
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAir-oxidation of phenolic resin aerogels: backbone reorganization, formation of ring-fused pyrylium cations, and the effect on microporous carbons with enhanced surface areas†
Hojat Majedi Far,
Suraj Donthula,
Tahereh Taghvaee,
Adnan Malik Saeed,
Zachary Garr,
Chariklia Sotiriou-Leventis* and
Nicholas Leventis *
*
Department of Chemistry, Missouri University of Science and Technology, Rolla, MO 65409, USA. E-mail: leventis@mst.edu; cslevent@mst.edu; Tel: +1-573-341-4391 Tel: +1-573-341-4353
First published on 2nd November 2017
Abstract
This paper is a thorough investigation of the chemical transformations during pyrolytic conversion of phenolic resins to carbons, and reports that all carbons obtained from main-stream phenolic resins including phloroglucinol–formaldehyde (FPOL), phloroglucinol–terephthalaldehyde (TPOL), resorcinol–formaldehyde (RF), and phenol–formaldehyde (PF) contain fused pyrylium rings and charge-compensating phenoxides. Those four phenolic resins were prepared via a fast HCl-catalyzed process as low-density nanostructured solids classified as aerogels, which, owing to their open porosity, allowed air circulation through their bulk. In that regard, the first step of this study was the air-oxidation of those phenolic resin aerogels at 240 °C. In FPOL and TPOL aerogels, that air-oxidation step kicked off a cascade of reactions leading to ring-fusion aromatization and formation of pyrylium O+-heteroaromatic rings in every repeat unit of the polymeric backbone. Despite the complexity of the process, those structural forms were well-defined, and were retained through pyrolytic carbonization (800 °C). Under the same conditions (240 °C/air), RF and PF aerogels did not undergo aromatization; instead, they just went through an autooxidation-like process that converted the –CH2– bridges between phenolic moieties into carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). Importantly, however, upon further stepwise pyrolysis under Ar, by 600 °C all four systems (TPOL, FPOL, RF and PF), irrespective of whether they had been previously oxidized or not, converged to a common chemical composition. Thereby, carbon produced by pyrolysis of phenolic resins at 800 °C always contains fused pyrylium rings. All chemical analysis relied on FTIR, solid-state 13C NMR, XPS and CHN analysis. The only and significant difference made by the low-temperature (240 °C) air-oxidation step was identified with the surface areas of carbons from aromatizable systems (TPOL and FPOL), which were higher than those from direct pyrolysis of as-prepared aerogels. Upon further reactive etching with CO2, those surface areas went as high as 2778 ± 209 m2 g−1. Those findings are directly relevant to high surface area carbons for gas sorption (e.g., capture and sequestration of CO2) and ion exchange materials.
O). Importantly, however, upon further stepwise pyrolysis under Ar, by 600 °C all four systems (TPOL, FPOL, RF and PF), irrespective of whether they had been previously oxidized or not, converged to a common chemical composition. Thereby, carbon produced by pyrolysis of phenolic resins at 800 °C always contains fused pyrylium rings. All chemical analysis relied on FTIR, solid-state 13C NMR, XPS and CHN analysis. The only and significant difference made by the low-temperature (240 °C) air-oxidation step was identified with the surface areas of carbons from aromatizable systems (TPOL and FPOL), which were higher than those from direct pyrolysis of as-prepared aerogels. Upon further reactive etching with CO2, those surface areas went as high as 2778 ± 209 m2 g−1. Those findings are directly relevant to high surface area carbons for gas sorption (e.g., capture and sequestration of CO2) and ion exchange materials.
1. Introduction
Phenolic resins are the condensation product of phenol or phenol derivatives with formaldehyde.1,2 If the reaction is run under sol–gel conditions, the reaction mixture turns into a gel, and if the pore-filling solvent is converted into a supercritical fluid that is vented off as a gas, phenolic resins take the form of highly porous, low-density, high surface area solids, classified as aerogels.3,4 Pyrolysis of phenolic resin aerogels under inert atmosphere, and most notably those of resorcinol–formaldehyde (RF),5–7 has been the primary source of carbon aerogels and xerogels.8–10It is known that the high open porosity of aerogels allows gasses to diffuse through their bulk with near open-air rates.11,12 That property is utilized in this report by inserting an air-oxidation step at the early stages of pyrolysis (<250 °C) of phenolic resin aerogels, and it is reported that certain of those materials undergo complete backbone aromatization yielding fused O+-heteroaromatic pyrylium rings along their entire skeletal framework. Subsequently, by using those fully characterized fused aromatic ring systems as a new point of departure, it was found that all high-temperature (>600 °C) pyrolysis products of mainstream phenolic resins include fused aromatic pyrylium rings, even without prior lower-temperature air-oxidation. However, phenolic aerogels that undergo air-oxidation-induced ring-fusion aromatization at lower temperatures, show a significant enhancement in the surface area of their carbons.
The impetus of this work stems from the pyrolytic carbonization of polyacrylonitrile (PAN), and of polybenzoxazines (PBOs). PAN is a linear polymer and the main source of graphite fiber for high performance fiber-matrix composites.13,14 Direct pyrolysis of PAN under inert atmosphere leads to complete decomposition. Carbonization of PAN (yield: 70% w/w (ref. 15 and 16)) requires a prior air-oxidation step at 200–300 °C that yields a new polymer consisting of a ladder-type structure with fused pyridine rings along its backbone.17 Similarly, polybenzoxazines (PBOs) are a special type of phenolic resins with Mannich type bridges (–CH2–N(R)–CH2–) between phenolic rings, instead of the typical –CH2– bridges in other phenolic resins.18–20 PBOs are heat-resistant and are pursued as low-cost alternatives to polyimides. PBOs are also carbonizable, but curiously the carbon yield of PBO aerogels (up to 61% w/w) was much higher than that from PBOs in bulk form (27% w/w).21 In analogy to PAN, the higher carbonization yield of PBO aerogels was traced to O2-induced aromatization and formation of fused N+-heteroaromatic pyridinium rings along the polymeric backbone.22 It was concluded that that process was facilitated by air circulating freely through the open porosity of PBO aerogels during a curing step up to 200 °C that, prior to that discovery, was thought only as necessary in order to complete polymerization of the benzoxazine monomer. The question then became whether oxidative ring-fusion aromatization could be forced upon more conventional phenolic resins, and if so, whether it would play a role in the carbonization process. That inquiry was pursued with aerogels derived from (Scheme 1) phenol (P), resorcinol (R) and phloroglucinol (POL) reacted with formaldehyde (F), as well as with aerogels derived from POL reacted with terephthalaldehyde (T). Reflecting their chemical composition, those phenolic resin aerogels are abbreviated as PF, RF, FPOL and TPOL, respectively. Collectively, all those four types of resins in aerogel form are referred to as RES. Again, since the scope of this work was to explore chemical transformations in phenolic resins brought about by air-oxidation, working with RES was advantageous, because their open porosity allowed unobstructed air-circulation through their bulk. The chemical composition of the four model systems along pyrolysis was followed with XPS, solid-state 13C NMR, FTIR and elemental analysis. The evolution of the skeletal framework and the pore structure was followed with SEM, and N2-sorption porosimetry. It was found that at 240 °C in air all four RES were oxidized, however only the two POL-systems, FPOL and TPOL, proceeded further to ring-fusion aromatization. Upon further pyrolysis under Ar, the chemical compositions of all four RES, either as-prepared, or after air-oxidation at 240 °C, converged at around 600 °C to a common chemical composition bearing two types of O: pyrylium O+, and charge compensating phenoxide O−. It was found that carbon aerogels from the aromatized versions of FPOL and TPOL had higher BET surface areas than carbons from direct pyrolysis of FPOL and TPOL. That property was magnified by reactive etching with CO2 at 1000 °C. Possible applications of those carbons include gas sorption and ion-exchange based separations.
 | ||
| Scheme 1 The monomers of this study. | ||
2. Results and discussion
2.1 Material synthesis
Aerogel synthesis and subsequent processing is summarized in Scheme 2. Processing details are described in the Experimental section. The exact formulations are given in Table S.I.1 (ESI, Appendix I†). The gelation of TPOL, FPOL, and RF was carried out at room temperature. PF gelled at 80 °C. In order to ensure crosslinking among polymeric chains, which is a necessary condition for phase separation of nanoparticles and formation of the aerogel network,23 the phenolic monomer-to-formaldehyde ratio for PF, RF and FPOL was set at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mol mol−1. On the other hand, since T bears two –CHO groups, it can play the role of the crosslinker itself, thus the POL:T mol ratio was set at 2:1. All gelation reactions were catalyzed with aq. HCl. Using Kanatzidis' work on phloroglucinol–terephthalaldehyde porous polymers as a point of departure,24 gelation of TPOL was carried out in 1,4-dioxane using a 16% w/w sol. FPOL and PF were also gelled from 1,4-dioxane sols. In order to use our previous developed time-efficient, HCl-catalyzed gelation of RF as a reference,25 gelation of that system was carried out in CH3CN using much less concentrated sols (3.3% w/w). The R:HCl mol/mol ratio was the same as before (8.4), and gelation at room temperature occurred in 2 h, consistent with our previous report.25 However, HCl-catalysis of the TPOL and FPOL sols was more efficient: gelation occurred in 1 min and 10 min, respectively, with a much lower catalyst concentration (POL:HCl ≈ 109 mol/mol). On the contrary, HCl-catalyzed gelation of PF was slow:26 using the same P:HCl mol/mol ratio as in RF (8.4), gelation took 24 h at 80 °C. All four wet-gels were aged, solvent-exchanged with acetone and were dried with liquid CO2 taken out at the end as a supercritical fluid (SCF). Post synthesis, RES aerogels were separated into two groups: the first one was pyrolyzed at 240 °C under Ar and the resulting samples are referred to as TPOL-240(Ar), RF-240(Ar), etc. The second group was pyrolyzed at 240 °C under air, and the corresponding samples are referred to as TPOL-240(air), RF-240(air), etc. Collectively, the two groups are referred to as RES-240(Ar), and RES-240(air), respectively. Next, as-prepared RES and RES-240(air) were pyrolyzed under Ar at 300 °C, 400 °C, 500 °C, 600 °C, 700 °C and 800 °C, using fresh samples at every successive pyrolysis temperature. That is, samples that were pyrolyzed at a given temperature, were characterized as will be described below, and were stowed; they were not pyrolyzed under Ar again at any other temperature. The samples from those pyrolytic experiments are abbreviated as C-RES-D-Θ and C-RES-O-Θ, where “C” stands as a reminder that those samples were obtained along carbonization; “D” stands for samples after direct pyrolysis of as-prepared RES; “O” stands for samples obtained after prior oxidation at 240 °C/air; and, “Θ” stands for the pyrolysis temperature. Finally, selected pyrolyzed samples, C-RES-D-Θ and C-RES-O-Θ, were subjected to reactive etching with CO2 at 1000 °C, and are abbreviated as EC-RES-D_(or_O)-Θ, whereas “EC” stands for “etched carbon.”
:2 mol mol−1. On the other hand, since T bears two –CHO groups, it can play the role of the crosslinker itself, thus the POL:T mol ratio was set at 2:1. All gelation reactions were catalyzed with aq. HCl. Using Kanatzidis' work on phloroglucinol–terephthalaldehyde porous polymers as a point of departure,24 gelation of TPOL was carried out in 1,4-dioxane using a 16% w/w sol. FPOL and PF were also gelled from 1,4-dioxane sols. In order to use our previous developed time-efficient, HCl-catalyzed gelation of RF as a reference,25 gelation of that system was carried out in CH3CN using much less concentrated sols (3.3% w/w). The R:HCl mol/mol ratio was the same as before (8.4), and gelation at room temperature occurred in 2 h, consistent with our previous report.25 However, HCl-catalysis of the TPOL and FPOL sols was more efficient: gelation occurred in 1 min and 10 min, respectively, with a much lower catalyst concentration (POL:HCl ≈ 109 mol/mol). On the contrary, HCl-catalyzed gelation of PF was slow:26 using the same P:HCl mol/mol ratio as in RF (8.4), gelation took 24 h at 80 °C. All four wet-gels were aged, solvent-exchanged with acetone and were dried with liquid CO2 taken out at the end as a supercritical fluid (SCF). Post synthesis, RES aerogels were separated into two groups: the first one was pyrolyzed at 240 °C under Ar and the resulting samples are referred to as TPOL-240(Ar), RF-240(Ar), etc. The second group was pyrolyzed at 240 °C under air, and the corresponding samples are referred to as TPOL-240(air), RF-240(air), etc. Collectively, the two groups are referred to as RES-240(Ar), and RES-240(air), respectively. Next, as-prepared RES and RES-240(air) were pyrolyzed under Ar at 300 °C, 400 °C, 500 °C, 600 °C, 700 °C and 800 °C, using fresh samples at every successive pyrolysis temperature. That is, samples that were pyrolyzed at a given temperature, were characterized as will be described below, and were stowed; they were not pyrolyzed under Ar again at any other temperature. The samples from those pyrolytic experiments are abbreviated as C-RES-D-Θ and C-RES-O-Θ, where “C” stands as a reminder that those samples were obtained along carbonization; “D” stands for samples after direct pyrolysis of as-prepared RES; “O” stands for samples obtained after prior oxidation at 240 °C/air; and, “Θ” stands for the pyrolysis temperature. Finally, selected pyrolyzed samples, C-RES-D-Θ and C-RES-O-Θ, were subjected to reactive etching with CO2 at 1000 °C, and are abbreviated as EC-RES-D_(or_O)-Θ, whereas “EC” stands for “etched carbon.”
 | ||
Scheme 2 Synthesis of acid-catalyzed phenolic aerogels (RES: TPOL, FPOL, RF, PF), air-oxidation, pyrolytic carbonization, and reactive etching. (![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) : products from direct pyrolysis of as-prepared RES; : products from direct pyrolysis of as-prepared RES; ![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) : products from pyrolysis of oxidized samples: Θ: pyrolysis temperature; C: carbon; EC: etched carbon). : products from pyrolysis of oxidized samples: Θ: pyrolysis temperature; C: carbon; EC: etched carbon). | ||
Certain key product yields along processing are cited in Table S.I.2 (ESI. Appendix I†). First, the yields of the air-oxidation products were quite high (w/w): TPOL-240(air), 90%; FPOL-240(air), 74%; RF-240(air), 82%; and, PF-240(air), 100%. Next, the yields of the D route at the terminal pyrolytic temperature (800 °C) of this study were on average a little higher than the yields of the route (% w/w vs. % w/w): C-TPOL-D-800/C-TPOL-O-800, 57/54; C-FPOL-D-800/C-FPOL-O-800, 54/50; C-RF-D-800/C-RF-O-800, 54/49; and, C-PF-D-800/C-PF-O-800, 61/49. However, by considering the errors in those yields (ESI, Table S.I.2†), the definitive edge of the D-route in terms of carbonization yields was rather only with the RF and PF systems. It is noted further that the pyrolytic yields of C-RES-O-Θ declined only a little in the 300 ≤ Θ≤ 600 °C range, compared with C-RES-D-Θ, whose yields declined steadily. This implies that the chemical changes brought about at 240 °C/air were stable for a few hundred °C afterwards. This point is important, because, as it will be shown in Section 2.3 below, above 600 °C, and processes converged chemically, thereby the chemical identity of the pyrolysis products at 240 °C/air dictates the common fate of all pyrolysis products afterwards.
2.2 Pyrolysis products at 240 °C/air versus 240 °C/Ar
Differential scanning calorimetry (DSC) of the four RES up to 300 °C under N2 showed no major heat exchanges. In air, however, all four RES showed strong exotherms with maxima in the 230–270 °C range (Fig. 1 and ESI, Appendix II Fig. S.II.1†). Based on those data, quantitative pyrolysis of all as-prepared RES was carried out at 240 °C, as described above. Consistent with our previous findings with RF,27 all RES-240(Ar) were chemically identical to as-prepared RES (see below). In contrast, RES-240(air) were chemically different from RES, and in fact they could be put in two groups: (a) TPOL-240(air) and FPOL-240(air), and (b) RF-240(air) and PF-240(air). Group (a), TPOL-240(air) and FPOL-240(air), bore fused aromatic pyrylium ions on their polymeric backbones, group (b) did not. Scheme 3 shows the structures of RES and RES-240(air) as inferred from the spectroscopic data below.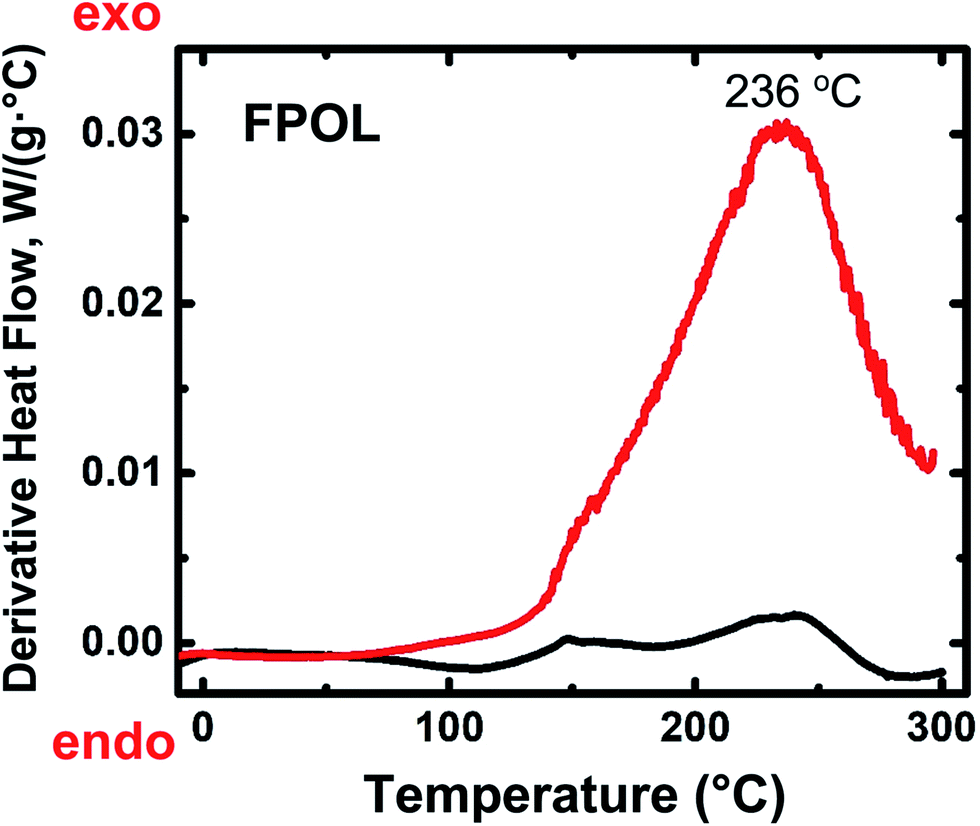 | ||
| Fig. 1 Representative differential scanning calorimetry (DSC) data for RES. Black line: under N2; red line: in air. (For TPOL, RF and PF, see ESI, Fig. S.II.1.†) | ||
 | ||
| Scheme 3 Structures of as-prepared RES aerogels, and their air-oxidation products. | ||
The FTIR spectra of all RES-240(Ar) (Fig. 2 and ESI, Appendix III Fig. S.III.1†) were identical to those of as-prepared RES. On the other hand, while RES-240(air) retained at least some of the –OH groups of their parent RES (note the absorptions with maxima in the 3394–3436 cm−1 range), they also showed a new absorption in the 1723–1737 cm−1 range, which was assigned to CO stretching, although those absorptions were not very strong by carbonyl standards. Pyrylium in-plane stretches were expected in the 1400–1650 cm−1 range,28,29 which overlaps with the stretches of other aromatic rings, thereby that region was not interpretably useful. On the contrary, the region below 1000 cm−1 was quite informative, especially in the case of TPOL-240(air). The out-of-plane (OOP) C–H bending at 833 cm−1 of the para-substituted ring of T,30 which was clearly present in both TPOL and TPOL-240(Ar), went missing from TPOL-240(air), in agreement with its involvement in a ring-fusion process (Scheme 3). Similarly, the out-of-plane C![[horiz bar, double dot above]](https://www.rsc.org/images/entities/char_e0f2.gif) C bending31,32 at 548 cm−1 was also missing from the oxidized product. In the latter region, the spectrum of FPOL-240(air) showed similar changes (see ESI, Fig. S.III.1†), consistent with ring fusion, while the spectra of RF-240(air) and PF-240(air) still showed the out-of-plane CC bending modes from isolated aromatic rings (see Fig. 2 and ESI, Fig. S.III.1†).
C bending31,32 at 548 cm−1 was also missing from the oxidized product. In the latter region, the spectrum of FPOL-240(air) showed similar changes (see ESI, Fig. S.III.1†), consistent with ring fusion, while the spectra of RF-240(air) and PF-240(air) still showed the out-of-plane CC bending modes from isolated aromatic rings (see Fig. 2 and ESI, Fig. S.III.1†).
 | ||
| Fig. 2 Representative FTIR data for RES as-prepared, and after treatment as indicated by the sample names. (For FPOL and PF see ESI, Fig. S.III.1.†) | ||
XPS gave more direct evidence for the formation of pyrylium rings. O 1s XPS spectra were obtained using compressed pellets made by mixing powders of the corresponding materials with Au dust (10% w/w), which dissipated electrostatic charges, and its 4f7/2 peak at 84.0 eV was used as an internal peak-position calibration standard. Three more compounds were also run in a similar fashion as external references: (a) 1-naphthol that gave a O 1s peak from the phenolic –OH group at 531.97 eV; (b) sodium phenoxide that gave a O 1s peak from the –O− group at 531.41 eV; and, (c) 2,6-diisopropyl-4-phenylpyrylium cation, which was available from other work in our laboratory,33 and showed a O+ peak at 534.26 eV. The O 1s spectra of the three references are shown in ESI, Appendix IV Fig. S.IV.1.† The O 1s XPS spectra of all as-prepared RES (Fig. 3 and ESI, Appendix IV Fig. S.IV.2†) showed, as expected, the presence of only a single type of oxygen (with maxima in the 531.52–532.66 eV range), which was assigned to phenolic type –OH.34–37 The XPS spectra of TPOL-240(air) and FPOL-240(air) showed three peaks: one at higher energies (533.62 eV and 535.24 eV, respectively), one more intense absorption in an intermediate energy range (532.64 eV and 533.45 eV, respectively), and one absorption at lower energies (531.39 eV and 531.61 eV, respectively). The intensity ratio of those three absorptions were 1:(1.4):1 in TPOL-240(air) and 1:(2.6):(1.3) in FPOL-240(air). Based on literature values, and on direct comparison with the O 1s XPS spectra of our three reference compounds (ESI, Fig. S.IV.1†), the high-energy absorptions of TPOL-240(air) and FPOL-240(air) were assigned to pyrylium O+;38 the intermediate range absorptions were assigned to phenolic type –OH;34–37 and, the lower energy absorptions were assigned to –O−,39,40 perhaps overlapping with some CO.34–37 Importantly, each of the O 1s XPS spectra of RF-240(air) and PF-240(air) consisted of only two absorptions, at 531.53/529.75 eV, and at 531.98/530.27 eV, respectively. Each of those high/low energy pairs were assigned to phenolic –OH and to CO, and the relative ratios were 1.8 in RF-240(air) and 2.2 in PF-240(air), i.e., they followed the trend of increasing phenolic –OH groups by going from RF to PF. Overall, the structures of Scheme 3 were all consistent with the O 1s XPS data.
 | ||
| Fig. 3 O 1s XPS data for TPOL and RF, and their air-oxidized versions as shown. (The spectra of FPOL are similar to TPOL, and of PF are similar to RF – see ESI, Fig. S.IV.2.†) | ||
All solid-state CPMAS 13C NMR spectra were run twice, once with a long cross-polarization (CP) contact time (3000 μs) and once with a shorter one (5 μs). The latter conditions forced carbons with no Hs to vanish. Fig. 4A compares the spectra of TPOL, TPOL-240(Ar) and TPOL-240(air); Fig. 4B compares the spectra of RF, RF-240(Ar) and RF-240(air). (Corresponding spectra for the FPOL and PF systems are shown in ESI, Appendix V Fig. S.V.1,† and they behaved pairwise similarly to the TPOL and RF systems, respectively.) For peak assignment refer to Scheme 3.
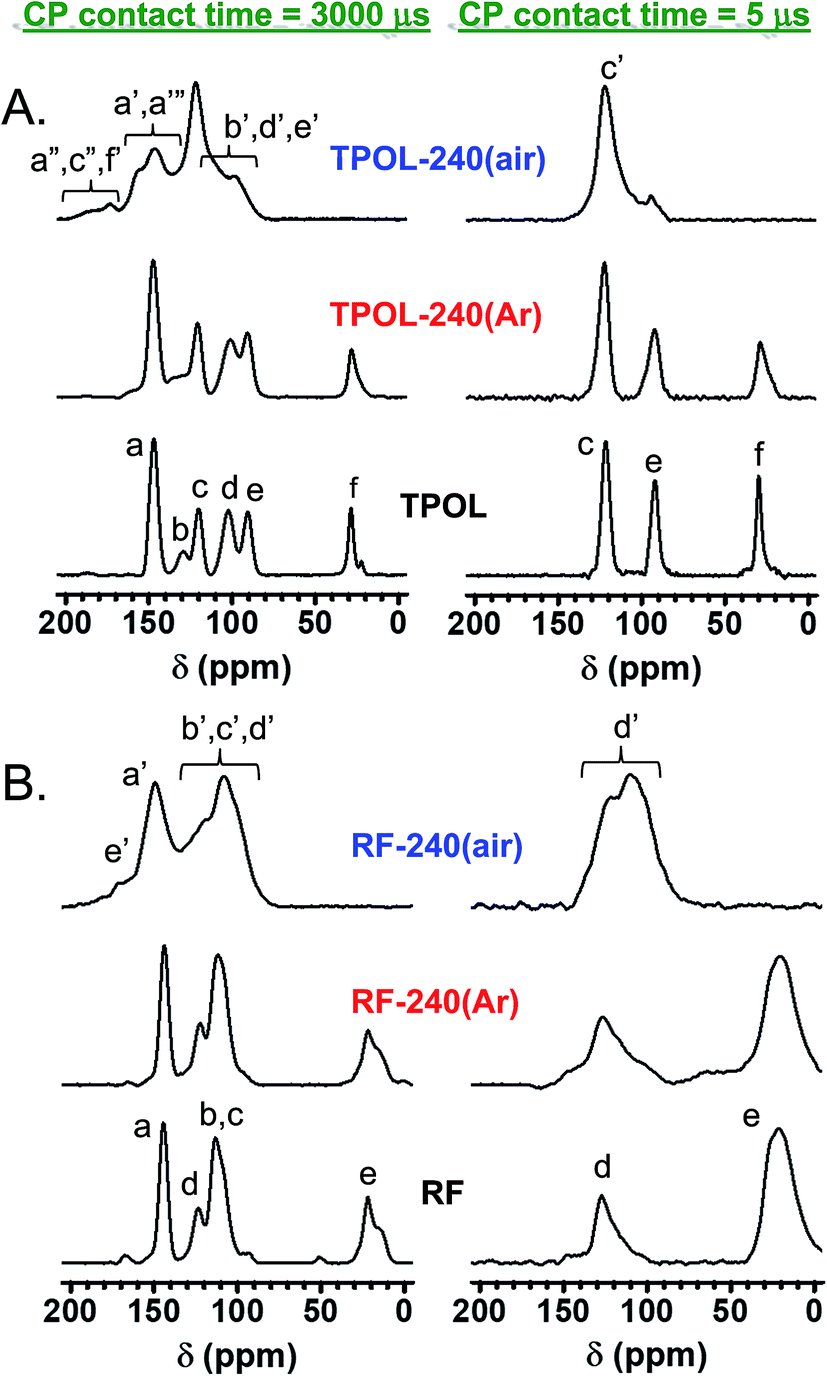 | ||
| Fig. 4 Solid-state CPMAS 13C NMR data for TPOL (A) and RF (B) and their pyrolyzed products at 240 °C under Ar or air, as shown. (Similar spectra for FPOL and PF are shown in ESI, Fig. S.V.1.†) | ||
In agreement with DSC and FTIR, the 13C NMR spectra of all RES-240(Ar) were similar to those of as-prepared RES aerogels. Using the aliphatic ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2 of the RF and PF as internal reference, the ratios of aromatic –H to H2 in the as–prepared (and the 240 °C/Ar-treated samples) were approximately 0.5:1 and 1:1, respectively, consistent with the 1:2 stoichiometry of R (or P) and F in their sols. FPOL and FPOL-240(Ar) had no aromatic –H, as expected from complete reaction of F with POL at their 2:1 stoichiometric ratio in the sol. On the other hand, consistent with the T:POL stoichiometry in the sol, and the expected structure (Scheme 3) TPOL had only one type of aromatic –H coming from POL (designated as “e”) and only one type coming from T (designated as “c”). The aliphatic H-to-“e”-to-“c” ratio was in the expected 1:1:2 value.
H2 of the RF and PF as internal reference, the ratios of aromatic –H to H2 in the as–prepared (and the 240 °C/Ar-treated samples) were approximately 0.5:1 and 1:1, respectively, consistent with the 1:2 stoichiometry of R (or P) and F in their sols. FPOL and FPOL-240(Ar) had no aromatic –H, as expected from complete reaction of F with POL at their 2:1 stoichiometric ratio in the sol. On the other hand, consistent with the T:POL stoichiometry in the sol, and the expected structure (Scheme 3) TPOL had only one type of aromatic –H coming from POL (designated as “e”) and only one type coming from T (designated as “c”). The aliphatic H-to-“e”-to-“c” ratio was in the expected 1:1:2 value.
After pyrolysis at 240 °C/air, no RES-240(air) showed any surviving aliphatic carbons from the methylene or methyne bridges (–CH2–, or –CH–) of the parent RES. All H-bearing aromatic carbons survived the oxidation process in both RF-240(air) and PF-240(air), and the spectra of those two materials were similar to those of their parent RES, and most importantly to one another; in fact, in terms of chemical shifts the two spectra were almost identical (compare Fig. 4B with ESI Fig. S.V.1B†). (It is noted in passing that the presence of two types of H-bearing aromatic carbons with about equal intensities in both RF-240(air) and PF-240(air), – refer to the low contact-time CP spectra – is attributed to the random 2,2- and 2,4-connectivity in adjacent repeat units expected of the phenolic moieties along the main chain.41) Now, it is noted that in PF-240(air) some H-bearing aromatic carbons would have survived the oxidation process irrespective of ring-fusion aromatization, however, in RF-240(air) the only chance for aromatic H's to survive is when no ring-fusion aromatization takes place. The latter realization together with the similarity of the 13C NMR, IR and XPS spectra of RF-240(air) and PF-240(air) render their structures similar to one another, and consistent with those shown in Scheme 3. On the contrary, as a result of ring-fusion aromatization the aromatic regions of TPOL-240(air) and FPOL-240(air) were more complicated than those of the corresponding as-prepared samples (Scheme 3). Based on the spectra of FPOL and FPOL-240(Ar), FPOL-240(air) was not expected and did not have any H-bearing carbons. On the other hand, the new broad downfield resonances of TPOL-240(air) at 189.5 and 174.2 ppm were assigned to the ortho (a′ and c′) and para (f′) carbons of the pyrylium ring, respectively. The meta carbons, d′ and b′, were expected significantly upfield,42–44 and were assigned to the resonance at 97.9 ppm. Curiously, the only surviving Hs in TPOL-240(air) were those coming from T; that is, even the “e” carbon of as-prepared TPOL had lost its H, thereby fused-aromatic systems had to be head-to-tail connected to one another, as shown in Scheme 3. That type of connectivity was attributed to electrophilic aromatic substitution reactions of one polynuclear aromatic system to another – see next section and refer to Scheme 4C below.
 | ||
| Scheme 4 Oxidation mechanism of RES demonstrated with TPOL, (A) Stage 1: initial oxidation leading to carbonyl group formation and change in connectivity along the polymer chain (repeat unit shown in bold; adjacent units included in order to show connectivity), (B) Stage 2: further oxidation and ring fusion aromatization (the Dewar-benzene-like canonical form in the first step was used for brevity), (C) Stage 3: interchain coupling at the e′-positions of two adjacent repeat units. | ||
2.3 The mechanism of ring-fusion aromatization
The elementary processes during oxidation and ring-fusion aromatization are illustrated via the most complicated system of this study, TPOL, in Scheme 4. In TPOL, those processes take place in three stages. In Stage 1, bridging –CH– groups are converted to carbonyls following a route akin to autooxidation (Scheme 4A):45,46 H-atom abstraction by O2 is followed by addition of OOH groups at the benzylic positions; subsequently, homolytic cleavage of the O–OH bond and of the adjacent C–C bond yields H2O2, a carbonyl group at the previously bridging position, and a phenyl radical. In addition to creating carbonyls, that process breaks down the polymer chain at several places, and disrupts crosslinking. At that point, polymer chains are free to relax to new positions, in which phenyl radicals couple and establish a new crosslinked configuration that remains similar in terms of connectivity to, but is more compact than the original TPOL. In Stage 2 (Scheme 4B), newly created carbonyl groups undergo 1,5-proton transfer tautomerization with the ortho OH groups of the POL moieties. In turn, tautomerized moieties undergo electrocyclic ring closure, to restore aromaticity. At that point, air (O2) oxidizes the newly created ether bridges (–O– + O2 → –O˙+– + O2˙−), and the byproduct of that oxidation (superoxide: O2˙−) acts as a Brønsted base, abstracts phenolic protons, and yields hydroperoxyl radicals (HO2˙−). The latter abstract H˙ atoms from the positions adjacent to –O˙+– and yield pyrylium rings.FPOL goes through Stages 1 and 2, yielding ring-fusion aromatization to pyrylium in analogy to TPOL (see ESI, Appendix VI†).
Based on the chemical characterization data of the previous section, RF and PF go through Stage 1, but oxidation stops there. It is noted, however, that in both of those systems the relative connectivity of at least one phenolic OH and the newly-formed bridging CO is similar to that in TPOL and FPOL; thereby, in principle, both RF and PF could undergo 1,5-proton transfer tautomerization, and thus become ready for electrocyclic ring closure. Again, after ring closure, both of those systems should be able to continue along the reaction pathways of Stage 2 with O2˙− and HO2˙−. But, they do not. It is speculated that Stage-1 oxidized PF and RF were unable to yield pyrylium, because the rings were not activated enough to undergo electrocyclic ring closure. On the contrary, the three OH groups of FPOL accommodate the loss of aromaticity involved with electrocyclic ring closure to the point that it is not very disfavored. A similar rationale holds true for TPOL.
Finally, TPOL, as opposed to FPOL, possesses hydrogen atoms in the “e” positions and continues with Stage 3, in which the tip of one fused aromatic system undergoes an electrophilic aromatic substitution with another (Scheme 4C). Elemental analysis of TPOL-240(air) gave (% w/w): C: 64.71 ± 0.16; H: 1.71 ± 0.34; O: 33.26 ± 0.07. (The % O was calculated via 100 − (% C + % H).) The theoretical elemental composition calculated from the repeat unit shown in Scheme 3 (i.e., the end-product of Stage 3, Scheme 4C) was (% w/w): C: 64.2; H: 1.6; O: 34.2. The agreement between the experimental and theoretical elemental compositions is considered remarkable given the complexity of the processes and the fact that no “purification” of the pyrolysis products was possible in the conventional synthetic organic sense. It is noted further that if oxidation of TPOL had stopped at the end of Stage 2, the expected elemental composition would have been (% w/w): C: 63.8; H: 2.13; O: 34.0, i.e., markedly different from the experimental results.
2.4 Chemical transformations along further pyrolysis of RES-240(air) versus RES
The materials from the 240 °C/air oxidation process were used for further investigation along pyrolysis of RES. For that, as-prepared, as well as 240 °C/air-treated aerogels (RES and RES-240(air), respectively) were pyrolyzed at 100 °C intervals, starting from 300 °C and ending at 800 °C (Scheme 2). At every pyrolysis temperature, we used fresh RES or RES-240(air) samples, as described in Section 2.1 above. The pyrolysis products are referred to as C-RES-D_(or_O)-Θ. The evolution of the chemical composition was monitored with elemental analysis, solid-state 13C NMR and FTIR. C-RES-O-800 and C-RES-D-800 were also characterized with XPS.Fig. 5 shows the elemental analysis data of all four systems along pyrolysis. Dashed lines follow the pyrolytic evolution of the 240 °C/air oxidized samples (RES-240(air)); solid lines follow the direct pyrolysis of as-prepared RES. All samples were analyzed three times, and in most cases error bars (one standard deviation) were within the respective symbols. The percent amount of oxygen was calculated via % O = 100 − (% C + % H). Relative to RES and RES-240(Ar), all RES-240(air) were deficient in H and richer in O, as expected. Between 300 °C and 500 °C RES-240(air) lost only a little additional H, while the amount of H in RES kept on decreasing steadily. At Θ ≥ 600 °C the C, H and O curves of each C-RES-D-Θ/C-RES-O-Θ pair converged – practically coincided. By 800 °C, all eight materials, independent of their RES-origin or the route ( or ), had produced carbons with the same, within error, CHO composition (C: 91.8 ± 1.8% w/w; H: 0.64 ± 0.40% w/w; O: 7.2 ± 1.7% w/w; errors are spreads of triplicate analysis). The common chemical features of the converging structures were investigated with solid-state 13C NMR and IR. The fate of oxygen was investigated with XPS.
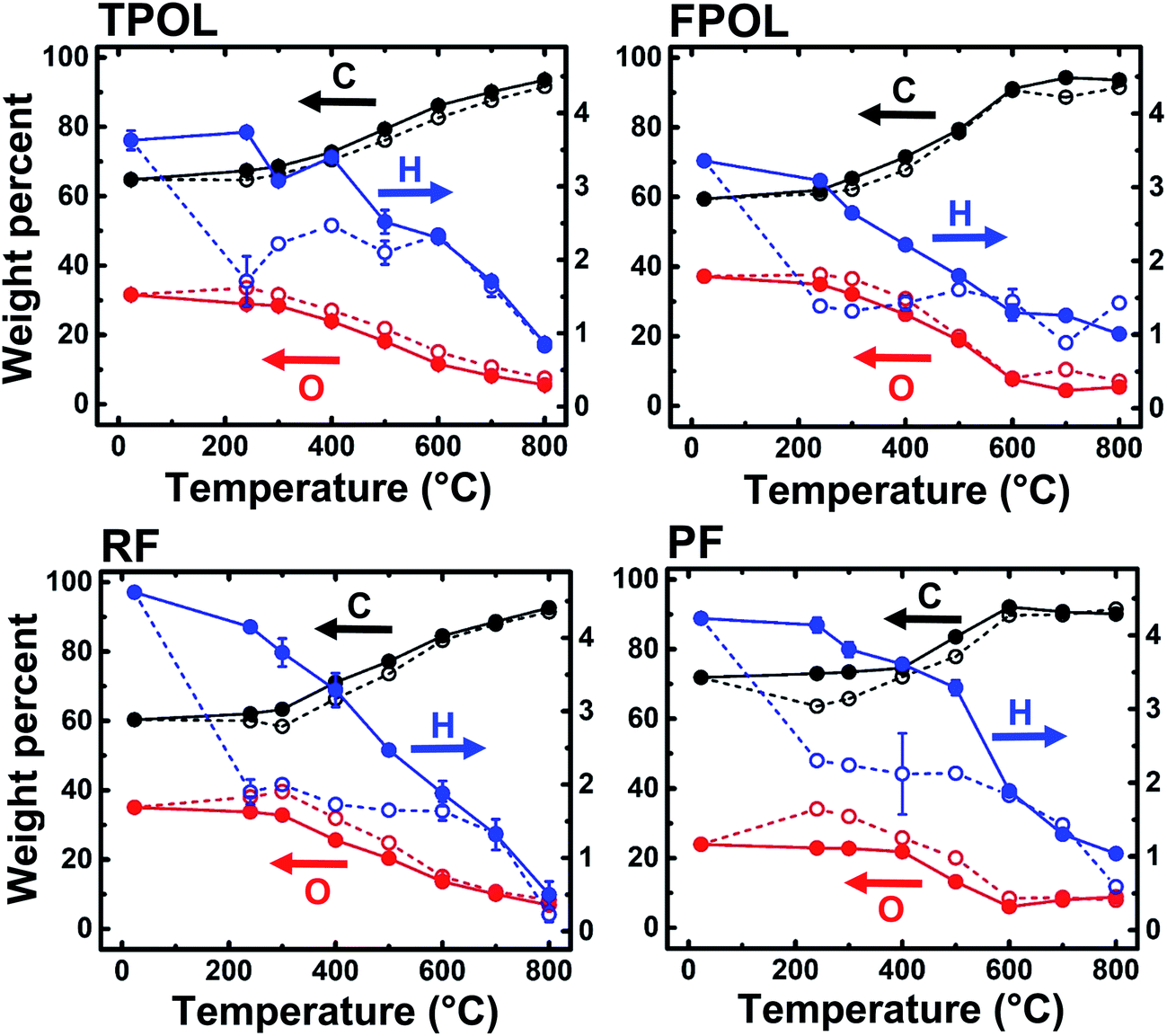 | ||
| Fig. 5 Elemental analysis data along pyrolysis under Ar towards porous carbons. Solid lines: pyrolytic evolution of elemental composition of as-prepared RES. Dashed lines: pyrolytic evolution of samples treated at 240 °C/air (RES-240(air)). (% weight of O by difference.) | ||
Fig. 6A and B show the CPMAS 13C NMR spectra along the pyrolytic evolution of the TPOL/TPOL-240(air), and the RF/RF-240(air) pair, respectively. Corresponding spectra for the FPOL/FPOL-240(air) and the PF/PF-240(air) pairs are shown in ESI, Appendix V Fig. S.V.2.† It is noted that the changes in the 300–600 °C range in all four RES-240(air) were not as drastic as the changes observed after pyrolysis of the corresponding as-prepared RES. That suggests that the 240 °C/air process accelerated the chemical changes that otherwise took place more progressively in the conventional pyrolytic carbonization of RES under inert atmosphere. Consistent with the elemental analysis data, by Θ = 600 °C the 13C NMR spectra of every C-RES-D-Θ/C-RES-O-Θ pair had converged to a common spectrum. More importantly though, by Θ = 600 °C, spectra became identical not only pairwise, but in fact all eight spectra, independent of their RES-origin or the route ( or ), were also identical to one another. Similar observations were made from the evolution of the FTIR spectra showing again that the spectra of all C-RES-D_(or_O)-Θ pairs became identical by 500 °C or 600 °C, depending on the RES (see ESI, Appendix III Fig. S.III.2A–D†). At 800 °C, the common FTIR spectrum of all eight C-RES-D_(or_O)-800 was quite simple, showing only two major absorptions at around 3396 cm−1 and 1619–1630 cm−1.
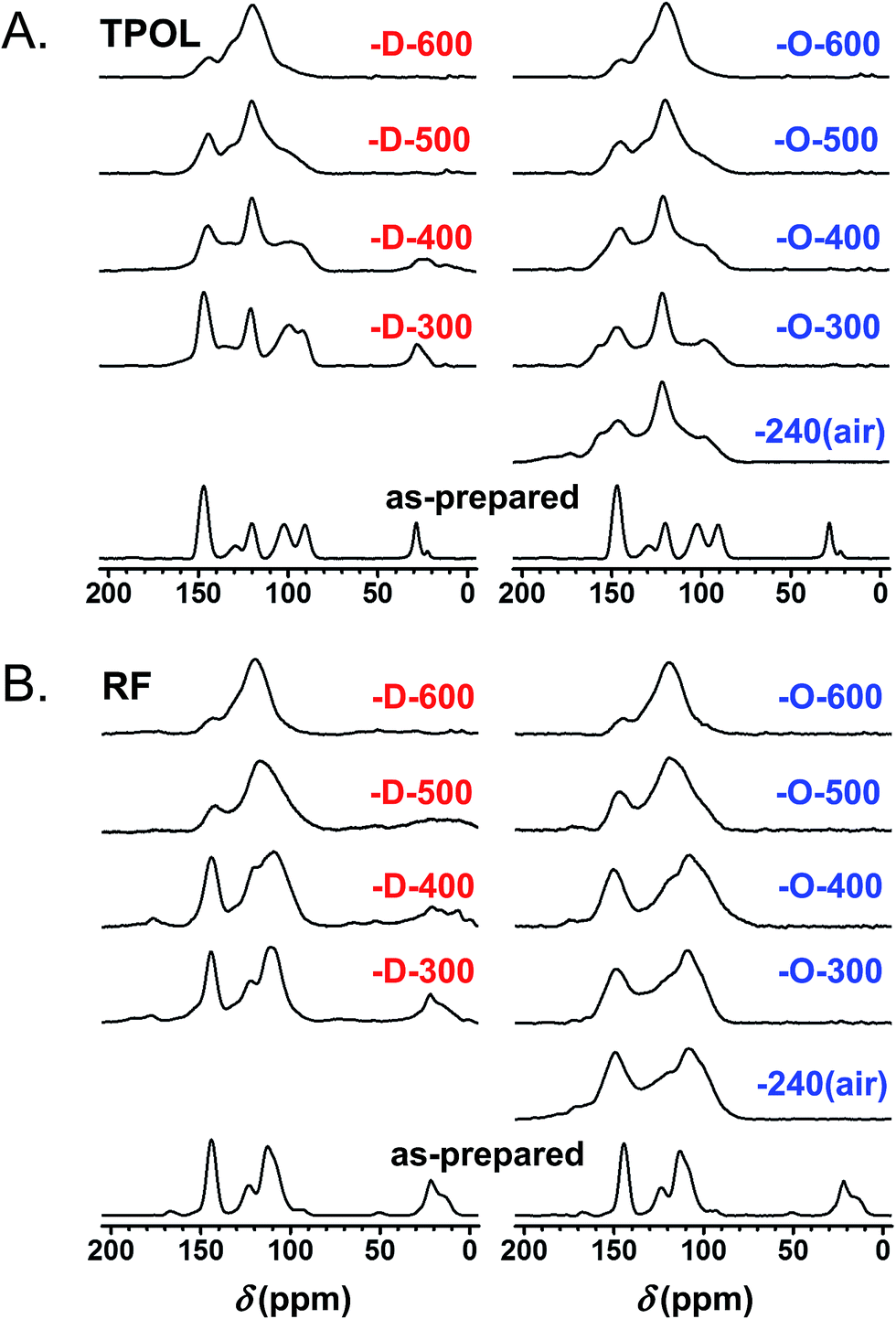 | ||
| Fig. 6 Solid-state 13C NMR data for TPOL (A) and RF (B) along pyrolysis as shown. designates samples from pyrolysis of TPOL and RF as-prepared. designates samples from pyrolysis of RES-240(air) (i.e., sample treated at 240 °C/air). (Similar spectra for FPOL and PF are given in ESI, Fig. S.V.2.†) | ||
Finally, Fig. 7 shows and compares the high-resolution O 1s XPS spectra of C-TPOL-D_(and_O)-800 (top) and of C-RF-D_(and_O)-800 (bottom). (The corresponding spectra of C-FPOL-D_(and_O)-800 and of C-RF-D_(and_O)-800 are shown in ESI, Appendix IV Fig. S.IV.3.†) The spectra of all eight C-RES-D- and C-RES-O- materials showed just two absorptions: one at 533.01–533.17 eV, which is consistent with the presence of pyrylium-type O+, and a second one of similar intensity at 531.28–531.69 eV, corresponding to charge compensating O−. (Please refer to the controls shown in ESI, Fig. S.IV.1.†) A common idealized structure for all pyrolysis products at 800 °C has been drawn by analogy to the structure of polyacrylonitrile-derived carbon at the same temperature range,47–49 and is shown in Scheme 5. The fused aromatic ring core of the graphitic sheets is made up of “aromatic repeat layers.” For example, the core of the structure of Scheme 5 consists of two such aromatic repeat layers color-coded in red and blue. Based on the average amount of O in all RES-derived carbons (7.2 ± 1.7% w/w), it is calculated that on average the graphitic core of all C-RES-D_(or_O)-800 materials consisted of 6.5 aromatic repeat layers.
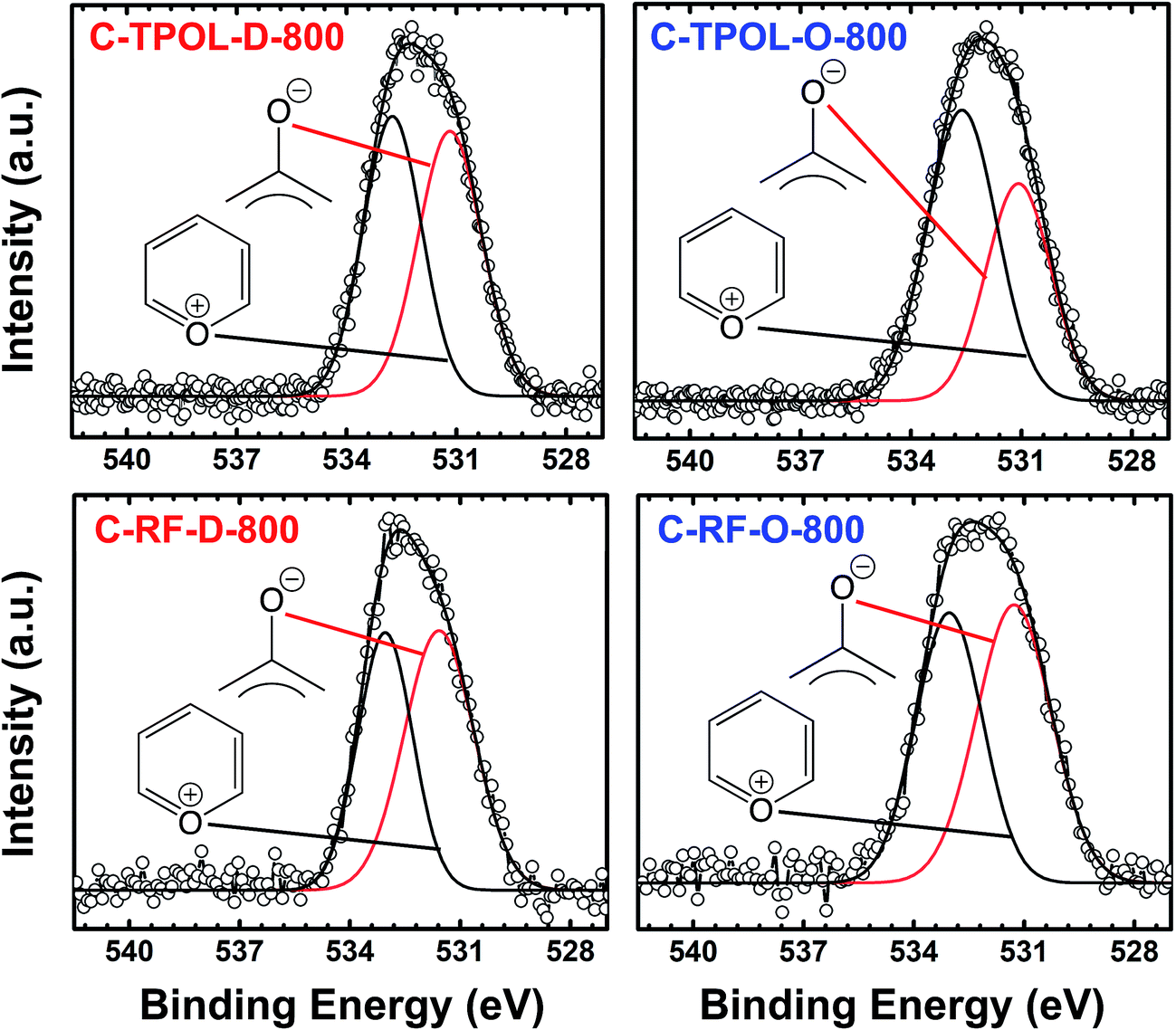 | ||
| Fig. 7 O 1s XPS data from the 800 °C-carbonized C-TPOL-D-800/C-TPOL-O-800 and C-RF-D-800/C-RF-O-800 pairs (top and bottom, respectively). (Similar data for the FPOL and PF systems are given in ESI, Fig. S.IV.3.†) | ||
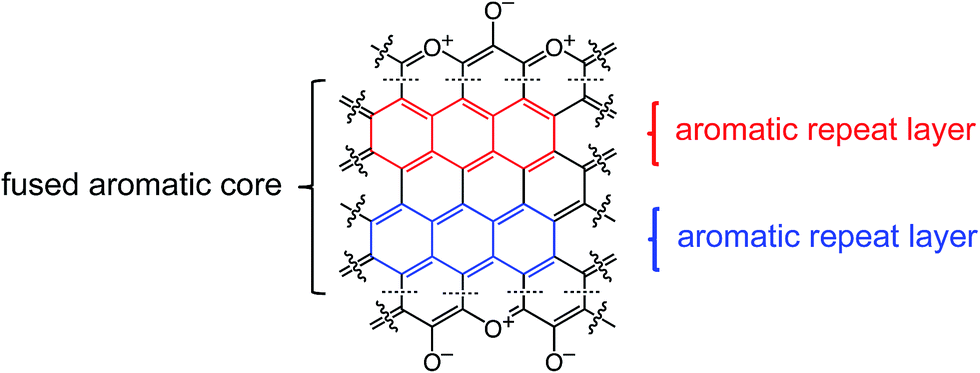 | ||
| Scheme 5 Idealized structure of carbons derived from pyrolysis of RES at 800 °C showing two repeat layers of the graphitic core, one in red, one in blue. | ||
In summary, air-oxidation of TPOL and FPOL gave two well-defined products (TPOL-240(air) and FPOL-240(air) – Scheme 3) with extensive ring-fusion aromatization along their backbone that survived for several hundred °C afterwards. Second, all pyrolyzed carbons converged to a common chemical composition irrespective of: (a) the starting phenolic system, (b) whether we inserted an early oxidation step in the carbonization process, or (c) whether the particular phenolic system undergoes ring-fusion aromatization (TPOL and FPOL), or just a simple oxidation of the –CH2– bridges (case of RF and PF). By considering those points together, we have concluded that ring-fusion aromatization, with formation of pyrylium and charge compensating phenoxides, is the common converging point along pyrolysis of all four main-stream phenolic resins of this study. Reasoning by analogy, we speculate that this is the common converging point along carbonization of all phenolic resins. The question then became whether low-temperature oxidative aromatization was of any practical use, namely whether it makes any difference in the properties of the resulting carbons. That is discussed in the next section, in conjunction with the evolution of the material properties along carbonization, and high-temperature reactive etching with CO2.
2.5 The evolution of material properties along pyrolytic carbonization and reactive etching
Bulk properties of interest include linear shrinkage, bulk density and porosity. Representative data exemplified with TPOL and RF are shown in Fig. 8. Tables with primary materials characterization data along pyrolysis and reactive etching are given in ESI, Appendix VII.† Cumulative plots of shrinkage, bulk density and porosity along pyrolysis of all four RES are given in ESI, Appendix VIII.†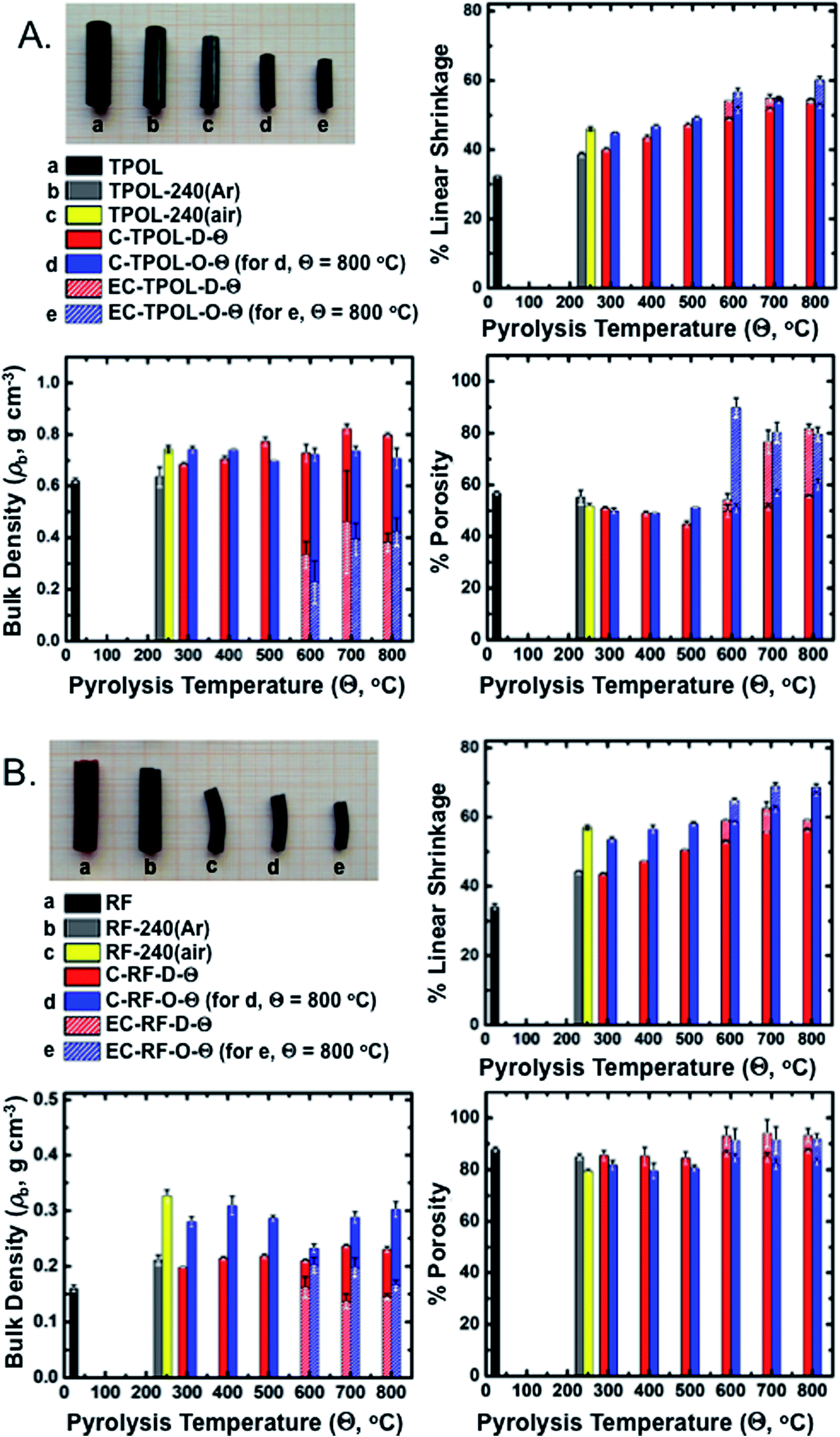 | ||
| Fig. 8 Photographs and general materials characterization data along pyrolysis and reactive etching of the TPOL (A) and the RF (B) systems. Linear shrinkage is relative to the molds. refers to products from direct pyrolysis of as-prepared RES. refers to products from pyrolysis of 240 °C/air-oxidized samples. Θ: pyrolysis temperature. Shaded segments of the bars indicate the values after reactive etching at 1000 °C of samples having been carbonized at Θ. | ||
Fig. 8A and B show photographs of as-prepared TPOL and RF, after Ar or air treatment at 240 °C, after carbonization at 800 °C, and finally after reactive etching at 1000 °C. At all stages of pyrolysis, samples shrunk relative to the as-prepared materials, but remained monolithic. The relative linear shrinkage was quantified by the bar graphs shown next to the photographs. Most of the shrinkage occurred during aerogel preparation: both TPOL and RF shrunk 32–34% relative to the molds. Treatment at 240 °C/Ar caused only a small additional shrinkage: 6% for TPOL, 10% for RF. Air-oxidation at 240 °C (yellow bars) caused an additional shrinkage relative to TPOL-240(Ar) and RF-240(Ar) (7.5%, and 12.8%, respectively) – by that point, the cumulative shrinkage is clearly noticeable in the photographs. Beyond that point, i.e., from 300 °C to 800 °C, samples either by direct () pyrolysis of TPOL and RF (red bars), or by pyrolysis of their 240 °C/air oxidized () derivatives (blue bars) kept on shrinking more with increasing pyrolysis temperature, preserving though, for the most part, the higher shrinkage caused by the 240 °C/air oxidation step (yellow bars). Reactive etching at 1000 °C/CO2 was conducted for all samples pyrolyzed at Θ ≥ 600 °C, and it caused only a small additional shrinkage relative to the respective parent carbons; said additional shrinkage is shown in Fig. 8 with shaded segments on top of the regular pyrolysis bars. Thus, for example, considering samples from the terminal pyrolysis temperature of this study (i.e., at Θ = 800 °C) the total shrinkage (from the molds) of EC-TPOL-O-800 was 60.2 ± 1.0% (vs. 52.8 ± 0.8% for C-TPOL-O-800), while EC-TPOL-D-800 shrunk less: 54.4 ± 0.4% (vs. 53.4 ± 0.1% for C-TPOL-D-800). Similarly, EC-RF-O-800 shrunk 68.6 ± 1.1% (vs. 66.8 ± 0.7% C-RF-O-800), while EC-RF-D-800 shrunk again less: 59.1 ± 0.2% (vs. 56.2 ± 0.5% for C-RF-D-800). The shrinkage of the two remaining systems, FPOL and PF, followed the same trends as a function of the pyrolysis temperature (Θ) as TPOL and RF (see ESI, Fig. S.VIII.1†).
Reflecting the lower concentration of its sol, as-prepared RF was less dense (0.160 ± 0.006 g cm−3) than TPOL, FPOL and PF (0.45–0.60 g cm−3). That trend followed the samples throughout pyrolysis, except reactive etching. Now, within each RES, the bulk densities, ρb, of all pyrolysis products were higher than those of the as-prepared samples. Upon closer inspection of ESI, Fig. S.VIII.2,† in general ρb did not follow the upward trend observed with shrinkage as a function of Θ. Instead, the ρb of all C-RES-O-Θ varied randomly within narrow zones throughout the pyrolysis range. At Θ ≤ 500 °C, the density of C-TPOL-O-Θ was slightly higher than the density of the samples, but that trend got reversed at Θ ≥ 600 °C (Fig. 8A). Along pyrolysis of FPOL, PF and RF the densities of the samples were always higher than the densities of the samples, and remained near the levels attained at 240 °C/air (ESI, Fig. S.VIII.2†). In FPOL and PF in particular, the densities of the samples kept on increasing with Θ, and by 800 °C the densities of the and samples had converged; on the other hand, no upward trend was noted in the densities of C-RF-D-Θ with Θ, thereby C-RF-O-800 remained more dense than C-RF-D-800 (Fig. 8B). Probably reflecting a more significant percent mass loss (see pyrolytic yields in ESI, Tables S.I.2–S.I.5†) than the small additional shrinkage during reactive etching, the densities of all etched samples were always lower than their parent carbons (notice the shorter shaded segments in the respective bars of Fig. 8). In particular, the densities of EC-RF-O-800 and EC-TPOL-O-800 were nearly 50% lower than those of C-RF-O-800 and C-TPOL-O-800. For EC-FPOL-D_(or_O)-800 and EC-PF-D_(or_O)-800, the density difference from the corresponding C-samples was smaller, around 15–20%.
Percent open porosities, Π, were calculated from the bulk (ρb) and skeletal densities (ρs) via Π = (ρs – ρb)/ρs. Skeletal densities are tabulated in ESI, Appendix VII† and are compared to one another in graph form in ESI, Appendix VIII Fig. S.VIII.3.† ρs-Values followed a common trend among all four RES: up to 500 °C, the ρs values of the samples were higher than the ρs values of the samples, then the two sets became equal and increased together from 1.2–1.3 g cm−3 to about 1.75–2.0 g cm−3 by 800 °C. Upon reactive etching, all ρs values increased again, moving in the 2.0–2.25 g cm−3 range. The last increase could be associated with closed micropores present in C-RES-D_(or_O)-Θ that became accessible after etching. Closed porosity notwithstanding, along carbonization, open porosities, both with respect to one another, and within each RES, followed the expected general trend established by ρb (see Fig. 8 and ESI, Fig. S.VIII.4). Reflecting mass loss in excess of differential shrinkage, by going from the C- to the EC-materials, porosities increased, most dramatically in the TPOL samples (Fig. 8A). Thus, although the porosities of the 800 °C-pyrolyzed and varieties of TPOL, FPOL and PF were all around 60% v/v, after reactive etching the porosities of EC-FPOL-D_(or_O)-800 and EC-PF-D_(or_O)-800 moved up in the 70–80% v/v range, while EC-TPOL-D_(or_O)-800 moved above the 80% v/v mark. (Owing, as discussed, to the lower starting density of RF, the porosities of C-RF-D_(or_O)-800 were already in the 83–87% v/v range, and the porosities of EC-RF-D_(or_O)-800) moved up in the 92–93% v/v range.)
Despite all the preceding analysis, there was no aspect of the macroscopic material properties that could be clearly and uniquely identified with the early ring fusion aromatization process that takes place in the TPOL and FPOL systems. The closest we came to such a difference with oxidized systems was the large drop in the bulk density and the simultaneous increase in porosity by going from C-TPOL-O-800 to EC-TPOL-O-800. However, that behavior was not observed in the FPOL system that also undergoes early ring fusion aromatization, and most importantly, even within the TPOL system itself, a similar drop in density and increase in porosity was also observed within the C-TPOL-D-800/EC-TPOL-D-800 pair. Thus, we turned into the nanostructure and the porous network of RES as the last stop for possible differences induced by early oxidative ring fusion aromatization.
2.6 The pyrolytic evolution of the nanostructure and the porous network
They were followed with SEM and N2 sorption porosimetry. The microporosity of selected samples was also probed with CO2 adsorption. Qualitatively, all systems behaved similarly, and typical primary data are exemplified with the TPOL system in Fig. 9. Data in similar format for the FPOL, RF and PF systems are shown in ESI, Appendix IX.†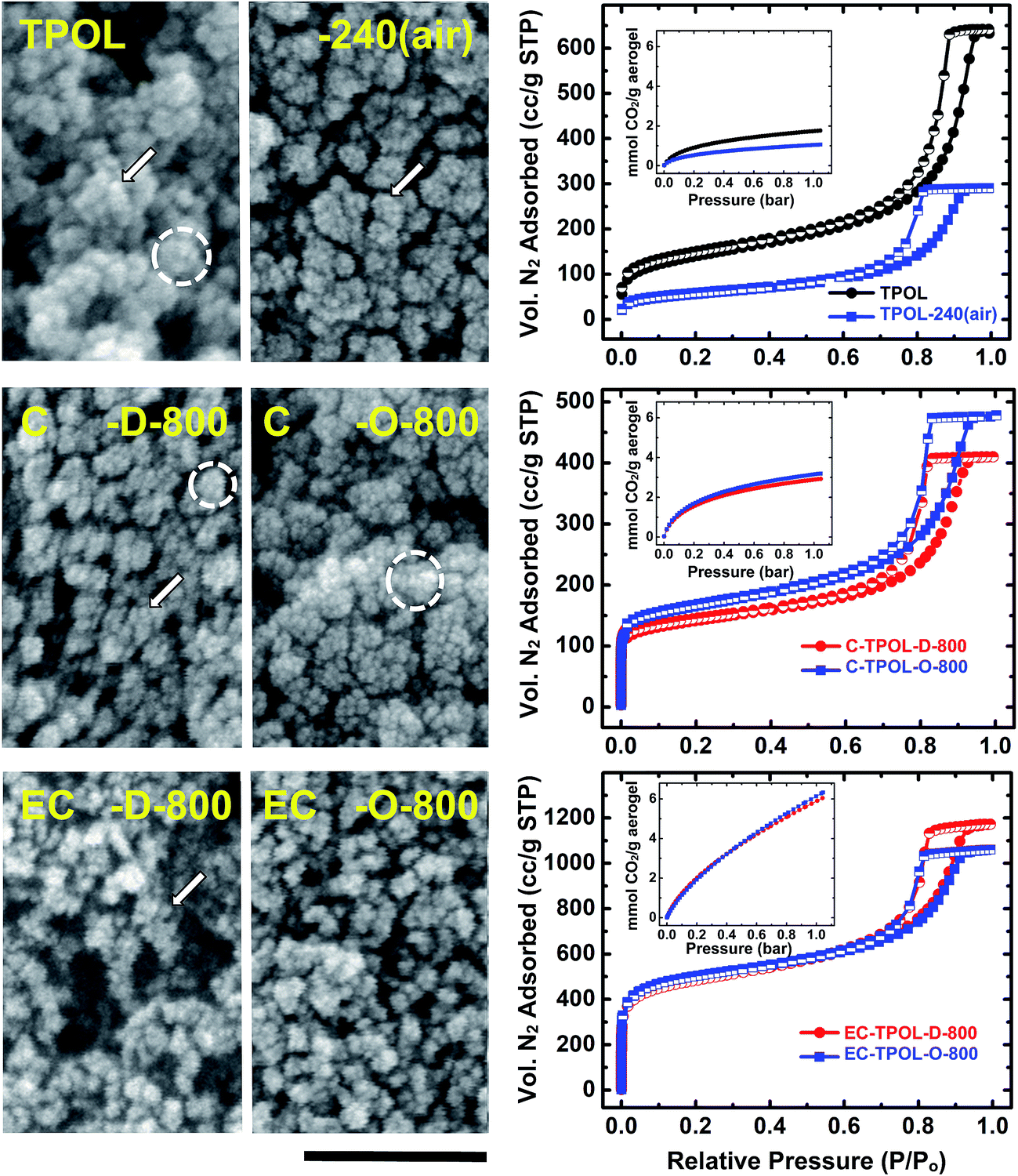 | ||
| Fig. 9 Representative SEM, N2 sorption isotherms (77 K) and CO2 sorption isotherms (0 °C – insets) exemplified by the TPOL system as follows: Top: TPOL and TPOL-240(air); Middle: carbonized C-TPOL-D-800 (red) and C-TPOL-O-800 (blue); Bottom: etched samples corresponding to the samples in middle row, EC-TPOL-D-800 (red) and EC-TPOL-O-800 (blue). SEM scale bar: 100 nm. (Corresponding data for the FPOL, RF and PF systems are shown in ESI, Appendix IX.†) | ||
All samples, at all stages of processing, consisted of smaller particles (arrows in Fig. 9) aggregating to larger ones (dashed circles), which in turn agglomerated to larger entities, and so on. The smallest particles discernible in SEM were on the order calculated for primary particles using gas sorption and skeletal density data (see ESI, Tables S.VII.1–S.VII.4†). Generally, there was an increase in size of primary particles in RES-240(air) (most pronounced in TPOL-240(air), where the particle size increased >2 × , from 8.4 nm to 18.4 nm) followed by a gradual decrease till C-RES-D_(or_O)-800. The primary particle size at that temperature was 5–7 nm in all pyrolyzed systems, irrespective of the or route. After reactive etching at 1000 °C, the most noticeable effect was that EC-RES-D_(or_O)-800 had more empty space.
At all stages of processing, all N2 sorption isotherms of RES, RES-240(air), C-RES-D_(or_O)-800 and EC-RES-D_(or_O)-800 (Fig. 9 and ESI, Fig. S.IX.1–3) were Type IV with broad saturation plateaus indicating that we are dealing with mesoporous materials. (Plateaus, albeit narrower, were observed even with the lower-density RF system – see ESI, Fig. S.IX.2.†) At low partial pressures (P/Po), all N2 sorption isotherms showed a quick rise that became more pronounced in the carbonized samples (C-RES-D_(or_O)-800), and even more pronounced in the carbonized and etched samples (EC-RES-D_(or_O)-800). That rapid increase in the volume of N2 adsorbed indicated microporosity that was confirmed and quantified with CO2 adsorption experiments (see insets in the isotherm frames of Fig. 9 and ESI, Fig. S.IX.1–3†): as the volume of N2 adsorbed at low P/Po increased, the total uptake of CO2 increased too.
The pore-size distributions along processing, of pores with sizes <300 nm are exemplified with the TPOL and RF systems in Fig. 10. Mesopore size distributions were obtained with the BJH method;50 micropore size distributions were obtained with the DFT method on the CO2 adsorption data.51,52 Data for the other two systems, FPOL and PF, are shown in ESI, Appendix X Fig. S.X.1.† In general, the behavior of RF and PF was almost identical to one another, while the behavior of FPOL was intermediate between that of TPOL and of RF/PF. A first observation in Fig. 10 is that the pore size distribution of as-prepared TPOL was different from that of the other three RES, implying an already quite rigid structure. By going from TPOL to TPOL-240(air) the mesopore size distribution moved to smaller diameters, while the micropore size distribution became more resolved. Both of those changes were consistent with molecular relaxation, contraction and further rigidization in a new, more compact molecular arrangement as described by the mechanism of Scheme 4. After ring fusion aromatization neither the mesopore, nor the micropore size distribution was affected by carbonization: the distribution profiles in C-TPOL-D_(or_O)-800 were practically the same as in TPOL-240(air). After etching, the smallest micropores were lost, the larger micropores became a little more narrow, and the mesopores remained unaffected. Now, it is noted that the pore size distributions of the carbonized and etched EC-TPOL-D_(or_O)-800 and EC-RF-D_(or_O)-800, and for this matter of all EC-RES-D_(or_O)-800, were practically identical irrespective of the system or the process ( or ), consistent with the common terminal pyrolytic fate of all RES. More importantly, however, the molecular flexibility of as-prepared RF and PF, that followed them in their oxidized structures, RF-240(air), PF-240(air), accounted for conformational minima right from the beginning, yielding microporous structures similar to those obtained from TPOL only after the chain relaxation processes described in Scheme 4. Consistent with those conclusions, the FPOL system, being more flexible than TPOL, realized a conformation minimum right from the as-prepared stage, just like RF and PF, thus its micropore profile was resolved just like in the latter two. However, FPOL undergoes oxidative ring fusion aromatization just like TPOL (see ESI, Appendix VI†). That process imposes contraction and rigidization, which in turn is expressed by the mesopore profile of FPOL-240(air) (and above) moving to smaller pores.
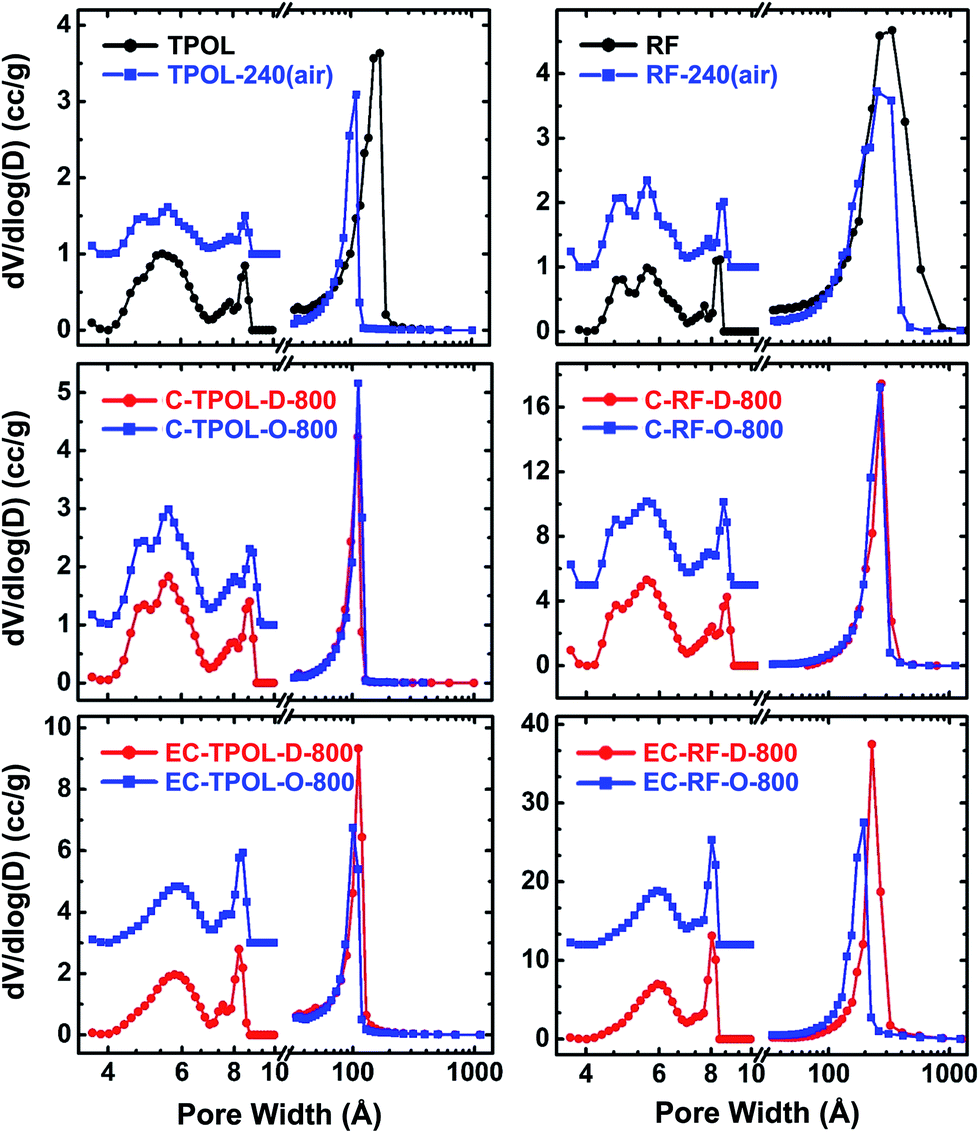 | ||
| Fig. 10 Pore size distributions in the meso and micropore range of samples as shown. Left: from the TPOL system; Right: from the RF system. Mesopore size distributions (curves at >10 Å) from the BJH equation applied to N2 sorption data. Micropore size distributions (curves at <10 Å, magnified 3× and offset for clarity) using a DFT model on CO2 sorption data and assuming slit pores. (Data for the FPOL and PF systems are given in ESI, Appendix X.†) | ||
Similarities or differences in the pore size distributions notwithstanding, the absolute pore volumes that correspond to those distributions paint a complementary view of the effects brought about by ring fusion aromatization, and pave the way towards explaining differences in surface areas. The micropore volumes (tabulated in ESI, Appendix VII†) were extracted from the CO2 adsorption isotherms by assuming slit-pore geometry and using the Horvath–Kawazoe method.53 Micropore volumes in all RES were very small to begin with (<0.08 cm3 g−1), and generally decreased even further in RES-240(air). The largest such decreases were observed with the aromatizable systems: from 0.07 cm3 g−1 to 0.04 cm3 g−1 in TPOL/TPOL-240(air), and from 0.08 cm3 g−1 to 0.06 cm3 g−1 in TPOL/TPOL-240(air). The largest decrease in the TPOL/TPOL-240(air) pair agrees with the molecular contraction expected from Scheme 4. Upon carbonization at 800 °C, the micropore volumes increased to 0.10–013 cm3 g−1 in all C-RES-D_(or_O)-800. Upon further reactive etching, the micropore volumes exactly doubled in all EC-RES-D_(or_O)-800 (all in the 0.22–0.27 cm3 g−1 range), except in EC-RF-D_(or_O)-800, where the micropore volume increased 3.5 × (to 0.44 cm3 g−1). Interestingly, in all RES, the sum of pore volumes in the range of 1.7–300 nm (by N2 sorption) and of micropores (by CO2 adsorption) was about equal to the total pore volume calculated from bulk and skeletal density data via Vtotal = (1/ρb) − (1/ρs), meaning that all materials lacked any significant macroporosity.54 (All relevant values are cited in ESI, Tables S.VII.1–S.VII.4.†) What is important to look at though, is the Vmicropore:Vtotal ratio, which can be calculated directly from the Vmicropore and VTotal data shown in ESI, Tables S.VII.1–S.VII.4.† After carbonization, but before etching, the percent micropore volumes of both C-PF-D_(or_O)-800, and especially of C-RF-D_(or_O)-800, were consistently behind the values of the corresponding C-TPOL-D_(or_O)-800 and C-FPOL-D_(or_O)-800. Curiously, after carbonization and etching the percent micropore volume of EC-PF-D_(or_O)-800 (16.25% and 18.2%, for and respectively) seemed to have caught up with those of EC-TPOL-D_(or_O)-800 (11.6% and 13.8%), and those of EC-FPOL-D_(or_O)-800 (19.7% and 24.1%, for and respectively). On the other hand, the percent micropore volumes of EC-RF-D_(or_O)-800 (6.8% and 8.0%, for and respectively) were consistently behind. Perhaps, that the out-of-line behavior of EC-PF-D_(or_O)-800 was related to the resilience of C-PF-D_(or_O)-800 to etching (refer to the etching yields in ESI, Table S.I.2†).
It is speculated that the trend towards higher micropore volumes in the aromatized systems is directly related to the molecular rigidization that fixes the relative position of the polymeric chains. In systems that cannot be aromatized by an early oxidation step (RF and PF), later aromatization (recall all systems converged chemically above 600 °C) may compete with other processes, e.g., melting of polymeric segments, which may have reduced microporosity. Most probably related to the link between higher micropore volumes and early oxidative ring-fusion aromatization, BET surface areas of carbons derived from TPOL-240(air) and FPOL-240(air) were consistently about 20% higher than the surface areas of carbons derived by direct pyrolysis of as-prepared TPOL and FPOL (Fig. 11). That edge identified in the surface areas of C-TPOL-O-Θ and C-FPOL-O-Θ over C-TPOL-D-Θ and C-FPOL-D-Θ followed the samples through etching. As shown in Fig. 11, considered as groups, the BET surface areas of EC-TPOL-O-(600 ≤ Θ ≤ 800) and EC-FPOL-O-(600 ≤ Θ ≤ 800) were higher than the BET surface areas of EC-TPOL-D-(600 ≤ Θ ≤ 800) and EC-FPOL-D-(600 ≤ Θ ≤ 800). The highest surface area was recorded for EC-TPOL-O-600 and was equal to at 2778 ± 209 m2 g−1 (see ESI, Table S.VII.1†). Micropore surface areas (using the Harkins and Jura method55,56) are included in Fig. 11, and for carbonized samples they were about 50% of the BET surface areas. The micropore surface areas of the carbonized and etched samples were between 30% and 50% of their BET surface areas. On the other hand, both the BET and the micropore surface areas of carbons and etched carbons from RF-240(air) and PF-240(air) were at all levels indistinguishable from those via direct pyrolysis of RF and PF (Fig. 11). Although in absolute terms the BET surface areas of the carbonized RF and PF systems were in the same range as those from TPOL and FPOL, their micropore areas were generally lower.
 | ||
| Fig. 11 BET surface area data (upper curves, full symbols), and micropore surface area data (lower curves, open symbols) of all samples. Black: as-prepared samples. Red: samples from direct pyrolysis of as-prepared samples at the temperature indicated; blue: samples from pyrolysis of samples oxidized in air at 240 °C. Dash-lines connect samples etched with CO2 at 1000 °C with the corresponding carbonized samples. All data points are averages of three samples from different batches, run at different times. Error bars mostly within the symbols. | ||
3. Conclusion
Aerogels became an enabling tool for the study of the carbonization process of four main-stream phenolic resins. It was found that low-temperature (240 °C) oxidation of FPOL and TPOL aerogels with air circulating through their bulk caused ring-fusion aromatization and formation of pyrylium heteroaromatic rings along their skeletal backbone. Other phenolic resins like RF and PF got oxidized too, but their oxidized forms did not proceed with aromatization at 240 °C. Eventually, upon further pyrolysis, either as-prepared samples, or samples from a prior 240 °C/air oxidation step, converged to a common carbon structure that includes fused pyrylium rings, and charge-compensating phenoxides. It is surprising that although pyryliym has been identified in biochar,38 it has been missed among the pyrolysis products of phenolic resins.41 The closest prior studies ever came to pyrylium had been to claim saturated five- and six-membered rings with oxygen between aromatic rings.57–61 On the hindsight, it seems that key to our findings was the fact that we compared the oxidative behavior of several systems of increasing complexity, which turned out to be beneficial, because it allowed us to track the fate of aromatic carbons using solid-state 13C NMR. Whenever an early oxidative ring-fusion aromatization could take place (cases of TPOL and FPOL), its effect was to rigidize the polymeric structure and create microporosity that contributed towards carbons with higher surface areas than those obtained by direct pyrolysis of as-prepared materials. Those findings are directly relevant to high surface area carbons for gas sorption (e.g., capture and sequestration of CO2 (ref. 62)) and for ion-exchange materials via the fixed O+ and O− sites on the carbon backbones, as has been proposed for biochar bearing pyrylium moieties.384. Experimental
4.1 Materials
All reagents and solvents were used as received without further purification. Terephthalaldehyde (T), phloroglucinol (POL), 12.1 N hydrochloric acid, and 1,4-dioxane were purchased from Acros Organics U.S.A. Resorcinol (R), formaldehyde (F, 37% w/w aqueous solution methanol stabilized), and acetonitrile (ACS reagent grade) were purchased from Aldrich Chemical Co. Phenol (P, crystals/certified ACS grade) was purchased from Fisher Scientific. Syphon grade CO2 and ultra-high purity argon were obtained from BOC Gases, Murray Hill, N.J. supplied locally by Ozarc Gases. Liquid nitrogen was purchased from Airgas, Rolla, Mo.4.2 Preparation of phenolic resin aerogels (RES)
All formulations and gelation points are listed in ESI, Appendix I Table S.1.†In a typical procedure, the predetermined amount of the phenolic monomer was dissolved in 2/3 of the predetermined amount of the appropriate solvent. Dissolution of POL in 1,4-dioxane was assisted by heating and stirring for 5 min at 70 °C under N2. In the case of TPOL, T was also mixed with POL and the two reagents were dissolved in 1,4-dioxane together as just described. In the cases of the three formaldehyde-based aerogels (FPOL, RF, PF), the appropriate amount of the commercially available formaldehyde solution was added at room temperature to the solution of the phenolic compound. (In the case of FPOL, the POL solution was allowed to cool back to room temperature before adding the formaldehyde solution.) Subsequently, the predetermined amount of the catalyst (12.1 N HCl) was dissolved in the remaining 1/3 of the predetermined amount of solvent, and the acid solution was added to the phenolic compound/aldehyde solution at room temperature to form the sol. All sols were stirred briefly and were poured in polypropylene molds (either polypropylene Scintillation Vials General Purpose, 6.5 mL, Sigma-–Aldrich Catalogue no. Z376825, 1.27 cm inner diameter; or, Wheaton polypropylene Omni-Vials, part no. 225402, ∼1 cm in diameter). The molds were sealed with their caps, wrapped with Parafilm™, and were kept for 24 h for gelation and aging. TPOL, FPOL and RF gelled at room temperature and aging was carried out at the same temperature. Gelation of PF was carried out at 80 °C (24 h); PF wet-gels were aged at 70 °C (12 h). Wet-gels were washed with acetone (3 × 8 h, using 4× the volume of the gel each time), and were dried in an autoclave with CO2 taken out at the end as a supercritical fluid.
4.3 Air oxidation of RES and preparation of RES-240(air)
RES aerogels were placed in a conventional convection oven at 240 °C for 5 h, and turned from brown or red to dark brown.4.4 Conversion of as-prepared RES aerogels, or air-oxidized RES-240(air) aerogels into carbon aerogels C-RES-D-Θ and C-RES-O-Θ
Aerogel monoliths, including as-prepared and those treated at 240 °C in air, were pyrolyzed in a tube furnace under flowing high purity Ar (300 mL min−1) at 300 °C, 400 °C, 500 °C, 600 °C, 700 °C, or 800 °C for 5 h. The temperature of the furnace was increased to the target temperature Θ at 2.5 °C min−1. New samples were used at each pyrolysis temperature. At the end of the heating period, cooling was controlled also at 2.5 °C min−1 under continuous flow of Ar. As-prepared RES aerogels pyrolyzed at a target temperature Θ are designated as C-RES-D-Θ. Air-oxidized aerogels, RES-240(air), pyrolyzed at a target temperature Θ are designated as C-RES-O-Θ.4.5 Reactive etching of C-RES-D-Θ and C-RES-O-Θ carbon aerogels into EC-RES-D-Θ and EC-RES-O-Θ
Either C-RES-D-Θ or C-RES-O-Θ aerogels prepared at Θ = 600 °C, Θ = 700 °C, or Θ = 800 °C were pyrolyzed further at 1000 °C for 3 h under flowing CO2 (300 mL min−1). For this, at the end of the 5 h pyrolysis-under-Ar period, the temperature of the furnace was increased to 1000 °C, the gas was switched to CO2 and pyrolysis continued for the prescribed time. At the end of this period, the flowing gas was switched back to Ar and the tube was cooled back to ambient temperature at 2.5 °C min−1.4.6 Methods
Drying of acetone-exchanged wet-gels with supercritical fluid (SCF) CO2 was carried out in an autoclave (SPIDRY Jumbo Supercritical Point Dryer, SPI Supplies, Inc. West Chester, PA). Samples submerged in acetone were loaded into the autoclave cooled at 14 °C. The pressure vessel was closed and liquid CO2 was allowed until it displaced all acetone, which was then drained out. Liquid CO2 was allowed in the vessel several more times until acetone was extracted out of the pores of the samples completely. The criterion for the latter was that CO2 released from the vessel formed dry ice powder. At that point, the temperature of the autoclave was raised to 40 °C and SCF CO2 was vented off as a gas.4.7 Physical characterization
Bulk densities (ρb) were calculated from the weight and the physical dimensions of the samples. Skeletal densities (ρs) were measured using helium pycnometry on a Micromeritics AccuPyc II 1340 instrument.4.8 Chemical characterization
CHN elemental analysis was conducted with a Perkin-Elmer Model 2400 CHN Elemental Analyzer, calibrated with acetanilide, urea, trans-stilbene, benzophenone, and glycine.Infrared (FTIR) spectra were taken in KBr pellets, on a Nicolet-FTIR Model 750 spectrometer.
Solid-state CPMAS 13C-NMR spectra were obtained with samples ground into fine powders on a Bruker Avance III 400 MHz spectrometer with a carbon frequency of 100 MHz, using a 7 mm Bruker MAS probe at a magic angle spinning rate of 5 kHz, with broadband proton suppression, and CP TOSS pulse sequence. The Total Suppression of Spinning Sidebands (TOSS) pulse sequence was applied by using a series of four properly timed 180° pulses on the carbon channel at different points of a cycle before the acquisition of the FID, after an initial excitation with a 90° pulse on the proton channel. The 90° excitation pulse on the proton and the 180° excitation pulse on carbon were set to 4.2 μs and 10 μs, respectively. Under those conditions, spectra of all RES, RES-240(Ar) and RES-240(air) were taken twice, once with the cross polarization contact time set at 3000 μs and once at 5 μs. Solid-state 13C NMR spectra were referenced externally to glycine (carbonyl carbon at 176.03 ppm). Chemical shifts are reported versus TMS (0 ppm).
XPS data were obtained with a Kratos Axis 165 photoelectron spectroscopy system. Samples for XPS were prepared by mixing powders of the aerogels (like those prepared for solid-state NMR) with Au powder (10% w/w) and pellets were compressed like those used for FTIR. (For highly conductive samples, the amount of Au powder could be reduced to 5% w/w.) Such pellets were placed flat on a conductive carbon tape that was then adhered to stainless steel sample holders. Samples were introduced into the analysis chamber one at a time and the chamber was evacuated at 10−8 Torr or lower. No ion sputtering was carried out on any of the samples. An Al monochromatic source at 150 watts was used for excitation. A charge neutralizer was used to reduce the effects of differential or sample charging. The analysis area was 700 × 300 microns. Elemental quantification calculations were based on broad survey results from single sweeps at higher sensitivity (pass energy = 80), and were carried out with Kratos Axis Vision processing software and its appropriate relative sensitivity factors for the particular XPS system. High resolution elemental scans where carried out at a lower sensitivity (pass energy = 20), using multiple sweeps to improve the signal-to-noise ratios. Deconvolution of C 1s and O 1s spectra of the samples were performed with Gaussian function using OriginPro 8.5.1 software.
4.9 Thermal analysis
Differential Scanning Calorimetry (DSC) was conducted both under N2 and in air with a TA Instruments Modulated Differential Scanning Calorimeter (MDCS) Model 2920 calibrated with a sapphire standard. Samples were used as powders (4–8 mg), and the MDSC instrument was run from −30 to 300 °C at 10 °C min−1.4.10 Structural characterization
Scanning electron microscopy (SEM) was conducted with Au-coated samples on a Hitachi Model S-4700 field-emission microscope.4.11 Pore structure analysis
The pore structure was probed with N2-sorption porosimetry at 77 K using either a Micromeritics ASAP 2020 or a TriStar II 3020 version 3.02 surface area and porosimetry analyzer. Before porosimetry, samples were outgassed for 24 h under vacuum at 80 °C. Data were reduced to standard conditions of temperature and pressure (STP). Total surface areas, σ, were determined via the Brunauer–Emmett–Teller (BET) method from the N2-sorption isotherms. Micropore surface areas were calculated via t-plot analysis of the isotherms using the Harkins and Jura Model.55,56 Pore size distributions were determined with the Barret–Joyne–Halenda (BJH) equation applied to the desorption branch of the N2-sorption isotherms.50 Micropore analysis was conducted either with N2-sorption at 77 K using a low pressure transducer (0.1 Torr) on the Micromeritics ASAP 2020 surface area and porosity analyzer, or with CO2-sorption up to 760 Torr (0.03 relative pressure) at 273 K (ice-water bath) using the Micromeritics Tristar II 3020 version 3.02 mentioned above. Micropore size distributions were calculated from the CO2 adsorption data using a DFT model,51,52 and micropore volumes were obtained using the Horvath–Kawazoe (HK) method,53 applied on the same data. Average pore diameters were calculated using the 4 × Vtotal/σ method, where Vtotal is the total pore volume per gram of sample, and can be calculated either via Vtotal = (1/ρb) − (1/ρs) or from the single highest volume of N2 adsorbed along the adsorption isotherm (at P/P0 ∼ 1). Average pore diameter values, calculated with Vtotal by both methods, are cited herewith; the two values converge for mostly mesoporous materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Army Research Office for financial support under Award Number W911NF-14-1-0369. We also thank the Materials Research Center of the Missouri University of Science and Technology for support with materials characterization.References
- A. Gardziella, L. A. Pilato and A. Knop, Phenolic Resins, Springer-Verlag, Berlin, Germany, 2nd edn, 2000 Search PubMed.
- C. P. R. Nair, Prog. Polym. Sci., 2004, 29, 401–498 CrossRef CAS.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4265 CrossRef CAS PubMed.
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- R. W. Pekala, J. Mater. Sci., 1989, 24, 3221–3227 CrossRef CAS.
- X. Lu, M. C. Arduini-Schuster, J. Kuhn, O. Nilsson, J. Fricke and R. W. Pekala, Science, 1992, 255, 971–972 CAS.
- R. W. Pekala and D. W. Schaefer, Macromolecules, 1993, 26, 5487–5493 CrossRef CAS.
- S. A. Al-Muhtaseb and J. A. Ritter, Adv. Mater., 2003, 15, 101–114 CrossRef CAS.
- W.-C. Li, A.-H. Lu and F. Schüth, Chem. Mater., 2005, 17, 3620–3626 CrossRef CAS.
- C. Scherdel and G. Reichenauer, Microporous Mesoporous Mater., 2009, 126, 133–142 CrossRef CAS.
- N. Leventis, I. A. Elder, D. R. Rolison, M. L. Anderson and C. I. Merzbacher, Chem. Mater., 1999, 11, 2837–2845 CrossRef CAS.
- N. Leventis, A.-M. M. Rawashdeh, I. A. Elder, J. Yang, A. Dass and C. Sotiriou-Leventis, Chem. Mater., 2004, 16, 1493–1506 CrossRef CAS.
- B. A. Newcomb, Composites, Part A, 2016, 91, 262–282 CrossRef CAS.
- L. Liu, C. Jia, J. He, F. Zhao, D. Fan, L. Xing, M. Wang, F. Wang, Z. Jiang and Y. Huang, Compos. Sci. Technol., 2015, 121, 56–72 CrossRef CAS.
- J. Li, X. Huang and L. Chen, J. Electrochem. Soc., 2000, 147, 2653–2657 CrossRef CAS.
- Y.-X. Wang and Q. Wang, J. Appl. Polym. Sci., 2007, 104, 1255–1259 CrossRef CAS.
- K. E. Perepelkin, Fibre Chem., 2003, 35, 409–416 CrossRef CAS.
- H. Ishida, in Handbook of Benzoxazine Resins, ed, H. Ishida and T. Agag, Elsevier, New York, 2011, pp. 3–81 Search PubMed.
- T. Takeichi, T. Kawauchi and T. Agag, Polym. J., 2008, 40, 1121–1131 CrossRef CAS.
- N. N. Ghosh, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2007, 32, 1344–1391 CrossRef CAS.
- P. Lorjai, S. Wongkasemjit, T. Chaisuwan and A. M. Jamieson, Polym. Degrad. Stab., 2011, 96, 708–718 CrossRef CAS.
- S. Mahadik-Khanolkar, S. Donthula, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2014, 26, 1303–1317 CrossRef CAS.
- A. G. Sadekar, S. S. Mahadik, A. N. Bang, Z. J. Larimore, C. A. Wisner, M. F. Bertino, A. K. Kalkan, J. T. Mang, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2012, 24, 26–47 CrossRef CAS.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2011, 23, 1818–1824 CrossRef CAS.
- S. Mulik, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2007, 19, 6138–6144 CrossRef CAS.
- C. Scherdel and G. Reichenauer, Microporous Mesoporous Mater., 2009, 126, 133–142 CrossRef CAS.
- N. Leventis, S. Mulik and C. Sotiriou-Leventis, Chem. Mater., 2008, 20, 6985–6997 CrossRef.
- Z. Yoshida, H. Sugimoto and S. Yoneda, Tetrahedron, 1974, 30, 2099–2105 CrossRef CAS.
- A. R. Katritzky, S. Sengupta and A. Dinculescu, J. Heterocycl. Chem., 1986, 23, 169–171 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., New York, 7th edn, 2005, pp. 87–88 Search PubMed.
- D. A. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Introduction to Spectroscopy, Cengage Learning, Belmont, CA USA, 4th edn, 2009, pp. 15–104 Search PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, Inc., New York, 3rd edn, 2001, pp. 157–167 Search PubMed.
- A. Thangavel, C. Sotiriou-Leventis, R. Dawes and N. Leventis, J. Org. Chem., 2012, 77, 2263–2271 CrossRef CAS PubMed.
- G. P. López, D. G. Castner and B. D. Ratner, Surf. Interface Anal., 1991, 17, 267–272 CrossRef.
- D. Briggs and G. Beamson, Anal. Chem., 1993, 65, 1517–1523 CrossRef CAS.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CAS.
- R. Ciriello, A. Guerrieri, F. Pavese and A. M. Salvi, Anal. Bioanal. Chem., 2008, 392, 913–926 CrossRef CAS PubMed.
- M. Lawrinenko and D. A. Laird, Green Chem., 2015, 17, 4628–4636 RSC.
- C. M. Kim, H. S. Jeong and E. H. Kim, Surf. Sci., 2000, 459, L457–L461 CrossRef CAS.
- L. Q. Wu, S. Q. Li, Y. C. Li, Z. Z. Li, G. D. Tang, W. H. Qi, L. C. Xue, L. L. Ding and X. S. Ge, Appl. Phys. Lett., 2016, 108, 021905 CrossRef.
- D. D. Werstler, Polymer, 1986, 27, 757–764 CrossRef CAS.
- P. Sándor and L. Radics, Org. Magn. Reson., 1981, 16, 148–155 CrossRef.
- A. M. Bello and L. P. Kotra, Tetrahedron Lett., 2003, 44, 9271–9274 CrossRef CAS.
- A. R. Katritzky, Y.-K. Yang, B. Gabrielsen and J. Marquet, J. Chem. Soc., Perkin Trans. 2, 1984, 857–866 RSC.
- J. Fossey, D. Lefort and J. Sorba, Free Radicals in Organic Chemistry, John Wiley & Sons, Inc, New York, 1995, p. 218 Search PubMed.
- J. March, Advanced Organic Chemistry, John Wiley & Sons, Inc, New York, 4th edn, 1992, pp. 705–706 Search PubMed.
- B. Saha and G. C. Schatz, J. Phys. Chem. B, 2012, 116, 4684–4692 CrossRef CAS PubMed.
- Q. Chen, X. Jiang, Y. Bin and M. Matsuo, Polym. J., 2007, 39, 568–578 CrossRef CAS.
- S. Naeem, V. Baheti, V. Tunakova, J. Militky, D. Karthik and B. Tomkova, Carbon, 2017, 111, 439–447 CrossRef CAS.
- P. A. Webb and C. Orr, Analytical Methods in Fine Particle Technology, Micromeritics Instrument Corporation, Norcross, GA, USA, 1997, pp. 80–81 Search PubMed.
- D. Lozano-Castelló, D. Cazorla-Amorós, A. Linares-Solano and A. M. Salvi, Carbon, 2004, 42, 1231–1236 Search PubMed.
- B. B. Saha, S. Jribi, S. Koyama and I. I. El-Sharkawy, J. Chem. Eng. Data, 2004, 56, 1974–1981 CrossRef.
- G. Horvath and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470–475 CrossRef CAS.
- With regards to macroporosity, the case of C-RF-D_(or_O)-800 and EC-RF-D_(or_O)-800 was particularly interesting (ESI, Table S.VII.3†) because, on one hand, owing to the low starting density of those materials, the Vtotal values were always much higher than the values of all other corresponding carbons, on the other hand RF-derived carbons lacked completely any pores above >300 nm.
- W. D. Harkins and G. Jura, J. Am. Chem. Soc., 1944, 66, 1362–1366 CrossRef CAS.
- P. A. Webb and C. Orr, Analytical Methods in Fine Particle Technology, Micromeritics Instrument Corporation, Norcross, GA, USA, 1997, pp. 67–68 Search PubMed.
- H. Jiang, J. Wang, S. Wu, Z. Yuan, Z. Huc, R. Wu and Q. Liu, Polym. Degrad. Stab., 2012, 97, 1527–1533 CrossRef CAS.
- L. Liu, J. Yang and Q. Meng, J. Sol–Gel Sci. Technol., 2013, 67, 304–311 CrossRef CAS.
- R. Banerjee, K. Patil and K. Khilar, Can. J. Chem. Eng., 2006, 84, 328–337 CrossRef CAS.
- K. Ouchi, Carbon, 1966, 4, 59–66 CrossRef CAS.
- Y. Yamashita and K. Ouchi, Carbon, 1981, 19, 89–94 CrossRef CAS.
- A. M. Saeed, P. M. Rewatkar, H. Majedi Far, T. Taghvaee, S. Donthula, C. Mandal, C. Sotiriou-Leventis and N. Leventis, ACS Appl. Mater. Interfaces, 2017, 9, 13520–13536 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Appendix I: formulation of RES, gelation times and pyrolytic yields. Appendix II: differential scanning calorimetry of the four RES. Appendix III: FTIR data. Appendix IV: high-resolution O 1s XPS data. Appendix V: solid-state CPMAS 13C NMR data. Appendix VI: air-oxidation mechanism of FPOL. Appendix VII: tables with cumulative materials characterization data of all samples. Appendix VIII: cumulative shrinkage, bulk and skeletal density, and porosity data in graph form. Appendix IX: SEM, N2 sorption and CO2 sorption analysis data. Appendix X: additional meso and micropore size distribution data. See DOI: 10.1039/c7ra10958j |
| This journal is © The Royal Society of Chemistry 2017 |