolated from the roots of E. fischeriana. Among these, the structures of 2 and 3 are closely related to the upper portion of co-occurring 4–13, suggesting that 2 and 3 might be the biogenetic precursors of the ent-abietane diterpenoids containing an additional five-membered lactone ring. The anti-tumor activities of isolated compounds were evaluated against HT-29, Panc-28, and Bel-7402 cell lines. We present herein the isolation and structural characterization of 1–14, as well as their bioactivities (Fig. 1).
 | ||
| Fig. 1 Chemical structures of 1–14. | ||
Results and discussion
Fischeriabietane A (1) was obtained as white amorphous powder. The molecular formula of 1 was detected as C21H32O3 by the HRESIMS m/z 315.2311 [M − H2O + H]+ (calcd 315.2319). The UV spectrum revealed the absorptions at λmax 203 and 264 nm. The strong IR absorption bands at 3132 cm−1, 1735 cm−1, and 1646 cm−1 suggested the presence of hydroxyl and α, β-unsaturated carbonyl groups. The 1H NMR spectrum (Table 1) showed three methyl singlets at δH 0.73 (3H, s, H3-20), 0.87 (3H, s, H3-19), 0.93 (3H, s, H3-18), one vinylic methyl at δH 1.96 (3H, d, J = 2.0 Hz, H3-17), one methoxyl at δH 3.74 (3H, s), one oxygenated methine at δH 4.35 (1H, brs, H-7), and one olefinic proton at δH 6.43 (1H, s, H-14). The 13C NMR spectrum (Table 2) displayed 21 carbon signals, including one carboxyl carbon, four olefinic carbons, one oxygenated methine, three tertiary and one vinyl methyls, and one methoxyl. Detailed analysis of the 1H and 13C NMR data suggested the structure of 1 share the similar skeleton with the known compound methyl-8β-hydroxy-12-oxo-ent-abietadi-13,15(17)-ene-16-oate,11 except the absence of the ketone group and the presence of an additional oxygenated methine (δH 4.35, H-7) in 1, the vinylic methylene at C-17 was also replaced by an vinylic methyl (δH 1.96, H3-17) in 1. In the HMBC spectrum (Fig. 2), the correlation from H3-17 (δH 1.96) to C-13 (δC 143.2) and C-16 (δC 170.3) verified the location of H3-17. Moreover, the correlations from H-7 (δH 4.35) to C-5/C-14 determined the hydroxyl group was located at C-7, which was confirmed by the 1H–1H COSY correlations (Fig. 2) between H-7 (δH 4.35) and H2-6 (δH 1.84, 1.62). The E-configuration of the double bond between C-13 and C-15 was deduced from the NOESY correlation between H3-17 and H-14 (Fig. 3). In addition, the NOESY correlations between H-5/H-7, H-5/H-9 suggested these protons were β-orientated, and that 7-OH and 20-CH3 were α-orientated. A comparison between experimental and calculated ECD data using the time-dependent density functional theory (TD-DFT) method at the B3LYP/6-31G (d) level was applied to determine the absolute configuration of 1. The experimental ECD spectrum showed the strong negative Cotton effect at 286.5 (Δε − 4.23) nm, which well matched with the calculated (5R,7R,9S,10R)-1 ECD curve (Fig. 5). Hence, the structure of 1 was defined as (E)-methyl-7β-hydroxy-ent-abieta-8(14),13(15)-diene-16-oate, and named fischeriabietane A.| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 1.09 m | 1.01 m | 1.30 m | 1.38 m | 1.26 m |
| 1.67 m | 1.73 m | 1.80 m | 1.87 m | 1.77 m | |
| 2 | 1.23 overlapped | 1.18 overlapped | 1.23 overlapped | 1.39 m | 1.17 m |
| 1.46 m | 1.48 m | 1.56 overlapped | 1.60 m | 1.47 overlapped | |
| 3 | 1.22 overlapped | 1.18 overlapped | 1.23 overlapped | 1.25 overlapped | 1.19 m |
| 1.44 m | 1.43 m | 1.46 m | 1.45 m | 1.41 m | |
| 5 | 1.60 m | 1.04, m | 1.10 dd (2.8, 12.6) | 1.25 overlapped | 1.06 dd (2.6, 12.3) |
| 6 | 1.62 m | 1.52 m | 1.53 m | 1.42 m | 1.47 overlapped |
| 1.84 m | 1.86 m | 1.83 m | 1.93 m | 1.74 m | |
| 7 | 4.35 brs | 1.62 m | 1.56 overlapped | 2.15 m | 1.56 m |
| 2.03 overlapped | 1.99 m | 2.48 m | 2.04 m | ||
| 9 | 2.32 m | 2.02 overlapped | 2.10 brs | 2.52 s | 1.83 d (3.5) |
| 11 | 1.34 m | 2.45 m | 4.08 d (2.1) | 4.20 s | 3.73 d (3.5) |
| 1.79 m | |||||
| 12 | 2.09 m | ||||
| 3.11 m | |||||
| 14 | 6.43 s | 3.72 brs | 4.00, brs | 6.48 s | 4.17 s |
| 17 | 1.96 d (2.0) | 2.15 s | 4.53 Abq (13.3) | 4.55 s | 4.61 d (2.4) |
| 18 | 0.93 s | 0.92 s | 0.95 s | 0.93 s | 0.93 s |
| 19 | 0.87 s | 0.84 s | 0.88 s | 0.82 s | 0.85 s |
| 20 | 0.73 s | 0.76 s | 0.95 s | 0.73 s | 0.73 s |
| OCH3 | 3.74 s | 3.81 s | 3.74 s | 3.28 s |
| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 39.3 | 39.4 | 38.9 | 38.7 | 39.1 |
| 2 | 19.0 | 18.6 | 18.5 | 19.1 | 18.4 |
| 3 | 42.2 | 41.9 | 41.8 | 41.8 | 41.7 |
| 4 | 33.1 | 33.5 | 33.7 | 33.8 | 33.8 |
| 5 | 47.0 | 54.2 | 53.7 | 55.0 | 53.5 |
| 6 | 29.8 | 21.4 | 21.1 | 25.1 | 21.0 |
| 7 | 73.1 | 35.1 | 34.4 | 37.8 | 35.1 |
| 8 | 148.8 | 62.7 | 65.9 | 154.1 | 65.9 |
| 9 | 47.6 | 49.1 | 61.6 | 55.8 | 59.2 |
| 10 | 39.0 | 39.9 | 39.6 | 41.8 | 39.7 |
| 11 | 21.7 | 35.3 | 73.2 | 57.3 | 74.3 |
| 12 | 27.5 | 196.3 | 203.4 | 86.8 | 107.5 |
| 13 | 143.2 | 143.3 | 144.8 | 150.2 | 154.2 |
| 14 | 125.7 | 58.8 | 62.9 | 112.5 | 57.4 |
| 15 | 121.6 | 129.4 | 134.2 | 121.2 | 133.2 |
| 16 | 170.3 | 171.7 | 166.7 | 170.7 | 168.8 |
| 17 | 15.0 | 16.7 | 58.8 | 55.8 | 56.6 |
| 18 | 33.6 | 33.9 | 33.7 | 33.8 | 33.5 |
| 19 | 22.2 | 21.9 | 22.0 | 21.6 | 22.2 |
| 20 | 14.6 | 15.4 | 15.8 | 15.9 | 15.1 |
| OCH3 | 51.6 | 52.7 | 52.9 | 51.7 |
 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of 1 and 5. | ||
 | ||
| Fig. 3 Key NOESY correlations of 1. | ||
Fischeriabietane B (2), obtained as white amorphous powder, had a molecular formula of C21H30O4 based on the HRESIMS m/z 347.2210 [M + H]+ (calcd 347.2217). The 1H NMR spectrum of 2 displayed one vinylic methyl at δH 2.15 (3H, s, H3-17), three methyl singlets at δH 0.76 (3H, s, H3-20), 0.84 (3H, s, H3-19), and 0.92 (3H, s, H3-18), one methoxyl at δH 3.81 (3H, s, –OCH3), and one oxygenated methine at δH 3.72 (1H, brs, H-14). The NMR data (Tables 1 and 2) of 2 resembled to those of 1, except that the hydroxyl and the double carbon bond (C-8, 14) were absent, and the extra ketone group (δC 196.3, C-12) was present in 2. In the HMBC spectrum, the correlations from H-9 (δH 2.02), H2-11 (δH 2.45), and H-14 (δH 3.72) to the carbonyl carbon C-12 (δC 196.3) indicated the ketone group was located at C-12. Comparing to the NMR data of 1, the chemical shifts of C-8 (δC 65.91) and C-14 (δH 3.72, δC 62.52) were lower, together with the correlations from H-14 (δH 3.72) to C-7/C-12/C-15 suggested the double carbon bond between C-8 and C-14 in 1 was replaced by an epoxy group in 2. In the NOESY spectrum, the correlation between H3-17 and H-14 confirmed the Z-configuration of double bond between C-13 and C-15. The NOE correlation between H3-20 and H-14 indicated the β-orientation of 8, 14-epoxy group. Therefore, the structure of 2 was established as (Z)-methyl-8β,14β-epoxy-ent-abieta-13(15)-ene-16-oate, and named fischeriabietane B.
Fischeriabietane C (3), obtained as white amorphous powder. Based on the HRESIMS spectrum ([M + H]+ 379.2106 m/z calcd for 379.2115), the molecular formula was deduced as C21H30O6. The comparison of the NMR data (Tables 1 and 2) with those of 2 indicated that 3 was an analogue, except that two additional hydroxyls (C-11, 17) were present in 3. In the HMBC spectrum, the correlations from H-11 (δH 4.08) to C-8/C-9/C-10/C-12, and from H2-17 (δH 4.53) to C-13/C-15/C-16 determined two additional hydroxyls were located at C-11 and C-17, respectively. In the NOESY spectrum, the correlations of H3-20/H-14, H3-20/H-11 established the β-orientations of 11-OH and the 8, 14-epoxy group. Thus, the structure of 3 was assigned as (Z)-methyl-11β, 17-dihydroxy-8β,14β-epoxy-ent-abieta-13(15)-ene-16-oate, and named fischeriabietane C.
Fischeriabietane D (4) was obtained as white amorphous powder, whose molecular formula was revealed as C20H26O4 by the HRESIMS m/z 331.1900 [M + H]+ (calcd 331.1904). The 1H NMR spectrum showed three methyl singlets at δH 0.73 (3H, s, H3-20), 0.82 (3H, s, H3-19), and 0.93 (3H, s, H3-18), one oxygenated methine at δH 4.20 (1H, s, H-11), one oxygenated methylene at δH 4.55 (2H, s, H2-17), and one olefinic proton at δH 6.48 (1H, s, H-14). The NMR data (Tables 1 and 2) of 4 was very similar to the known compound ent-11β-hydroxyabieta-8(14),13(15)-dien-16,12β-olide,12 except the presence of one extra hydroxyl (C-17) and epoxy group (C-11, 12) in 4. In the HMBC spectrum, the correlations from H2-17 (δH 2.45) to C-15/C-16 suggested the hydroxyl was located at C-17 (δC 55.8). Besides, the HMBC correlations from H-11 (δH 4.20) to C-8/C-12, from H-9 (δH 2.52) and H-14 (δH 6.48) to C-12 (δC 86.8) determined the epoxy group was assigned at C-11 and C-12. In the NOE difference experiments of 4, the NOE enhancements was observed between H-11 and H3-20 defined the structure of 4 was 17-hydroxy-11β,12β-epoxy-ent-abieta-8(14),13(15)-diene-16,12-olide, and named fischeriabietane D.
Fischeriabietane E (5) had a molecular formula of C21H30O6, as determined by the ion peak at m/z 379.2108 [M + H]+ (calcd 379.2115) in the HRESIMS spectrum. The 1H and 13C NMR data (Tables 1 and 2) represent very similarity to those of the known compound 7-deoxylangduin B,13 except the absence of the double carbon bond between C-8 and C-14, and the presence of an additional methoxyl (δH 3.28; δC 51.7) in 5. The conversion of the chemical shifts of C-8 and C-14 (δC 157.2 to 65.9; δC 114.7 to 57.4) suggested the double carbon bond was replaced by the epoxy group in 5, which was confirmed by the HMBC correlations (Fig. 3) from H-14 (δH 4.17) to C-7 (δC 35.1), C-8 (δC 65.9), C-12 (δC 107.5), and C-13 (δC 154.2), from H-9 (δH 1.83) to C-8 (δC 65.9) and C-14 (δC 57.4). Additionally, the HMBC correlations from –OCH3 (δH 3.28) to C-12 (δC 107.5) suggested the location of the methoxyl was C-12. In the NOESY spectrum (Fig. 4), the correlations of H3-20/H-14 and H3-20/H-11 revealed the β-orientations of 11-OH and 8, 14-epoxy group, and the α-orientation of H3-20.
 | ||
| Fig. 4 Key NOESY correlations of 5. | ||
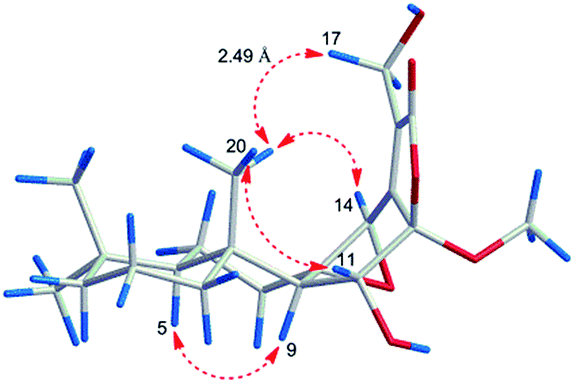
However, the relative configuration of C-12 was ambiguous. An assumption was provided with the orientation of –OCH3. When the –OCH3 was β-orientated, the spatial distance between H3-20 and H2-17 was 2.49 Å, which was enough to construct an NOE effect. To the contrary, the spatial distance between H3-20 and H2-17 was 6.34 Å, which is difficult to construct an NOE effect. The NOESY correlation (Fig. 4) between H3-20 and H2-17 suggested the orientation of –OCH3 should be set as β. To determine the absolute configuration of 5, the comparison between the experimental and calculated ECD curves was performed. The experimental spectrum showed a positive Cotton effect at 252.5 (Δε + 1.73) nm, which was consistent with the calculated curves of 5R,8S,9R,10R,12R,14R (Fig. 5). Finally, the structure of 5 was established as 12β-methoxy-11β,17-dihydroxy-8β,14β-epoxy-ent-abieta-13(15)-ene-16,12-olide, and named fischeriabietane E.
 | ||
| Fig. 5 Experimental ECD and calculated ECD spectrum of 1 and 5. | ||
By the comparison of spectroscopic data with literature, the known compounds isolated from the roots of E. fischeriana were identified as 11β-hydroxy-8,14-epoxy-ent-abieta-13(15)-en-16,12-olide (6),12 8β,14β-epoxy-13,15-abiatene-16,12α-olide (7),14 jolkinolide B (8),15 17-hydroxyjolkinolide B (9),15 jolkinolide A (10),15 17-hydroxyjolkinolide A (11),16 yuexiandajisu D (12),17 ent-11β-hydroxyabieta-8(14),13(15)-dien-16,12β-olide (13),16 13β-hydroxy-ent-abiet-8(14)-en-7-one (14),18 respectively.
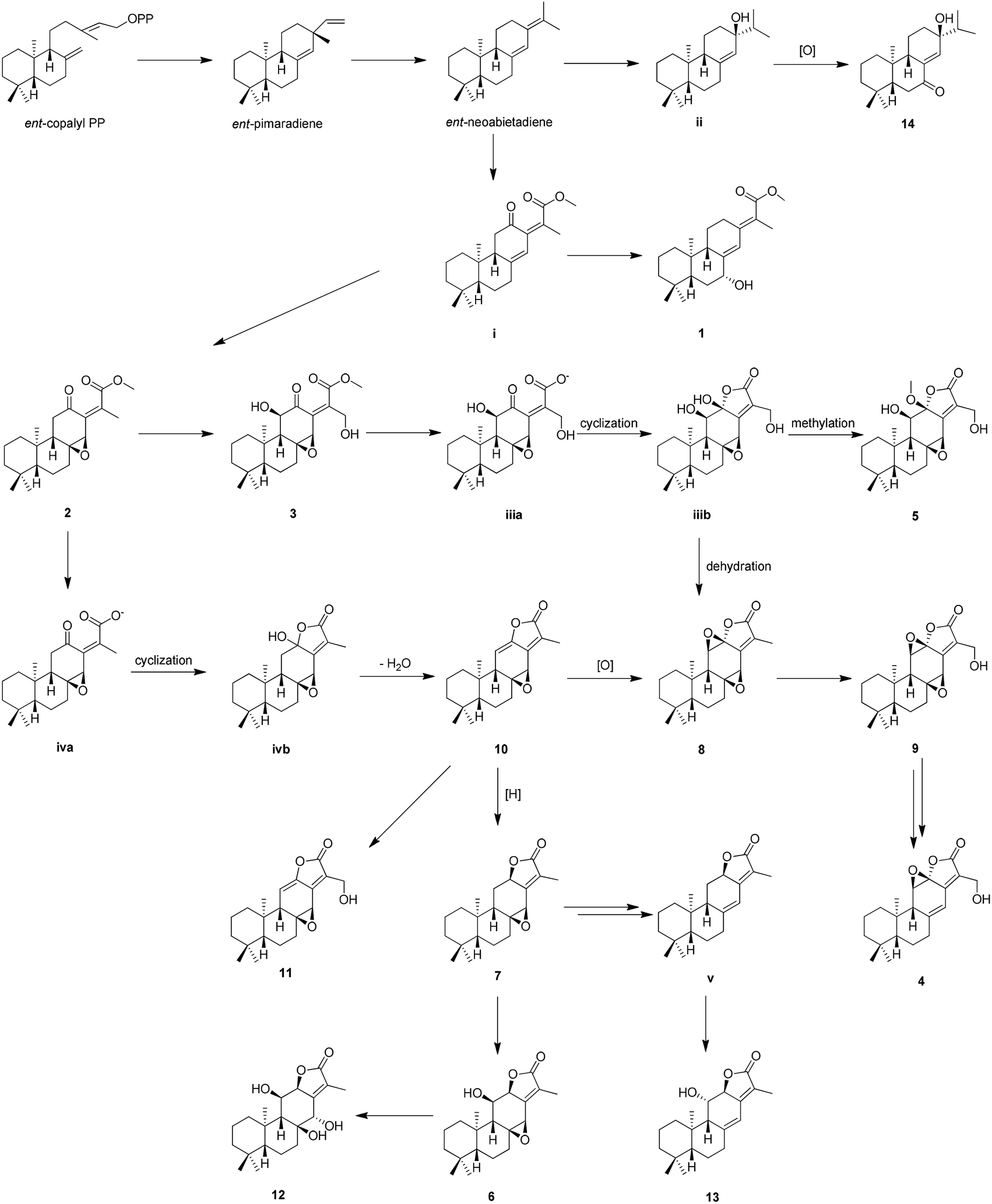
The proposed biosynthetic routes of 1–14 were illustrated in Scheme 1. All the isolates may be originally biosynthesized from the ent-neoabietadiene, the oxidation of which could led to the generation of 14 and intermediate i, respectively. Then, the intermediate i might be oxidized and hydroxylated to obtain 1–3. Notably, 1–3 represent a highly oxidized ent-abietane diterpenoid skeleton, 2 and 3 may undergo a series of intramolecular cyclization, oxidation, and dehydration reactions to produce the ent-abietane diterpenoids bearing an additional five-membered lactone ring, including 4–13.
 | ||
| Scheme 1 The plausible biosynthetic pathways for 1–14. | ||
In the biological assay, 1–14 were evaluated for their cytotoxicity activities on Panc-28, Bel-7402, and HT-29 cell lines. Compound 3 showed inhibitory activities with IC50 values of 12.9 ± 3.3 μM and 20.7 ± 4.2 μM against Bel-7402 and Panc-28 cell lines, respectively. Meanwhile, 2 exhibited inhibitory activity with IC50 values of 47.2 ± 4.6 μM against Panc-28 cells. (Methotrexate was used as the positive control and IC50 values of 27.3 ± 5.1, 31.7 ± 6.8, and 36.2 ± 9.4 μM for the Panc-28, Bel-7402, and HT-29 cells, respectively). From the above results, the ent-abietane diterpenoids containing two carbonyl groups generally showed better activities than others, and two carbonyl units may be the key moiety that taking effect against the tumor cells.
Experimental section
General experimental procedures
UV spectra were measured with a JASCO V-650 spectrometer, and the optical rotations were obtained using a JASCO P-2000 spectrometer, together with the ECD spectra were recorded on a JASCO J-815 spectrometer (JASCO, Easton, MD, U.S.A.). IR spectra were obtained on a Nicolet iS5 spectrometer (Thermo Scientific, Waltham, MA, U.S.A.). NMR measurements were carried out on a Bruker 400/600 MHz NMR instruments (Bruker-Biospin, Billerica, MA, U.S.A.). HRESIMS spectra were obtained on a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific, Waltham, MA, U.S.A.). An Agilent 1100 series system (Agilent Technologies, CA, U.S.A.) with an ES Sonoma C18 column (250 × 4.6 mm, 5 μm, CA, U.S.A.) was used for HPLC analyses. Preparative HPLC was performed on a Shimadzu LC-6AR instrument equipped with a SPD-20A detector using an YMC-Pack ODS-A column (250 mm × 20 mm, 5 μm, Kyoto, Japan). Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., People's Republic of China), Rp-18 (50 μm, YMC, Japan) and Sephadex LH-20 (Pharmacia Fine Chemicals, Uppsala, Sweden) were used for column chromatography (CC). TLC was performed using GF254 plates (Qingdao Marine Chemical Inc., People's Republic of China).Plant material
The roots of E. fischeriana were collected in Lindian, Heilongjiang, People's Republic of China, in August 2015, and identified by Professor Hai Ju of China–Japan Friendship Hospital. A voucher specimen (20150818) is deposited in the Institute of Clinical Medical Sciences, China–Japan Friendship Hospital, Beijing, People's Republic of China.Extraction and isolation
The dried roots of E. fischeriana (46.0 kg) were extracted thrice with EtOH–H2O (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, v/v, 450 L × 3) at 75 °C for 2 h each time to give a residue (2.0 kg), which was then suspended in H2O (20 L) and partitioned with petroleum ether, CH2Cl2 and EtOAc (thrice with each 30 L), successively. The CH2Cl2 extract (550.0 g) was subjected to a silica gel CC with a gradient of petroleum ether–EtOAc (100:1 to 0:100, v/v) to afford nine fractions, Fr.1–9. Fr.2 (10.0 g) was subjected to an Rp-C18 column by the gradient of MeOH–H2O (30:100 to 100:0, v/v) to produce seven fractions Fr.2.1–7. Fr.2.3 (3.5 g) was recrystallized from MeOH to obtain 8 (1.2 g), and Fr.2.4 (0.2 g) was applied to a semi-preparative HPLC (MeOH–H2O, 80:20, v/v), to obtain 3 (23.0 mg), 10 (35.0 mg), and 11 (26.0 mg). Fr.3 (45.0 g) was separated by an Rp-C18 column with an elution gradient from 20:80 to 100:0 (MeOH–H2O) to afford ten fractions, Fr.3.1–10. Fr.3.3 (11.3 g) was further separated by a silica gel CC with a cyclohexane–EtOAc system (5:1, 4:1, 3:1, v/v) and semi-preparative HPLC (MeOH–H2O, 70:30, v/v), to yield 4 (6.0 mg), 6 (20.5 mg), 7 (35.0 mg), and 13 (15.0 mg). By the same method, Fr.3.6 (3.5 g) was separated to yield 2 (20.5 mg). 9 (50.5 mg) was gained in the form of crystal from Fr.3.5 (1.4 g). Fr.4 (40.0 g) was chromatographed on a column of Rp-C18 and eluted by a gradient of MeOH–H2O (30:70 to 100:0, v/v) to give five fractions, Fr.4.1–5. Fr.4.2 (0.7 g) was repeatedly subjected to an Rp-C18 column with MeOH–H2O (60:40, v/v) and semi-preparative HPLC (MeCN–H2O, 50:50, v/v), to obtain 1 (15.5 mg), 5 (8.3 mg), and 14 (1.0 mg). Fr.4.2 (0.2 g) was purified by semi-preparative HPLC (MeCN–H2O, 55:45, v/v) to yield 12 (2.0 mg).
:5, v/v, 450 L × 3) at 75 °C for 2 h each time to give a residue (2.0 kg), which was then suspended in H2O (20 L) and partitioned with petroleum ether, CH2Cl2 and EtOAc (thrice with each 30 L), successively. The CH2Cl2 extract (550.0 g) was subjected to a silica gel CC with a gradient of petroleum ether–EtOAc (100:1 to 0:100, v/v) to afford nine fractions, Fr.1–9. Fr.2 (10.0 g) was subjected to an Rp-C18 column by the gradient of MeOH–H2O (30:100 to 100:0, v/v) to produce seven fractions Fr.2.1–7. Fr.2.3 (3.5 g) was recrystallized from MeOH to obtain 8 (1.2 g), and Fr.2.4 (0.2 g) was applied to a semi-preparative HPLC (MeOH–H2O, 80:20, v/v), to obtain 3 (23.0 mg), 10 (35.0 mg), and 11 (26.0 mg). Fr.3 (45.0 g) was separated by an Rp-C18 column with an elution gradient from 20:80 to 100:0 (MeOH–H2O) to afford ten fractions, Fr.3.1–10. Fr.3.3 (11.3 g) was further separated by a silica gel CC with a cyclohexane–EtOAc system (5:1, 4:1, 3:1, v/v) and semi-preparative HPLC (MeOH–H2O, 70:30, v/v), to yield 4 (6.0 mg), 6 (20.5 mg), 7 (35.0 mg), and 13 (15.0 mg). By the same method, Fr.3.6 (3.5 g) was separated to yield 2 (20.5 mg). 9 (50.5 mg) was gained in the form of crystal from Fr.3.5 (1.4 g). Fr.4 (40.0 g) was chromatographed on a column of Rp-C18 and eluted by a gradient of MeOH–H2O (30:70 to 100:0, v/v) to give five fractions, Fr.4.1–5. Fr.4.2 (0.7 g) was repeatedly subjected to an Rp-C18 column with MeOH–H2O (60:40, v/v) and semi-preparative HPLC (MeCN–H2O, 50:50, v/v), to obtain 1 (15.5 mg), 5 (8.3 mg), and 14 (1.0 mg). Fr.4.2 (0.2 g) was purified by semi-preparative HPLC (MeCN–H2O, 55:45, v/v) to yield 12 (2.0 mg).
Structure characterization
ε) 203 (4.42), 264 (4.15) nm; ECD (MeOH) λmax (Δε) 287 (−4.23) 359 (+0.15) nm; IR (KBr) νmax 3132, 2955, 2925, 2876, 2853, 1736, 1647, 1458, 1400, 1244, 1153, 1111, 1035, 892, 721, 578 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS (m/z): 315.2311 [M – H2O + H]+ (calcd for C21H32O3, 315.2319).ε) 205 (4.16), 251 (4.39) nm; IR (KBr) νmax 3349, 2944, 2831, 2318, 2050, 1452, 1110, 1031 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS (m/z): 347.2210 [M + H]+ (calcd for C21H31O4, 347.2217).ε) 209 (3.86), 238 (3.77) nm; IR (KBr) νmax 3415, 3131, 3008, 1635, 1400, 1125 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS (m/z): 379.2106 [M + H]+ (calcd for C21H31O6, 379.2115).ε) 204 (3.89), 286 (3.95) nm; IR (KBr) νmax 3352, 2947, 2836, 2505, 1450, 1413, 1116, 1031 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS (m/z): 331.1900 [M + H]+ (calcd for C20H27O4, 331.1904).ε) 235 (4.01) nm; ECD (MeOH) λmax (Δε) 253 (+1.73) nm; IR (KBr) νmax 3127, 3012, 2848, 1748, 1636, 1400, 1114 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS (m/z): 379.2108 [M + H]+ (calcd for C21H31O6, 379.2115).Cytotoxic assay
The isolated compounds were evaluated the cytotoxicity against the human pancreatic cancer cell line Panc-28, human lung adenocarcinoma cell line Bel-7402, and human fibrosarcoma cell line HT-29 with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphen-yl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) method,19 with methotrexate as a positive control. The purities of all isolates and methotrexate were more than 98%. The cells (Panc-28, Bel-7402, and HT-29) were purchased from Cell Resource Center of Peking Union Medical College, Beijing, People's Republic of China. Panc-28 and Bel-7402 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum in a humidified atmosphere with 5% CO2 in air at 37 °C, and HT-29 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum in a humidified atmosphere with 5% CO2 in air at 37 °C. The cancer cells were seeded in 96-well cell culture plates at a density of 3500 cells per well. After 24 hours incubation, the cancer cells were treated with different concentrations of 1–14 and being cultured for another 24 h. Then, MTS (20 μL per well) were immediately added to the culture medium and incubated for 4 hours, formazan production was measured using a microplate reader (Biotech, power wave, USA) at 490 nm. The cell growth inhibition and half inhibitory concentration (IC50) values were then calculated.ECD calculation
Conformational search was performed by using the MMFF molecular mechanics force field. Further optimization of the structures in methanol was performed using the Gaussian 09 program. The optimized structures were shown in ESI Tables 1 and 2.† Optimization was confirmed by computation of frequency. Conformational distribution of the optimized structures was investigated at B3LYP/6-31G (d) and suggested the major conformers (>98%). Energies of the geometric conformations in MeOH were calculated at the B3LYP/6-311+G (d) level. Solvent effects were considered by using the conductor-like polarizable continuum model (CPCM). The hybrid B3LYP functionals were chosen to run the TDDFT calculations, solving for 20 states for per molecule. The overall theoretical ECD spectra were obtained according to the Boltzmann weighting of each conformers. The ECD spectra were generated using the program SpecDis with a half-bandwidth of 0.30 eV (for 1 and 5). After hypsochromic-shifting −9 nm (for 1), and −5 nm (for 5) by UV correction, in the 190–400 nm region, the theoretically calculated ECD spectra of 5R,7R,9S,10R-1 and 5R,8S,9R,10R,12R,14R-5 were in good agreement with the experimental ECD spectra of 1 and 5 (Fig. 5).Conclusions
In summary, five new ent-abietane diterpenoids (1–5), together with nine analogues (6–14) were isolated from the roots of E. fischeriana. Notably, 2 and 3 could be considered as the biosynthetic precursors for the ent-abietane diterpenoids with an additional five-membered lactone ring. In addition, the assays of anti-tumor activities showed 2 and 3 had the moderate inhibitory against Bel-7402 and Panc-28 cells. Our findings add to the diversity and complexity of ent-abietane diterpenoids, which may provide reference for both the synthetic chemists and pharmacologists.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81473119, No. 81473590), ‘Ten Diseases & Ten Drugs’ Project of Beijing Municipal Science & Technology Commission (No. Z171100001717009). We appreciate Dr Huan-Li Xu (School of Basic Medical Sciences, Capital Medical University) for the guidance in the pharmacological experiment.Notes and references
- Editorial committee of the Flora of China, Flora of China, Science Press, Beijing, 2004, p. 89 Search PubMed
.
- Q. W. Shi, X. H. Su and H. Kiyota, Chem. Rev., 2008, 108, 4295–4327 CrossRef CAS PubMed
- Jiangsu New Medical College, Dictionary of Chinese Materia Medica, Shanghai Science and Technical Press, Shanghai, 1977, pp. 1898–1900 Search PubMed
- J. W. Lee, C. Jin, Q. Lee, H. Jang, D. Lee, H. J. Lee, J. W. Shin, S. B. Han, J. T. Hong, Y. Kim, M. K. Lee and B. Y. Hwang, J. Nat. Prod., 2016, 79, 126–131 CrossRef CAS PubMed
- T. X. Zhou, G. H. Bao, Q. G. Ma, G. W. Qin, C. T. Che, Y. Lv, C. Wang and Q. T. Zheng, Tetrahedron Lett., 2003, 44, 135–137 CrossRef CAS
- L. L. Pan, P. L. Fang, X. J. Zhang, W. Ni, L. Li, L. M. Yang, C. X. Chen, Y. T. Zheng, C. T. Li, X. J. Hao and H. Y. Liu, J. Nat. Prod., 2011, 74, 1508–1512 CrossRef CAS PubMed
- X. Z. Kuang, W. Li, Y. Kanno, N. Yamashita, K. Nemoto, Y. Asada and K. Koike, J. Nat. Med., 2016, 70, 120–126 CrossRef CAS PubMed
- H. Y. Xu, Z. W. Chen, J. C. Hou, F. X. Du and J. C. Liu, Oncol. Rep., 2013, 29, 212–218 CrossRef CAS PubMed
- Y. Wang, X. Q. Ma, S. S. Yan, S. S. Shen, H. L. Zhu, Y. Gu, H. B. Wang, G. W. Qin and Q. Yu, Cancer Res., 2009, 69(18), 7302–7310 CrossRef CAS PubMed
- H. L. Yang, Y. Li, P. F. Huo, X. O. Li, D. L. Kong, W. Mu, W. Fang, L. X. Li, N. Liu, L. Fang, H. J. Li and C. Y. He, Int. Immunopharmacol., 2015, 26, 119–124 CrossRef CAS PubMed
- T. Morgenstern, M. Bittner, M. Silva, P. Aqueveque and J. Jakupovic, Phytochemistry, 1996, 41, 1149–1153 CrossRef CAS
- H. B. Wang, W. Chen, Y. Y. Zhang, X. Y. Wang, L. P. Liu, L. P. Tong and Y. Chen, Fitoterapia, 2013, 91, 211–216 CrossRef CAS PubMed
- C. J. Wang, Q. L. Yan, Y. F. Ma, C. P. Sun, C. Wang and X. C. Ma, J. Nat. Prod., 2017, 80, 1248–1254 CrossRef CAS PubMed
- X. D. Zhang, W. Ni, H. Yan, G. T. Li, H. M. Zhong, Y. Li and H. Y. Liu, Chem. Biol., 2014, 11, 760–766 CAS
- G. F. Liu, Y. Q. Fu, Z. Q. Yang, H. Q. Zhao and X. M. Fan, Zhongyao Tongbao, 1988, 13, 35–56 CAS
- C. T. Che, T. X. Zhou, Q. G. Ma, G. W. Qin, I. D. Williams, H. M. Wu and Z. S. Shi, Phytochemistry, 1999, 52, 117–121 CrossRef CAS
- H. M. Shi, I. D. Williams, H. H.-Y. Sung, H. X. Zhu, N. Y. Ip and Z. D. Min, Planta Med., 2005, 71, 349–354 CrossRef CAS PubMed
- Y. B. Wang, R. Huang, H. B. Wang, H. Z. Jin, L. G. Lou and G. W. Qin, J. Nat. Prod., 2006, 69, 967–970 CrossRef CAS PubMed
- R. Yuan, H. L. Xu, X. H. Liu, Y. Tian, C. Li, X. L. Chen, S. N. Su, I. Perelshtein, A. Gedanken and X. K. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 31806–31812 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: 1D NMR, 2D NMR, HRESIMS, and ECD spectra. See DOI: 10.1039/c7ra11982h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |