
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tandem intramolecular cyclisation/1,3-aryl shift in N-(4,4-diethoxybutyl)-1-arylmethanimines (Kazan reaction): synthesis of 3-benzylidene-1-pyrrolines†
A. V. Smolobochkina,
A. S. Gazizov *a,
A. S. Melyashovaa,
J. K. Voroninaa,
A. G. Strelnika,
S. Z. Vatsadzeb,
A. R. Burilova,
M. A. Pudovika,
O. A. Fedorovac and
O. G. Sinyashina
*a,
A. S. Melyashovaa,
J. K. Voroninaa,
A. G. Strelnika,
S. Z. Vatsadzeb,
A. R. Burilova,
M. A. Pudovika,
O. A. Fedorovac and
O. G. Sinyashina
aA.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center, Russian Academy of Sciences, Arbuzova str., 8, Kazan, Russian Federation. E-mail: agazizov@iopc.ru
bLomonosov Moscow State University, Chemistry Department, 1, GSP-1, 1-3 Leninskiye Gory, Moscow, 119991, Russian Federation
cA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, GSP-1, V-334, Vavilova str. 28, Moscow, 119991, Russian Federation
First published on 1st November 2017
Abstract
In this article, a novel tandem reaction, which transforms N-(4,4-diethoxybutyl)imines into 3-arylidene-1-pyrrolines, is described. The substrate scope of the starting acetals includes arenes with electron-donating and withdrawing groups. The X-ray study of products confirmed the E-stereochemistry of the double bonds formed. The best yields (99%) are found in boiling xylene in the presence of TsOH (or 2-nitroresocinol) during 40 (50) hours. The study of substituents effect on the course of the reaction revealed that cascade process takes place, combining acid-catalyzed intramolecular cyclization of N-(4,4-diethoxybutyl)imines and unusual 1,3-sigmatropic shift of the aryl fragment. Cyclic imines that are formed in high/excellent yields are of interest both from the viewpoint of their biological activity and synthetic usefulness.
Introduction
A pyrrolidine core is involved in many known drug preparations,1,2 as well as biologically active natural compounds, and the development of the methods for the synthesis of these compounds is an everlasting challenge for synthetic organic chemists.1,3,4 Most of the strategies for the synthesis of substituted pyrrolidines employ intramolecular C–N bond-forming reactions for the construction of the heterocyclic ring.5,6 Development of methods of synthesis of substituted pyrrolidines via one-pot heterocyclic ring closure with the simultaneous formation of an exocyclic carbon–carbon bond still remains a major challenge. Imines represent one of the classes of starting compounds widely used in these reactions. As an example, the synthesis of pyrrolidine derivatives by the intramolecular Mannich reaction of 2-amino-4-nitrobutyric7 and γ-aminobutyric acids8,9 and cyclization of N-(3-chloropropyl)imines in the presence of lithium10 can be mentioned. In these reactions, imines act as electrophiles, which represent the counterparts of carbonyl compounds. At the same time, the presence of nucleophilic nitrogen atom with lone electron pair provides their reactions with electrophilic particles, as exemplified by the formation of acyliminium salts.11,12 Examples of such intramolecular reactions, which give rise to pyrrolidine ring, is the acid-mediated cyclization of N-(1-phosphoryl-4-chlorobutyl)imines13 and cyclization of γ,δ-alkenylimines under the action of electrophilic reagents.14–17 An approach to the synthesis of proline derivatives from ketals and acetals of 2-((diphenylmethylene)amino)-5-oxopentanoic and 2-((diphenylmethylene)amino)-5-oxohexanoic acids under the action of trifluoroacetic acid described in the recent work should particularly be mentioned.18However, very few of these methods allow for simultaneous intramolecular C–N bond formation and intermolecular formation of a carbon–carbon bond.19–22 Existing methods require harsh reaction conditions or toxic or expensive reagents, such as Pd complexes.23 There is the only example of synthesis of α-substituted pyrrolidine via reaction of 1-(4-oxobutyl)urea derivative with thiophenol in acidic media.24
We have recently developed a new approach to the synthesis of substituted heterocyclic systems from nitrogen-containing acetals, which is summarized in reviews.25,26 This approach is based on the combination of two processes, namely, intramolecular cyclization of the starting acetal to give heterocyclic ring and further intermolecular functionalization of the intermediate. Using this approach, we succeeded in the synthesis of imidazolidine-2-one27 and 2-arylpyrrolidine derivatives28 with various substituents at nitrogen atom (Scheme 1A). Intermediate compound in these reactions was cyclic iminium cation, whose reaction with aromatic nucleophile afforded corresponding aryl-substituted heterocycles. In the frames of this work, we assumed that the application of N-(4,4-diethoxybutyl)imines to these reactions would also lead to the formation of cyclic iminium cations. The presence of two different electrophilic centers and nucleophilic aryl fragment in the compound would open possibilities for various intramolecular processes and, thus, for the synthesis of 3-substituted pyrrolidine derivatives (Scheme 1B).
 | ||
| Scheme 1 Intramolecular ring closure in nitrogen-containing acetals. | ||
Herein, we report the results of our studies on the acid-catalyzed cyclization of N-(4,4-diethoxybutyl)imines. The mechanism of pyrrolidine ring formation in this case appeared to be different from the mechanism we previously proposed.27 It turned out that new cascade reaction takes place, which combines acid-catalyzed intramolecular cyclization of N-(4,4-diethoxybutyl)imines followed by unusual 1,3-sigmatropic shift of aryl fragment. The result of these cascade transformations is the formation of 3-arylidene-1-pyrroline derivatives (Scheme 1C).
Results and discussion
We have initially studied intramolecular cyclization of N-(4,4-diethoxybutyl)-1-phenylmethanimine 1a (R = Ph) in the presence of trifluoroacetic acid (Scheme 2). The product of this reaction was (E)-3-phenylidene-1-pyrroline 2a, which was isolated as trifluoroacetate in 56% yield. Until recently, the only method for the synthesis of these compounds was the reaction of benzaldehyde derivatives with 1-pyrroline29 or pyrrolidine.30 | ||
| Scheme 2 Intramolecular cyclization of N-(4,4-diethoxybutyl)-1-arylmethanimines 1. | ||
In the next step, we studied the effect of aromatic substituent on the course of this reaction (Scheme 2). Initial imines 1a–r were prepared from corresponding aldehydes and (4,4-diethoxy)butane-1-amine (see ESI†). The results are summarized in Table 1.
| No. | Cmpd | R | Yielda, % |
|---|---|---|---|
| a According to NMR spectroscopy data, the yield of individual compound is given in parentheses.b Isolated as free base.c Compound 1n was not isolated, see ESI.d Reaction did not proceed. | |||
| 1 | (E)-2a | Ph | 80 (56, 52b) |
| 2 | (E)-2b | 4-MeO–C6H4 | 92 (50) |
| 3 | (E)-2c | 4-Propargyloxy–C6H4 | 89 (43) |
| 4 | (E)-2d | 4-Br–C6H4 | 69 (26, 68b) |
| 5 | (E)-2e | 4-Cl–C6H4 | 73 (40) |
| 6 | (E)-2f | 4-HO–C6H4 | ∼99 (70) |
| 7 | (E)-2g | 4-NO2–C6H4 | 79 (37) |
| 8 | (E)-2h | 4-Me2N–C6H4 | 66 (19) |
| 9 | (E)-2i | 3-F–C6H4 | 77 (42) |
| 10 | (E)-2j | 3-I–C6H4 | 85 (41) |
| 11 | (E)-2k | 2-HO–C6H4 | 58 (19) |
| 12 | (E)-2l | 2-HO–5-Cl–C6H3 | 51 (15) |
| 13 | (E)-2m | 1-Naphthyl | 61 (31) |
| 14 | (E)-2nc | 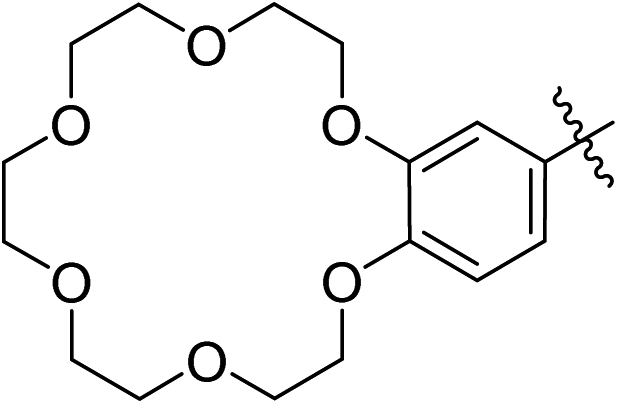 |
∼99 (97b) |
| 15 | 2o | 3,5-t-Bu-4-HO-C6H2 | —d |
| 16 | 2p | Antracen-9-yl | —d |
| 17 | 2q | 4-Pyridyl | —d |
| 18 | 2r | 3-Pyridyl | −87 (84b) |
The introduction of electron donor substituent at p-position of aromatic fragment slightly increases the yield of products (E)-2b,c,f,n (Table 1, entries 2, 3, 6, 14). An exception is compound (E)-2h (Table 1, entry 8) bearing dimethylamino group at p-position. This is presumably related to the protonation of nitrogen atom in acidic medium, which changes its behavior from electron donor to electron acceptor. The presence of electron acceptor nitro groups at p-position (Table 1, entry 7) or halogens (Table 1, entries 4, 5) gives a slight decrease in the yield of pyrrolines (E)-2d,e,g, in analogy with the presence of halogens at m-position of aromatic ring (Table 1, entries 9, 10). In this case, the yield of m- and p-substituted products is comparable. In contrast, the introduction of electron donor hydroxy group at o-position leads to the drastic decrease in the yield of products (E)-2k,l as compared to their p-substituted counterpart (E)-2f (Table 1, entries 11, 12 and no. 6). If bulky tert-butyl substituents are attached to hydroxyl group, the reaction does not proceed (Table 1, entry 15). As follows from the data, the substituents at p- and m-positions of phenyl fragment affect weakly the yield of target compounds. The presence of substituents at o-position restricts the reaction, though in this case target compounds can be prepared in reasonable yields. However, with the substitution of hetero- and polyaromatic fragments for phenyl, target compounds are not observed (Table 1, entries 16–18). The exceptions are compounds (E)-2m,r (Table 1, entries 13, 18), which were isolated in 31% and 84% yield, correspondingly.
It should also be noted that exclusively (E)-isomers of compounds 2a–r were obtained and no (Z)-isomers were detected in either case. The structure of the compounds was proved by two-dimensional spectroscopy data; in addition, the structure of compounds (E)-2a,b,i was confirmed by X-ray study (Fig. 1). In all crystals practically planar 3-aryliden-1-pyrrolinium cations (deviation from plane 0.046(2) Å in 2a, 0.033(2) Å in 2b and 0.084(7) Å in 2i) are associated with the trifluoroacetate anion by classical NH⋯O hydrogen bonds (distances N⋯O are 2.698(3) Å in 2a, 2.694(4) Å in 2b, and 2.688(9) Å in 2i).
 | ||
| Fig. 1 The molecular structure of compound 2b*CF3COOH. Ellipsoids are provided with a probability of 50%. | ||
Further we discovered that pyrrolinium salts 2a–r undergo slowly (E)-(Z) isomerization in solution. This isomerization was studied in more detail using compounds 2k as an example. Isomeric structure of compound 2k was determined by the set of homo- and heterocorrelation experiments (1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC,31,32 and 1D DPFGNOE33). Initially proton spin systems were established from 1H–1H COSY experiment. Then, the entire structure of fragments and their relationship was determined by the combination of 1H–13C HSQC/HMBC correlations. Unambiguous assignment of the signals of (E) and (Z) forms was made on the basis of observed nuclear Overhauser effects (NOEs) (Fig. 2, key NOEs are indicated by arrows).
 | ||
| Fig. 2 1H NMR spectra of compound 2k in CD3OD at 303 K at various time intervals, arrows indicate key NOEs. | ||
At the same time, by using of 2a,d,n we showed that as free base the compounds are stable in (E)-form for a long time. In accordance with these data, we suggest that the key factor for (E)-(Z) isomerization of 1-pyrrolines 2 is the presence of positive charge at nitrogen atom (Scheme 3). It should be noted that analogous (E)-(Z) isomerization of 3-arylidene-1-pyrrolidines under UV irradiation is mentioned in the literature.29
 | ||
| Scheme 3 (E)-(Z) isomerization of pyrroline 2k. | ||
In the next step of investigations, we studied the effect of reaction conditions and catalyst in the case of imine 1a (Table 2).
| No. | Solvent | Catalyst | Temp. | Time, h | Yield, % |
|---|---|---|---|---|---|
| a Yield of individual compound.b Product was isolated as trifluoroacetate.c Reaction did not proceed.d Content of the product in the reaction mixture according to NMR spectroscopy data.e The product was not purified from catalyst. | |||||
| 1 | CHCl3 | CF3COOH (1 eq.) | 20 °C | 6 | 56a,b |
| 2 | C6H6 | — | Reflux | 60 | —c |
| 3 | C6H6 | TsOH (0.01 eq.) | Reflux | 30 | ∼5d |
| 4 | o-Xylene | TsOH (0.01 eq.) | Reflux | 30 | ∼30d |
| 5 | o-Xylene | TsOH (0.01 eq.) | Reflux | 60 | ∼50d |
| 6 | AcOH | — | 20 °C | 20 | ∼30d |
| 7 | AcOH | — | Reflux | 20 | ∼40d |
| 8 | o-Xylene | PhCO2H (0.1 eq.) | Reflux | 80 | ∼60d |
| 9 | o-Xylene | 2-Nitroresorcinol (0.1 eq.) | Reflux | 50 | ∼99d,e |
| 10 | o-Xylene | TsOH (0.1 eq.) | Reflux | 40 | ∼99d (81a) |
| 11 | o-Xylene | Al2O3 (0.1 eq.) | Reflux | 40 | ∼74d (50a) |
As follows from table, the reaction at room temperature in the presence of 1 equiv. of trifluoroacetic acid allows isolating the target compound in 56% yield (Table 2, entry 1). According to NMR spectroscopy data, the competitive hydrolysis of initial imine 1a is observed. The use of stronger p-toluenesulfonic acid at 0.01 equiv. of the amount of initial imine gave only trace amounts of compound 2a (Table 2, entry 3). An increase in the temperature and time of reaction resulted in a significant increase in the amount of the product (Table 2, entries 4, 5). The reaction does not proceed without catalyst even after long-term heating at elevated temperatures (Table 2, entry 2).
We also studied the possibility of this reaction in the presence of weak acids. Long-term heating of imine 1a in the presence of benzoic acid afforded target compound 2a in 60% yield (Table 2, entry 8). During the reaction in acetic acid, the content of pyrroline 2a in the reaction mixture decreases regardless of the temperature of reaction (Table 2, entries 6, 7). It is interesting that the employment of 2-nitroresorcinol as a catalyst gives the target compound almost quantitatively according to NMR spectroscopy data. Nevertheless, we did not succeed in the complete purification of the product of reaction from catalyst in this case (Table 2, entry 9). The use of heterogeneous catalyst represented by acidic aluminum oxide provided the isolation of individual pyrroline 2a in 50% yield (Table 2, entry 11). The reaction in boiling o-xylene in the presence of 0.1 equiv. of p-toluenesulfonic acid was most optimal (Table 2, entry 10). Target (E)-3-phenylidene-1-pyrroline 2a was isolated in 81% yield in this case.
The proposed mechanism of the reaction is given in Scheme 4. According to scheme, the first step of the reaction is the protonation of nitrogen atom with the formation of iminium salt A. Subsequent elimination of ethanol molecule leads to enol derivative B. Further intramolecular cyclization with π-electrons of double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond gives cyclic intermediate C. The combination of these steps can be considered intramolecular Mannich reaction, which is analogous to the previously described cyclization of imines of γ-aminobutyric acid under the action of Lewis acid.8 In the next step of reaction, intermediate D is formed as a result of [1,3]-sigmatropic rearrangement, which is accompanied by the migration of aryl fragment. The driving force of this rearrangement is presumably the formation of more stable iminium cation. It should be noted that the reactions accompanied by 1,3-migration of phenyl group are quite rare. Until recently, only one example of 1,3-migration of phenyl group in pyrrole derivatives is known.34 In the final step of reaction, (E)-3-arylidene-1-pyrroline 2 is formed through the elimination of ethanol molecule.
C bond gives cyclic intermediate C. The combination of these steps can be considered intramolecular Mannich reaction, which is analogous to the previously described cyclization of imines of γ-aminobutyric acid under the action of Lewis acid.8 In the next step of reaction, intermediate D is formed as a result of [1,3]-sigmatropic rearrangement, which is accompanied by the migration of aryl fragment. The driving force of this rearrangement is presumably the formation of more stable iminium cation. It should be noted that the reactions accompanied by 1,3-migration of phenyl group are quite rare. Until recently, only one example of 1,3-migration of phenyl group in pyrrole derivatives is known.34 In the final step of reaction, (E)-3-arylidene-1-pyrroline 2 is formed through the elimination of ethanol molecule.
 | ||
| Scheme 4 Proposed pathway for the formation of 3-arylidene-1-pyrrolines 2. | ||
It was already mentioned that nitrogen atom in imines may act as nucleophile in the reactions with carbocations. Assuming this fact, the formation of compounds 2 can be rationalized by acid-catalyzed intramolecular cyclization of N-(4,4-diethoxybutyl)imine 1 with the formation of 1-pyrroline and benzaldehyde derivative18 (Scheme 5A). The similar pathway includes initial N-(4,4-diethoxybutyl)imine 1 hydrolysis followed by intramolecular cyclization of 4,4-diethoxybutan-1-amine (Scheme 5B). The last stage of both processes is the previously described29,35 reaction of 1-pyrroline with aldehyde.
 | ||
| Scheme 5 Alternative routes of the formation of 3-arylidene-1-pyrrolines 2. | ||
To make a final decision among these mechanisms, we first studied the possibility of formation of 1-pyrroline from 4,4-diethoxybutan-1-amine according to the Scheme 5, pathway B. It was shown that 4,4-diethoxybutan-1-amine does not undergo intramolecular cyclization in reaction conditions, regardless of the solvent, catalyst used and its amount.
Further, we investigated the reaction of 1-pyrroline with benzaldehyde. The previously described36 pyrroline trimer (1,6,11-triazatetracyclo-[10.3.0.0. (ref. 2 and 6)0 (ref. 7 and 11)]pentadecane) was used to generate 1-pyrroline in situ. Although the reaction of this compound with aromatic aldehydes in neutral conditions is described,29,35 no data is present on its acid-catalyzed variant. We carried out this reaction in chloroform at room temperature at the presence of 1 eq. of trifluoroacetic acid and in refluxing o-xylene with 0.1 eq. of p-toluensulfonic acid as catalyst. In both cases reaction resulted in a complex mixture of products and only traces of target 3-arylidene-1-pyrroline 2a was observed.
Additionally, we tried to exclude the possibility of N-(4,4-diethoxybutyl)imine 1 cyclization according to the Scheme 5, pathway A. In line with this mechanism, the presence of the lone pair at nitrogen atom is a necessary condition for the reaction to occur. Thus, we have decided to study the intramolecular cyclization of iminium salt 3, which has no lone pair at nitrogen atom. However, treatment of the compound 1a with methyl iodide in acetonitrile at room temperature surprisingly led to the formation of 1-pyrroline derivative 4. Reaction proceeded in almost quantitative yield even without acid catalyst (Scheme 6). Although the NMR spectrum of the reaction mixture showed the presence of iminium salt 3 alongside with compound 4 and starting acetal 1a, we didn't succeeded in isolation it in pure form due to its rapid intramolecular cyclization.
 | ||
| Scheme 6 Intramolecular cyclization of N-alkyliminium salt 3. | ||
It is evident that in this case, that the implementation of the latter reaction mechanism is impossible and the data obtained so far indicate the mechanism of the formation of 1-pyrrolines 2 depicted in Scheme 4.
It should be noted that 3-arylidene-1-pyrroline salts 2 can be considered intermediate compounds in Mannich reaction. To demonstrate their synthetic potential, we carried out the reaction of compounds (E)-2a,d with acetone and 4-chlororesorcinol, respectively, and 2-substituted pyrrolidine derivatives 5,6 were prepared (Scheme 7).
 | ||
| Scheme 7 Synthesis of 2-substituted pyrrolidine derivatives. | ||
The structure of compound 6 was additionally confirmed by X-ray study (Fig. 3). The structure of the cation in this compound is not planar due to the quaternization of the nitrogen atom. Anion and cation are binded by hydrogen bond. The crystal structure is formed by hydrogen bonds between cations and anions, and represents infinite chains along the axis 0a, connected by CH⋯O interactions.
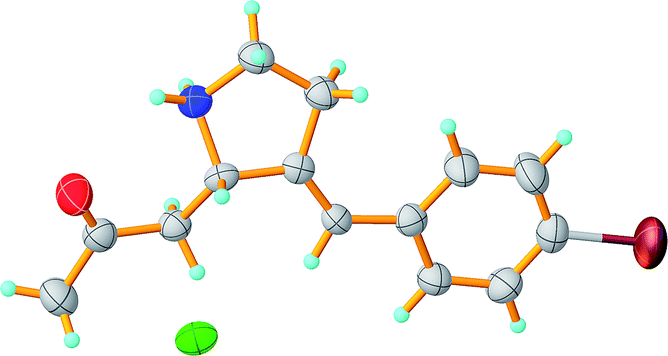 | ||
| Fig. 3 The molecular structure of compound 6*HCl in crystals. Ellipsoids are provided with a probability of 50%. | ||
Conclusions
Thus, we have discovered a novel tandem reaction, which transforms N-(4,4-diethoxybutyl)imines to 3-arylidene-1-pyrrolines, and we suggest to call this new process as “Kazan reaction”. The reaction proceeds according to consecutive intramolecular Mannich reaction and [1,3]-sigmatropic rearrangement of aryl fragment and does not require expensive and/or highly toxic reagents. Cyclic imines are formed in high/excellent yields and are of interest both from the viewpoint of their biological activity and synthetic value.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (Grant No. 16-13-10023).Notes and references
- P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS.
- E. De Clercq, Nat. Rev. Drug Discovery, 2006, 5, 1015–1025 CrossRef CAS PubMed.
- C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405 RSC.
- V. A. Efimov, A. V. Aralov and O. G. Chakhmakhcheva, Russ. J. Bioorg. Chem., 2010, 36, 663–683 CrossRef CAS.
- M. Piehon and B. Figadsre, Tetrahedron: Asymmetry, 1996, 7, 927–964 CrossRef.
- A. Mitchinson and A. Nadin, J. Chem. Soc., Perkin Trans. 1, 2000, 2862–2892 RSC.
- H. Y. Kim, J.-Y. Li, S. Kim and K. Oh, J. Am. Chem. Soc., 2011, 133, 20750–20753 CrossRef CAS PubMed.
- S. Suresh and M. Periasamy, Tetrahedron Lett., 2004, 45, 6291–6293 CrossRef CAS.
- N. Kise, K. Ohya, K. Arimoto, Y. Yamashita, Y. Hirano, T. Ono and N. Ueda, J. Org. Chem., 2004, 69, 7710–7719 CrossRef CAS PubMed.
- M. Yus, T. Soler and F. Foubelo, J. Org. Chem., 2001, 66, 6207–6208 CrossRef CAS PubMed.
- M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Russ. Chem. Rev., 2017, 86, 1 CrossRef CAS.
- B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431–1628 CrossRef CAS PubMed.
- O. A. Ramírez-Marroquín, I. Romero-Estudillo, J. L. Viveros-Ceballos, C. Cativiela and M. Ordóñez, Eur. J. Org. Chem., 2016, 2016, 308–313 CrossRef.
- D. Schley and J. Liebscher, Eur. J. Org. Chem., 2007, 2007, 2945–2957 CrossRef.
- D. De Smaele and N. De Kimpe, J. Chem. Soc., Chem. Commun., 1995, 8, 2029 RSC.
- N. de Kimpe, M. Boelens, J. Piqueur and J. Baele, Tetrahedron Lett., 1994, 35, 1925–1928 CrossRef CAS.
- N. De Kimpe and M. Boelens, J. Chem. Soc., Chem. Commun., 1993, 916–918 RSC.
- T. Kano, T. Kumano, R. Sakamoto and K. Maruoka, Org. Biomol. Chem., 2013, 11, 271–278 CAS.
- R. C. Larock, H. Yang, S. M. Weinreb and R. J. Herr, J. Org. Chem., 1994, 59, 4172–4178 CrossRef CAS.
- H. Yorimitsu, K. Wakabayashi, H. Shinokubo and K. Oshima, Bull. Chem. Soc. Jpn., 2001, 74, 1963–1970 CrossRef CAS.
- Y. Tamaru, M. Hojo and Z. Yoshida, J. Org. Chem., 1988, 53, 5731–5741 CrossRef CAS.
- Y. Tamaru and M. Kimura, Synlett, 1997, 749–757 CrossRef CAS.
- M. B. Bertrand and J. P. Wolfe, Tetrahedron, 2005, 61, 6447–6459 CrossRef CAS.
- L. E. Overman and J. P. Wolfe, J. Org. Chem., 2001, 66, 3167–3175 CrossRef CAS PubMed.
- A. S. Gazizov, A. R. Burilov, M. A. Pudovik and O. G. Sinyashin, Russ. Chem. Rev., 2017, 86, 75–98 CrossRef CAS.
- A. V. Smolobochkin, A. S. Gazizov, A. R. Burilov and M. A. Pudovik, Chem. Heterocycl. Compd., 2016, 52, 753–765 CrossRef CAS.
- M. S. Khakimov, A. S. Gazizov, A. R. Burilov, M. A. Pudovik and A. I. Konovalov, Russ. J. Gen. Chem., 2009, 79, 1163–1166 CrossRef CAS.
- A. S. Gazizov, A. V. Smolobochkin, A. R. Burilov and M. A. Pudovik, Chem. Heterocycl. Compd., 2014, 50, 707–714 CrossRef CAS.
- D. Sampedro, A. Migani, A. Pepi, E. Busi, R. Basosi, L. Latterini, F. Elisei, S. Fusi, F. Ponticelli, V. Zanirato and M. Olivucci, J. Am. Chem. Soc., 2004, 126, 9349–9359 CrossRef CAS PubMed.
- S. Mandal, S. Mahato and C. K. Jana, Org. Lett., 2015, 17, 3762–3765 CrossRef CAS PubMed.
- A. E. Derome, Modern NMR Techniques for Chemistry Research, Pergamon, Cambridge, 1988 Search PubMed.
- Atta-ur-Rahman, One and Two Dimensional NMR Spectroscopy, Elsevier, Amsterdam, 1989 Search PubMed.
- K. Stott, J. Stonehouse, J. Keeler, T.-L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199–4200 CrossRef CAS.
- W. M. L. Cheung and M. P. Sammes, J. Chem. Res., Synop., 1991, 236–237 CAS.
- Y. Nomura, T. Bando, Y. Takeuchi and S. Tomoda, Bull. Chem. Soc. Jpn., 1983, 56, 3199–3200 CrossRef CAS.
- K. Ogawa, Y. Nomura, Y. Takeuchi and S. Tomoda, J. Chem. Soc., Perkin Trans. 1, 1982, 3031–3035 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data and copies of NMR spectra for all compounds. CCDC 1544114–1544117. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11216e |
| This journal is © The Royal Society of Chemistry 2017 |