
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of an Au/B-ZnO/PET–GR flexible composite structure in photocatalysts and supercapacitor electrodes
Wei Wang ,
Taotao Ai*,
Yuanyuan Fan and
Wenhu Li
,
Taotao Ai*,
Yuanyuan Fan and
Wenhu Li
School of Materials Science and Engineering, Shaanxi University of Technology, Hanzhong 723000, Shaanxi, P. R. China. E-mail: aitaotao0116@126.com
First published on 13th November 2017
Abstract
The Au/B-ZnO/PET–GR composite structure with different boron doping concentrations was prepared on a flexible double-layer substrate of PET–GR using the hydrothermal technique assisted by the ion sputtering method. With the increase of B doping content, the morphology of nano-ZnO evolved from nanorods (NRs) to nanosheets (NSs). The degradation rate of the Au/3% B-ZnO/PET–GR catalyst achieved the maximum value of 38.11%, and the reaction of degrading an RhB solution followed first order reaction kinetics. The photocatalytic activity of the composite structure was effectively increased by the proper incorporation of B atoms, and the photocatalytic mechanism of the samples was analyzed by energy band theory. The equivalent series resistance and specific capacitance at a scan rate of 5 mV s−1 of Au/3% B-ZnO/PET–GR electrode were 3.1 Ω and 62.96 F g−1. The effects of the substrate and metal layer on the electrochemical and photocatalytic properties of the composite structure were also investigated.
1 Introduction
The one-dimensional zinc oxide (ZnO) nanomaterial is an excellent semiconductor photocatalyst because of its wide band-gap, high-efficiency electron mobility, high specific area and strong redox ability under ultraviolet (UV) irradiation with a wavelength of less than 387 nm.1,2 Compared with titanium dioxide (TiO2), it has an extensive absorption range of the UV spectrum and greater capacity to degrade some organic matter, and is considered as the most promising alternative to replace TiO2 nanomaterials. ZnO/graphene composites were synthesized by M. Ahmad et al. using a simple one-step method and their efficiency of degrading methylene blue was almost 100% under the irradiation of visible light.3 However, single ZnO nanoparticle can not satisfy the requirements of high degradation rates. This problem is solved by the following effective paths, including the combination of different species of semiconductor photocatalysts, ZnO nanoparticles modified by doping transition metal ions, the establishment of metal–semiconductor, semiconductor–semiconductor, multi-heterojunction semiconductor and other multi-level structure.4,5 Among them, taking n-ZnO/p-substrate heterojunction as a photocatalyst is a hotspot of research. Moreover, as a member of the family of transition metal oxide, ZnO has high electrochemical activity, pseudo-capacitive and is able to undergo rapid reversible oxidation/reduction, insertion/deinsertion and adsorption/desorption reactions in the electrochemical environment, which is deemed to be the supercapacitor material with bright prospect.6–8 The electrochemical performance of ZnO electrode is likely to be further improved by three means, such as increasing specific area of ZnO by controlling its microstructure, modifying the crystal surface with metal ions, loading it on the conductive carbon material.9,10 The ZnO/graphene supercapacitor material with the specific capacitance of 192 F g−1 was prepared by Liu et al.11 using a one-pot method at room temperature. In this paper, Au/B-ZnO/PET–GR composite structures with different B doping concentration were fabricated on PET–GR (polyethylene terephthalate–graphene) flexible substrate at a low temperature (90 °C). The effect of B doping content on the microstructure of ZnO was studied, and the growth mechanism of ZnO nanoparticles with different morphologies was investigated. The Au/B-ZnO/PET–GR structures were used as photocatalyst and electrochemical working electrode to explore their application in the field of photocatalysis and supercapacitor.2 Experimental
2.1 Synthesis of Au/B-ZnO/PET–GR composite structure
Au/B-ZnO/PET–GR composite structures were prepared using the hydrothermal method. Zinc nitrate hexahydrate (Zn(NO3)2·6H2O) of 0.5 mol L−1 and hexamethylenetetramine (C6H12N4, HMT) of 0.5 mol L−1 were dissolved in the deionized water to confect the precursor aqueous solution of 30 mL, and boric acid with the concentration of 0%, 1%, 3%, 5% and 7% (defining the doping concentration of 7% was 0.07 mol L−1) was added to the mixed solution, respectively. The reaction solution after stirring for 30 minutes was transferred to high pressure reactors. The ZnO seed layer was sputtered onto PET–GR substrate for 15 seconds using ion sputtering equipment at the conditions of the sputtering current of 10 mA and vacuum degree of 0.1 mbar. Among them, the PET–GR flexible double-layer substrate refers to the film formed by coating a graphene layer on the surface of polyethylene terephthalate (PET). In general, graphene layers were able to prepare by chemical vapor deposition (CVD) and reduction of graphite oxide.12,13 And the graphene layers were capable of transferring by the matrix etching method, the “roll-to-roll” transfer technique and the “electrochemical transfer” technique.14,15 Afterwards, the substrate adhered with the ZnO seed crystal was sandwiched with specimen holder and placed vertically into the autoclave, which was completely submerged in the as-prepared mixed solution. The reactor was sealed, diverted to electric oven and allowed to heat at a temperature of 90 °C for 5 hours. Cooling to room temperature (about 20–25 °C), the sample after washing with deionized water was allowed to dry in air. Following on, the intact Au/B-ZnO/PET–GR composite structure was obtained by sputtering the metal Au layer for 30 seconds on the surface of previously prepared structure at the conditions of sputtering current of 8 mA and vacuum degree of 0.1 mbar.2.2 Measurements and characterization
The phase composition and morphology of ZnO nanostructures were analyzed by X-ray diffractometer (XRD) (Rigaku, D/Max-2200PC) and scanning electron microscope (SEM) (JEOL, JSM-6700F). The photocatalytic activity of Au/B-ZnO/PET–GR composite was evaluated by photocatalytic degradation of rhodamine B solution (RhB), and the specific test steps were as follows: at room temperature, RhB solution of 25 mL at the amount-of-substance concentration of 10 mg L−1 was added to the quartz tube, Au/B-ZnO/PET–GR sample was suspended with fine copper wire so that it was at half of the liquid level below, and to ensure that the sample was parallel to the light source to increase irradiation area. The high pressure mercury lamp with power of 500 W (main wavelength of 365 nm) was selected as UV light source and it was 1 meter away from the sample. Magnetic stirring was employed to enhance mass transfer throughout whole reaction process. After 30 minutes of dark treatment, RhB solution of 2.5 mL was sampled at intervals of 20 minutes, and deionized water of 2.5 mL was added to prepare solution under test. The catalytic efficiency of photocatalyst was evaluated by monitoring the absorbance of RhB solution using UV-vis spectrophotometer (UV-5600PC), the maximum absorption wavelength of RhB was 554 nm. The electrochemical performance of the Au/B-ZnO/PET–GR composite structure was characterized on CHI660E electrochemical workstation, including cyclic voltammetry (CV) and A. C. impedance spectroscopy (EIS). The measurement was carried out using three-electrode system and completed at room temperature with 0.5 mol L−1 Na2SO4 solution as the electrolyte. The platinum wire, saturated calomel and as-fabricated composite structure were served as the counter, reference and working electrode, respectively. The frequency range of EIS testing was from 0.01 to 1 × 105 Hz and conducted at the open-circuit voltage with sine wave amplitude of 5 mV.3 Results and discussion
The crystal structure of the as-prepared Au/B-ZnO/PET–GR composite is analyzed by XRD patterns, as shown in Fig. 1a. The diffraction peaks at 31.8°, 34.6°, 36.5°, 47.2°, 56.8°, 62.8°, 66.4°, 68° and 69.2° are indexed to (100), (002), (101), (102), (110), (103), (200), (112) and (201) crystal faces of ZnO with hexagonal wurtzite structure (JCPDS 89-1397), except for the peak located near 53.8° is attributed to the PER–GR substrate. When the content of H3BO3 is too large (greater than or equal to 5%), the diffraction peak intensity of the products is obviously reduced, and the peaks of two single gold are observed, suggesting that the excessive incorporation of H3BO3 solution leads to a decrease in the crystallinity of the samples. The diffraction peaks of Au in the XRD patterns of the samples with B doping concentration of 0%, 1% and 3% are not obvious, which may be due to the following reasons: the Au content on the surface of the sample obtaining by ion sputtering is less, when the crystallinity of the product ZnO is very good, the diffraction peak of the ZnO crystal is particularly high, so the diffraction peak intensity of Au is very low relative to the peak of ZnO. Of course, the characteristic peaks of Au certainly exists in the above three XRD patterns, but the weak diffraction peak of Au is obscured by strong peaks of ZnO crystal. The lattice constants of the un-doped and 3% B-doped ZnO structures are calculated by the interplanar spacing formula for hexagonal system, and the results are a = 3.2759 Å, c = 5.2011 Å and a = 3.2777 Å, c = 5.3955 Å, respectively, which is due to the substitution doping or interstitial doping of B atoms, as well as the bending deformation of the flexible substrate cause the internal stress of ZnO crystal to change, and then giving rise to lattice distortion.16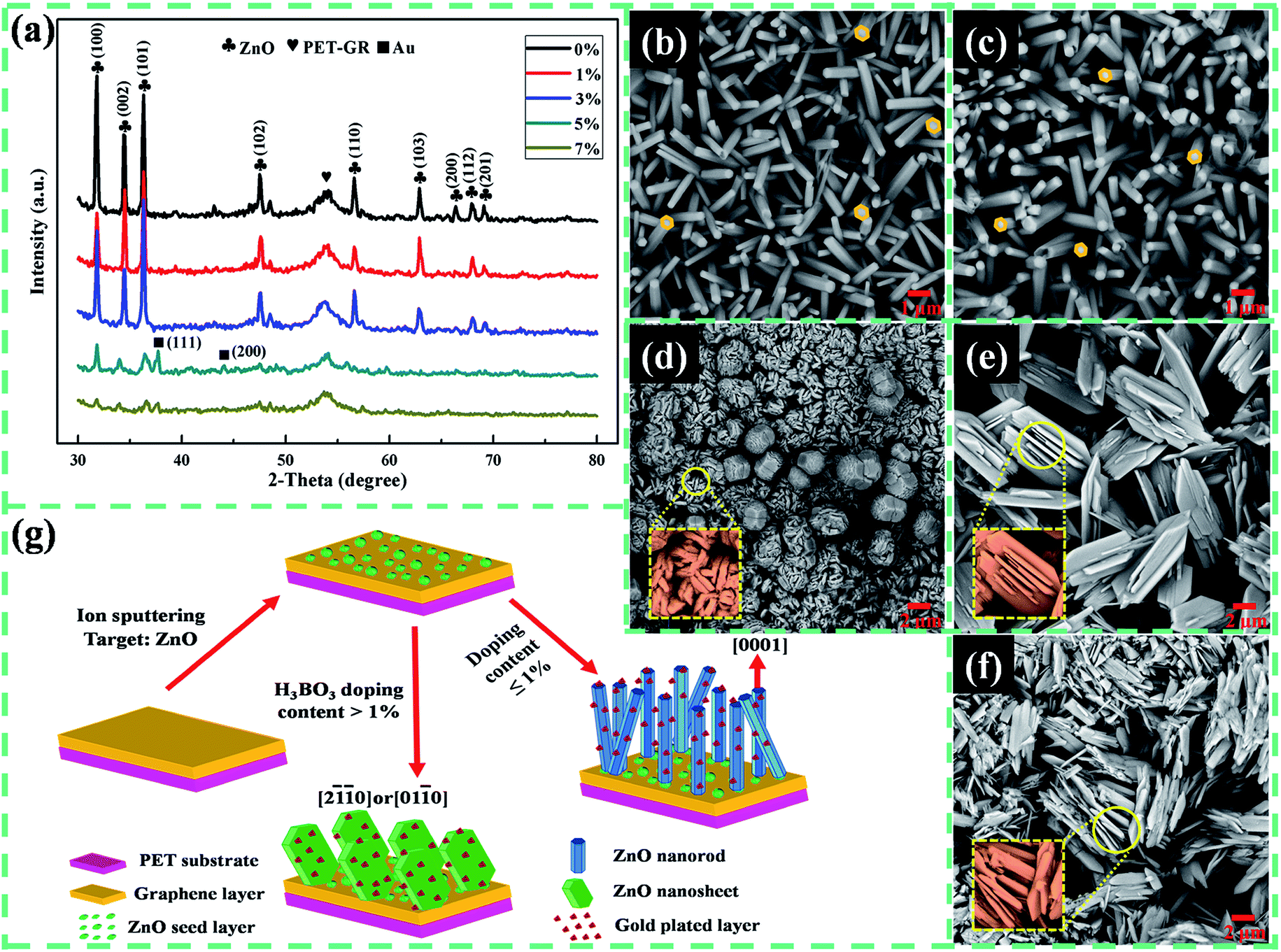 | ||
| Fig. 1 (a) XRD patterns of Au/B-ZnO/PET–GR composite structure, SEM images of BZO nanostructures grown on PET–GR substrate at different B doping concentrations: (b) 0%; (c) 1%; (d) 3%; (e) 5%; (f) 7% and (g) growth mechanism of BZO NRs/PET–GR and BZO NSs/PET–GR. | ||
The SEM images of ZnO with different B doping content grown on the double-layer flexible substrate PET–GR reveal that the incorporation of an appropriate amount of B atoms has little effect on the crystal morphology of ZnO, and show nanorod structures with a hexagonal cross section (Fig. 1b and c). All nanorods are distributed at higher densities on the substrate. The growth habit of ZnO crystal is transformed along with the further increase of the B doping amount. ZnO nanosheets with a length of 1.2 μm and a thickness of 300 nm are obtained (Fig. 1d) when the B doping content is 3%, which is homogeneously and tightly paved on the substrate. There is a handful of solid spherical ZnO formed by the stacking of many nanosheets. Subsequently, the doping quantity of B increased to 5%, the morphology of ZnO is further evolved, and form the nanosword with a length of 10.345 μm, a width of 3.207 μm and a height of 0.621 μm (Fig. 1e). Actually, it is also a kind of nanosheets. It is worth noting that in the direction perpendicular to the nanosword, a few pieces of nanosheets with different sizes embed in them, similar to the backbone of an aircraft. After the B incorporation amount continues to increase, the sample presents lamella structure (Fig. 1f), but the distribution of grain size is more extensive and uneven, and large-scale accumulation of crystalline grain is observed.
Fig. 1g demonstrates the growth mechanism of B-doped ZnO (BZO) nanostructures prepared by hydrothermal method. In the hydrothermal environment, the crystal habit of ZnO is closely related to the crystal structure, which is determined by the growth rate of each lattice plane. The ionic polar structure of ZnO with hexagonal wurtzite structure is composed of three parts: O atoms are arranged in the hexagonal closest packed way, Zn atoms exist in tetrahedral void, and half of the eight tetrahedral lattice voids are occupied, eventually leading to ZnO exhibiting polar crystal characteristics. ZnO has two polar faces, which are Zn polar faces with positive charge (0001) and O surfaces with negative charge (000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). Since the (0001) lattice plane has the lowest surface free energy, its growth rate is greatest. The growth rate of each crystal face of ZnO is V(0001) > V(101) > V(100) > V(000).17 Therefore, ZnO crystal grows rapidly along the (0001) direction, and finally the nanorod structure with hexagonal cross section is formed. After H3BO3 solution is gradually added, it reacts with coating agent C6H12N4 attached to the non-polar surface of the ZnO crystal, so that the HMT is decomposed, its shielding effect on the non-polarized plane is weakened, and the growth rate of these surfaces becomes faster, resulting in the growth of ZnO along the directions of [000] and [0001] or [20] and [010]18 and evolving into the morphology of nanosheets.
). Since the (0001) lattice plane has the lowest surface free energy, its growth rate is greatest. The growth rate of each crystal face of ZnO is V(0001) > V(101) > V(100) > V(000).17 Therefore, ZnO crystal grows rapidly along the (0001) direction, and finally the nanorod structure with hexagonal cross section is formed. After H3BO3 solution is gradually added, it reacts with coating agent C6H12N4 attached to the non-polar surface of the ZnO crystal, so that the HMT is decomposed, its shielding effect on the non-polarized plane is weakened, and the growth rate of these surfaces becomes faster, resulting in the growth of ZnO along the directions of [000] and [0001] or [20] and [010]18 and evolving into the morphology of nanosheets.
The photocatalytic activity of composite structure is studied by the model of photocatalytic degradation of organic dyes RhB, the results are described in Fig. 2a. The degradation rate is given by the formula ξ = (1 − C/C0) × 100%, where C and C0 are the dye concentration at time t = t1 and t = 0. After 90 minutes of UV irradiation, the RhB solution without catalyst is hardly degraded (degradation rate is about 5%), indicating that the self-degradation efficiency of RhB is relatively low. When Au/3% B-ZnO/PET–GR catalyst is added, the degradation rate of the sample reaches the maximum of 38.11%, which is 1.62 times that of the un-doped composite structure (23.49%), illustrating that the photocatalytic activity of the composites is enhanced by the incorporation of proper amount of B atoms. However, the degradation rates of the Au/5% B-ZnO/PET–GR and Au/7% B-ZnO/PET–GR are all lower than 20%, which is caused by the poor crystallinity of ZnO crystals after excessive B incorporation.
 | ||
| Fig. 2 Photocatalytic properties of Au/B-ZnO/PET–GR structures. (a) The efficiency of Au/BZO/PET–GR doped with different concentrations of B atoms to degrade RhB solution under UV light, (b) kinetics of the RhB degradation catalyzed by pure and 3% B-doped Au/ZnO/PET–GR, (c) photocatalytic degradation curves of Au/B-ZnO/PET and B-ZnO/PET–GR composite structures under two doping concentrations, (d) energy band diagram of Au/BZO/PET–GR composite: e−, electron; h+, hole; Ec1 (Ec2) and Ev1 (Ev2), conduction band and valence band of PET–GR substrate (B-ZnO); EVAC, vacuum level; Eb, built-in electric field; Wm (Ws), Au (ZnO) work function; (EF)m and (EF)s, Fermi level of Au and ZnO. | ||
According to Langmuir–Hinshelwood kinetic model (ln(C/C0) = Ka × t + b, Ka is the reaction rate constant),19 the relationship between ln(C/C0) and irradiation time is explored by means of linear fitting (Fig. 2b). The ln(C/C0) − T curves of un-doped and 3% B-doped composite structures exist an excellent linear correlation, and their correlation coefficients (R2) are 0.976 and 0.985, which are expressly close to 1. The photocatalytic degradation reaction of RhB by the Au/ZnO/PET–GR and Au/3% B-ZnO/PET–GR follows the first order reaction kinetics, and their dynamic equations are ln(C/C0) = −0.00277t − 0.01278 and ln(C/C0) = −0.0052t + 0.00034. The rate constant of B-ZnO nanostructure is larger than that of the un-doped sample, judging that the photocatalytic degradation capacity of the former is stronger, which is consistent with the results in Fig. 2a. Au/B-ZnO/PET and BZO/PET–GR catalyst are prepared by the same method in order to investigate the effect of substrates and additional metal layer on the photocatalytic properties for composite structure, the results are shown in Fig. 2c. The degradation rates of pure and 3% B-doped Au/B-ZnO/PET grown on a single layer substrate are 10.2% and 15.31%, respectively. This is because the substrate PET belongs to resin with good electrical insulating property and is unable to promote the transmission of electrons like graphene layer with high conductivity. In addition, the efficiencies of pure and 3% B-doped B-ZnO/PET–GR without sputtering Au metal layers are 18.88% and 27.49%, are also lower than that of Au/BZO/PET–GR catalyst.
The working mechanism of photocatalytic reaction of Au/B-ZnO/PET–GR composite structure is analyzed by band theory, the schematic diagram without regard to the interface state demonstrates in Fig. 2d. The intrinsic graphene is generally identified as zero band-gap semiconductor. However, due to the presence of small amounts of impurities and interaction of graphene-substrates, the graphene at room temperature is often a p-type semiconductor with very narrow band-gap.20 A p–n heterojunction is formed when it is in close contact with the n-type B-ZnO semiconductor. In the case of UV irradiation, valence electrons are excited by photons to the conduction band, generating the electron–hole pairs, that is, holes (electrons) appear on the valence band (conduction band) of B-ZnO (PET–GR). Since there is a built-in electric field (Eb) from ZnO side toward substrate side at the interface of heterojunction, it is produced by the diffusion movement of electrons or holes, giving rise to the migration of photogenerated electrons (holes) from conduction band (valence band) of substrate (ZnO) to conduction band (valence band) of ZnO (substrate). Thus, the electron–hole pairs are separated. Photogenerated holes, as a strong oxidizing agent, are able to oxidize hydroxide ions (OH−) in water into hydroxyl radicals (˙OH). And hydroxyl radicals are powerful oxidants in nature that are second only to fluorine. The electrons are capable of converting oxygen into superoxide radicals with oxidizing property, which in turn reacts with hydrogen ions to form hydroxyl radicals. Chemical equations are described as: h+ + OH− → ˙OH, e− + O2 → ˙O2−, ˙O2− + e− + 2H+ → 2˙OH, h+/˙O2−/˙OH + RhB → CO2 + H2O. When B atoms as a donor impurity are incorporated into n-type ZnO, the concentration of donor impurity in the n-region of heterojunction increases, and more electrons are ionized comparing to the case of un-doped, the diffusion movement of electrons from n-region to p-region is further enhanced. As a result, the built-in electric field larger than that of un-doped is formed, causing the photogenerated electron–hole pairs to be further separated. Therefore, the composite structure doped with 3% B atoms has better photocatalytic performance compared to pure Au/B-ZnO/PET–GR. It is also possible to improve the photocatalytic activity by sputtering an appropriate thickness of metal layer over ZnO nanostructure. Because the work function of metal Au (5.1 eV)21 is greater than that of ZnO semiconductor (4.84 eV),22 that is Wm > Ws, and the Fermi level of ZnO ((EF)s) is higher than that of Au ((EF)m) before they touch. When the equilibrium is reached, the Schottky barrier is formed at the interface, where the electrons of ZnO semiconductor flow to Au metal, thereby facilitating the separation of electron–hole pairs. Besides, the surface plasmon resonance effect of precious metal is also likely to be another reason for enhancing the photocatalytic activity of composite structure.23,24
The cyclic voltammetry measurement results suggest that the CV curves of Au/3% B-ZnO/PET–GR electrodes at different scan rate is not the regular rectangle exhibited by the ideal supercapacitor (Fig. 3a), indicating that the capacitive performance of the composite structure in the electrolyte is mainly derived from the redox reaction of pseudo-capacitance. A pair of Faraday redox peaks in the CV curve are observed near the potential windows of −0.5 and 0.8 V, which is caused by the following reason that the potential window is larger than the decomposition window of aqueous electrolyte considering the low electrolyte concentration of 0.5 M, thus, the oxidation and reduction peaks are more likely to be the decomposition of electrolytes rather than the sodium intercalation. The specific capacitance of the composite structure is calculated according to the following equation:  , where m is the electrode mass, K represents the scan rate, V2 and V1 are the upper and lower limits of potential window, and I(V) expresses the voltammetric current. When the scan rate is 5 mV s−1 and 20 mV s−1, the specific capacitance of 62.96 F g−1 and 40.64 F g−1 for Au/3% B-ZnO/PET–GR electrode are gained. Fig. 3b shows a graph of specific capacitance versus scan rate. As the scan rate increases from 5 mV s−1 to 20 mV s−1, the specific capacitance tends to decrease, but its retention rate attains 64.55%. On account of that at a large scan rate, the electrolyte ions are less likely to enter the interior of electrode and can't be diffused and absorbed,25 resulting in charge exchange between ions and electrode materials mainly occur on the surface of electrode, where only the external active surface is used for charge storage. However, in the case that scan rate is small, the exchange of electric charge lies in electrode’ inside and all of outer active surface.
, where m is the electrode mass, K represents the scan rate, V2 and V1 are the upper and lower limits of potential window, and I(V) expresses the voltammetric current. When the scan rate is 5 mV s−1 and 20 mV s−1, the specific capacitance of 62.96 F g−1 and 40.64 F g−1 for Au/3% B-ZnO/PET–GR electrode are gained. Fig. 3b shows a graph of specific capacitance versus scan rate. As the scan rate increases from 5 mV s−1 to 20 mV s−1, the specific capacitance tends to decrease, but its retention rate attains 64.55%. On account of that at a large scan rate, the electrolyte ions are less likely to enter the interior of electrode and can't be diffused and absorbed,25 resulting in charge exchange between ions and electrode materials mainly occur on the surface of electrode, where only the external active surface is used for charge storage. However, in the case that scan rate is small, the exchange of electric charge lies in electrode’ inside and all of outer active surface.
 | ||
| Fig. 3 Electrochemical performance of Au/B-ZnO/PET–GR composite electrodes measured in 0.5 M Na2SO4 electrolyte solution. (a) CV curves of Au/BZO/PET–GR electrode doped with 3% B at different scan rates, (b) diagrams of the scan rate versus the specific capacitance of Au/3% B-ZnO/PET–GR, (c) comparison of CV curves at a scan rate of 5 mV s−1 between Au/3% B-ZnO/PET–GR, 3% B-ZnO/PET–GR and Au/3% B-ZnO/PET electrodes, and the inset is the result of its corresponding capacitance, (d) electrochemical impedance spectroscopy of Au/B-ZnO/PET–GR and Au/ZnO/PET–GR electrodes, and the inset is a partially enlarged picture. | ||
The CV curves and specific capacitance of 3% B-ZnO/PET–GR and Au/3% B-ZnO/PET electrodes are also obtained to research the effect of substrate and metal layer on electrochemical nature for composite electrodes. As can be seen from Fig. 3c, the CV curves of two electrodes are similar in shape to that of Au/3% B-ZnO/PET–GR electrode, their capacitive properties are also determined by the Faraday pseudo-capacitance. But, the specific capacitance is significantly different, and its values of 60.89 F g−1 and 49.27 F g−1 for 3% B-ZnO/PET–GR and Au/3% B-ZnO/PET are gained, which are less than that of Au/3% B-ZnO/PET–GR electrode and are also smaller than that of 3DCS/polyaniline (three-dimensional CNT sponge/polyaniline) nanocomposite fibers (242.9 F cm−1) prepared by Li et al.26 It is speculated that the substrate plays a dominant role in factors which affect the electrochemical performance of the composite structure. This is chiefly due to the graphene layer with electron mobility of 2 × 105 cm2 V−1 s−1 and conductivity of 1 × 108 Ω m−1 provides the network of high conductivity for charge transfer in the process of charging and discharging.27,28 In addition, the graphene layer as a buffer layer is conducive to mutual contact of ZnO with entire substrate. When the substrate is only single-layer PET, there is a large number of defects in its interface with ZnO, and the contact is not especially tight, thus extending the path of ion diffusion and migration, which is not beneficial to electrochemical nature of electrode.
The typical electrochemical impedance spectra of Au/ZnO/PET–GR and Au/B-ZnO/PET–GR electrodes are described in Fig. 3d, and the inset is a partially enlarged picture. The equivalent series resistance (ESR) of 3.5 Ω and 3.1 Ω for above two electrodes are acquired from X-axis intercept of Nyquist diagram. The smaller ESR facilitates to lessen the energy loss of supercapacitor. In the high frequency region of EIS, a small section of capacitive loops is discovered, which is not a complete semicircle, indicating that the existence of electrode dispersion effect. This is related to the nonuniformity of interface properties between electrode and electrolyte or absorbed layer on electrode surface. Furthermore, the circular-arc radius of high-frequency impedance curve of B-ZnO is obviously smaller than that of pure ZnO, which deems that the interfacial resistance and charge transfer impedance of Au/B-ZnO/PET–GR electrode are smaller than those of the latter. Therefore, the proper amount of B doping are capable of decreasing the grain boundary resistance, promoting the charge transmission and then improving the electrochemical performance of composite electrode.
4 Conclusions
In summary, Au/B-ZnO/PET–GR composite structures were successfully prepared on flexible double-layer substrate by a simple and practicable method. The growth habit of ZnO nanocrystals was vastly affected by B atoms which were doped into the ZnO lattice, and then the growth rate of each crystal face changed, resulting in the transition of the morphology of ZnO from the nanorods to nanosheets. Au/B-ZnO/PET–GR exhibited favorable photocatalytic activity, the degradation rate increased from 23.49% to 38.11% due to the doping of B atoms. The main active materials in the degradation process of RhB solution were ˙OH, ˙O2− and holes. Au/3% B-ZnO/PET–GR showed pseudo-capacitance characteristics, and the graphene layer in the PET–GR substrate provided a high conductivity network for electrode charge transfer. The photocatalytic and electrochemical properties of composite structures were influenced by the variation of substrate and the lack of metal layer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (NSFC, No. 51671116), the Project Supported by Key Project of Natural Science Basic Research Plan in Shaanxi Province of China (No. 2016JZ016), Key Scientific Research Project of Natural Science Foundation of Education Department of Shaanxi Provincial Government (No. 16JS018), and the Scientific Research Startup Program for Introduced Talents of Shaanxi University of Technology, China (No. SLGQD16-04), Postgraduate Innovation Fund Project of Shaanxi University of Technology, China (No. SLGYCX1729).References
- O. Akhavan, ACS Nano, 2010, 4, 4174–4180 CrossRef CAS PubMed.
- C. Tian, Q. Zhang, A. Wu, M. Jiang, Z. Liang, B. Jiang and H. Fu, Chem. Commun., 2012, 48, 2858–2860 RSC.
- M. Ahmad, E. Ahmed, Z. L. Hong, J. F. Xu, N. R. Khalid, A. Elhissi and W. Ahmed, Appl. Surf. Sci., 2013, 274, 273–281 CrossRef CAS.
- T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, L. Servant, M. M. Müller, H. Kleebe, J. Ziegler and W. Jaegermann, Inorg. Chem., 2012, 51, 7764–7773 CrossRef PubMed.
- J. B. Zhong, J. Z. Li, J. Zeng, X. Y. He, W. Hu and Y. C. Shen, Mater. Res. Bull., 2012, 47, 3893–3896 CrossRef CAS.
- X. Dong, Y. Cao, J. Wang, M. B. Chan-Park, L. Wang, W. Huang and P. Chen, RSC Adv., 2012, 2, 4364–4369 RSC.
- S. Li, J. Wen, X. Mo, H. Long, H. Wang, J. Wang and G. Fang, J. Power Sources, 2014, 256, 206–211 CrossRef CAS.
- S. Li, J. Wen, X. Mo, H. Long, H. Wang, J. Wang and G. Fang, J. Power Sources, 2014, 256, 206–211 CrossRef CAS.
- Y. Li, X. Zheng, H. Si, M. Li, Y. Jiang, Y. Liu, X. Yan and Y. Zhang, J. Mater. Chem. A, 2016, 4, 17704–17710 CAS.
- X. Zheng, X. Yan, Y. Sun, Y. Li, M. Li, G. Zhang and Y. Zhang, J. Mater. Chem. A, 2016, 4, 17981–17987 CAS.
- Y. Liu, Y. Li, Y. Yang, Y. Wen and M. Wang, Scr. Mater., 2013, 68, 301–304 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and S. R. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- S. Bae, H. Kim, Y. Lee, X. Xu, J. Park, Y. Zheng, J. Balakrishnan, L. F. Tian, H. R. Kim, Y. Song, Y. Kim, K. S. Kim, B. Ozyilmaz, J. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed.
- H. H. Kim, Y. Chung, E. Lee, S. K. Lee and K. Cho, Adv. Mater., 2014, 26, 3213–3217 CrossRef CAS PubMed.
- M. Lorenz, R. Bottcher, S. Friedlander, A. Poppl, D. Spemann and M. Grundmann, J. Mater. Chem. C, 2014, 2, 4947–4956 RSC.
- J. Wu, M. Zhang, X. Xu, M. Liu and B. Fang, J. Chin. Ceram. Soc., 2009, 37, 1038–1043 CAS.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829–R858 CrossRef CAS.
- L. Yang, S. Dong, J. Sun, J. Feng, Q. Wu and S. Sun, J. Hazard. Mater., 2010, 179, 438–443 CrossRef CAS PubMed.
- S. Y. Zhou, G. H. Gweon, A. V. Fedorov, P. N. First, W. A. de Heer, D. H. Lee, F. Guinea, A. H. Castro Neto and A. Lanzara, Nat. Mater., 2007, 6, 770–775 CrossRef CAS PubMed.
- N. K. Reddy, M. Devika and C. W. Tu, RSC Adv., 2014, 4, 37563–37568 RSC.
- H. Wu, M. Xue, J. Ou, F. Wang and W. Li, J. Alloys Compd., 2013, 565, 85–89 CrossRef CAS.
- B. J. Niu, L. L. Wu, W. Tang, X. T. Zhang and Q. G. Meng, CrystEngComm, 2011, 13, 3678–3681 RSC.
- Z. Han, L. Wei, Z. Zhang, X. Zhang, H. Pan and J. Chen, Plasmonics, 2013, 8, 1193–1202 CrossRef CAS.
- X. Li, Z. Wang, Y. Qiu, Q. Pan and P. Hu, J. Alloys Compd., 2015, 620, 31–37 CrossRef CAS.
- Y. Li, Z. Kang, X. Yan, S. Cao, M. Li, Y. Liu, S. Liu, Y. Sun, X. Zheng and Y. Zhang, J. Mater. Chem. A, 2017, 5, 22559–22567 CAS.
- Y. Haldorai, W. Voit and J. J. Shim, Electrochim. Acta, 2014, 120, 65–72 CrossRef CAS.
- W. Qian, P. A. Greaney, S. Fowler, S. Chiu, A. M. Goforth and J. Jiao, ACS Sustainable Chem. Eng., 2014, 2, 1802–1810 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |