
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biofunctionalisation of p-doped silicon with cytochrome c553 minimises charge recombination and enhances photovoltaic performance of the all-solid-state photosystem I-based biophotoelectrode†
Julian David Janna Olmos ab,
Philippe Becquetc,
Dominik Grontd,
Jarosław Sare,
Andrzej Dąbrowskie,
Grzegorz Gawlike,
Marian Teodorczyke,
Dorota Pawlakef and
Joanna Kargul*a
ab,
Philippe Becquetc,
Dominik Grontd,
Jarosław Sare,
Andrzej Dąbrowskie,
Grzegorz Gawlike,
Marian Teodorczyke,
Dorota Pawlakef and
Joanna Kargul*a
aSolar Fuels Laboratory, Centre for New Technologies, University of Warsaw, Banacha 2C, 02-097 Warsaw, Poland. E-mail: j.kargul@uw.edu.pl
bFaculty of Biology, University of Warsaw, Miecznikowa 1, 02-096 Warsaw, Poland
cCarinthia University of Applied Sciences, Europastrasse 4, 9524 Villach, Austria
dLaboratory of Theory of Biopolymers, Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
eInstitute of Electronic Materials Technology, Wólczyńska 133, 01-919 Warsaw, Poland
fLaboratory of Materials Technology, Centre for New Technologies, University of Warsaw, Banacha 2C, 02-097 Warsaw, Poland
First published on 11th October 2017
Abstract
Surface-directed passivation of p-doped silicon (Si) substrate was achieved by its biofunctionalisation with hexahistidine (His6)-tagged cytochrome c553 (cyt c553), a soluble electroactive photosynthetic protein responsible for electron donation to photooxidised photosystem I (PSI). Five distinct variants of cyt c553 were genetically engineered by introducing the specific linker peptides of 0–19 amino acids (AA) in length between the cyt c553 holoprotein and a C-terminal His6-tag, the latter being the affinity ‘anchor’ used for the specific immobilisation of this protein on the semiconductor surface. Calculation of 2D Gibbs free energy maps for the five cyt c553 variants showed a significantly higher number of thermodynamically feasible conformations of immobilised cyt c variants containing longer linker peptides. Here we show that the distinct cyt c553-based Si bioelectrodes display some characteristics of the p–n-type diodes, albeit varying in the level of dark saturation current J0 considered as the charge recombination parameter. These combined bioinformatic and electrochemical analyses indicate that the cyt c553 variants with longer linker peptides, up to 19AA in length, allow for more structural flexibility of immobilised cyt c553 in terms of both, orientation and distance of the haem group with respect to the Si surface, and promote the efficient biopassivation of the semiconductor substrate. Incorporation of the specifically immobilised 19AA cyt c553 variant into the all-solid-state biophotoelectrodes containing light harvesting PSI module enhanced biophotovoltaic performance of the PSI biophotoelectrode compared to the analogous device devoid of cyt c553.
1. Introduction
To date, the most technologically advanced photovoltaics devices are those based on silicon (Si). By taking advantage of the current state of the art of Si-based photovoltaics we are presently able to generate electricity from sunlight relatively cheaply. Currently, more than 90% of the global photovoltaics market is based on Si technology and assuming a growth rate for this technology of 40% p.a., Si solar cells carry the great potential to contribute significantly towards meeting the ever increasing energy demand of mankind.1,2 As Si is the second most abundant and evenly distributed element in the earth's crust there is no risk of its shortage in the foreseeable future.1 To date, Si-based solar cells reach the highest reported power conversion efficiencies among single-junction solar cells. In fact, the second highest most efficient single-junction solar cell is the black Si solar cell with a reported efficiency of 27.6 ± 1.2%.3,4One of the key factors for improving the power conversion efficiency of solar cells is to minimise wasteful processes of charge recombination by optimisation of the nanostructuring of the working modules and their interfaces. To this end, semiconductor surface passivation is of the particular importance, as it often results in significant minimisation of surface charge recombination, leading to the better overall performance of semiconductor-based solar cells.5 To date, several passivation approaches have been applied mainly based on elaborate chemical and electric field effects.6,7 Recently, a new generation of passivated Si solar cells with an efficiency of 22.1% has been reported by Savin et al.8 who employed atomic layer deposition of Al2O3 on the surface of the nanostructured black Si to significantly decrease surface charge recombination by extending minority carrier lifetimes to the millisecond timescale. More recently, it has been shown that the key strategy for the enhancement of the power conversion efficiency is based on including two passivation layers; a top passivation layer on top of the photoactive substrate and a bottom passivation layer between Si and the photoactive substrate.4 Such an approach resulted in an increase of power conversion efficiency to over 26%.4
Functionalisation of semiconductor electrode materials with natural light harvesting proteins such as photosystem I (PSI) is a particularly attractive concept, as such a ‘green’ biohybrid solar technology utilises the combination of relatively cheap and stable semiconducting material, such as Si, together with the highly efficient light-harvesting/charge separating natural pigment/protein complex which absorbs well in the blue-red-far red part of the electromagnetic spectrum, and operates with the impressive internal quantum yield close to unity.9 However, the efficiency of the reported to date biohybrid PSI-based photoelectrodes is still limited to values below 1% mainly due to the recombination of photo-excited electron–hole pairs at the interface of the electrode modules, rather than by the PSI photochemical activity or stability. Other obstacles to obtain high power conversion efficiency of PSI-based biophotoelectrodes are poor electronic contact between the photoactive module and the electrode, as well as insufficient and heterogeneous PSI loading onto the electrode. Nevertheless, it has been recently shown that high energy photoactivated electrons that are generated in PSI upon light absorption can be recovered in biophotoelectrodes even faster than in natural photosynthesis itself,10 highlighting the great potential of PSI-based biohybrid solar technology once it is optimised.
Previously metal electrodes have commonly been used in PSI-based photobioelectrodes;11–13 however, application of semiconducting electrode materials allowed for a significant improvement of photocurrent and photovoltage outputs.14–18 Recently, the p-doped Si was found to be an excellent platform for electron donation to the photo-oxidised PSI19,20 due to well-matched valence and conduction bands for Si (0.5 V and −0.6 V vs. SHE, respectively), the P700 primary electron donor and the terminal electron acceptor, the iron–sulphur cluster FB (0.43 V and −0.58 V vs. NHE, respectively).20
Here we report that increasing the distance and structural flexibility of the haem group of immobilised cytochrome c553 (cyt c553) protein, the mobile redox-active electron donor of PSI, upon its specific immobilisation on the p-doped Si electrode leads to efficient passivation of the semiconductor surface, as measured by minimisation of the dark saturation current (J0) also known as the semiconductor surface recombination parameter. Importantly, incorporation of the most structurally flexible 19AA peptide linker cyt c553 variant as the conductive layer into the all-solid-state PSI-based biophotoelectrodes significantly enhanced the power conversion efficiency (20-fold) compared to the analogous device devoid of cyt c553, demonstrating feasibility of this molecular approach for improving the overall performance of PSI-based biophotoelectrodes.
2. Results and discussion
Optimisation of the electron transfer between an appropriate semiconductor substrate and PSI photoactive module is essential for improving the performance of such biophotoelectrodes.16,21,22 However, most of the PSI biosolar systems reported to date have relied on mass transfer through the use of artificial electron mediators (ref. 21, 23 and 24 and references therein), resulting in the significantly decreased performance of such devices.10,13,25–28 To overcome this limitation, our approach was to rationally engineer efficient direct electron transfer (DET) at the interface between the semiconductor surface and the (photo)electroactive PSI module by using the specifically oriented conductive layer of redox active cyt c553 in the all-solid-state device configuration. The 9 kDa cyt c553 protein functions in vivo as the natural electron donor to the photooxidised P700+ reaction centre of PSI upon the highly specific protein–protein interaction.21,29 The domain-specific interaction between cyt c553 and the PSI complex has been recently applied in the plasmonic nanostructures of silver nanowires, where it led to the spectacular enhancement of optical properties of PSI,30 as well as improvement of photocurrent output in various types of biophotoelectrodes.31–332.1 Linker peptide design and structural modelling of the cyt c553 variants
The electroactive protein used in our study, that constituted the biological conductive layer of the PSI-based biophotoelectrode, is cyt c553 from an extremophilic red microalga Cyanidioschyzon merolae, whereas the electrocompatible semiconductor substrate is heavily p-doped Si. The cyt c553 protein is a structural and functional homologue of cyt c6 which is a soluble redox active protein serving in vivo as the native electron donor to the photo-oxidised P700+ reaction centre of PSI.21,34 Application of the highly robust redox active proteins as modules of biophotoelectrodes, such as the photosynthetic proteins from C. merolae used in this study, carries the great potential to improve the long-term performance and stability of such novel, mediatorless all-solid-state biodevices due to inherently increased stability of extremophilic biological components across a wide range of extreme conditions.17,35,36The nuclear genome of C. merolae contains a single gene petJ which encodes cyt c553 protein that is characterised spectroscopically by the specific α peak at 553 nm in its reduced state. The X-ray analysis of the red algal cyt c553 protein showed that it is a Class I c-type cyt in which the redox-active haem prosthetic group covalently binds to Cys34 and Cys37. The central Fe atom of the haem group displays octahedral coordination with His18 and Met58 axial ligands.37
In the process of exploring alternative routes for the improvement of electron transfer at the solid-state interface of photovoltaic devices, it is important to design a rational approach for prediction of the optimal conformation of the immobilised redox active components that would allow for the efficient DET at the interfaces between the working modules. To this end, four distinct linker peptides of 5–19 amino acids (AA) in length were designed between the cyt c553 holoprotein and a C-terminal His6-tag (see Table 1), the latter used to anchor the cyt protein to the Ni–NTA-functionalised p-doped Si electrode surface. The control His6-tagged cyt c553 variant, termed 0AA, constituted the His6-tagged cyt c553 holoprotein without any additional peptide linker. Overall, the rationale in the design of the four peptide linker variants of cyt c was to choose AA residues in the peptide sequence that would allow for a variable degree of conformational freedom of motion of the immobilised cyt c553. The 4 peptide linkers were designed based on their rigidity versus flexibility, as well as using a specific sequence known to naturally form a semi-α-helix in the case of the 19-AA linker peptide (see Fig. 1). The inclusion of hydrophobic (Ala) and highly hydrophobic (Leu and Val) AA residues aimed to induce rigidity to the specifically immobilised cyt c553.
| Cyt c553 variant | AA sequence of linker peptides | Linker length (AA no.) | Molecular weight (Da) | Theoretical pI |
|---|---|---|---|---|
| 0AA | Holoprotein-HHHHHH | N/A | N/A | N/A |
| 5AA | Holoprotein-GSGLE-HHHHHH | 5 | 461.4 | 4.00 |
| 10AA | Holoprotein-GSGSGSGSLE-HHHHHH | 10 | 836.8 | 4.00 |
| 12AA | Holoprotein-GSSVDKLAAALE-HHHHHH | 12 | 1160.2 | 4.37 |
| 19AA (semi α-helix) | Holoprotein-AEAAAKEAAAKEAAAKALE-HHHHHH | 19 | 1814.0 | 4.95 |
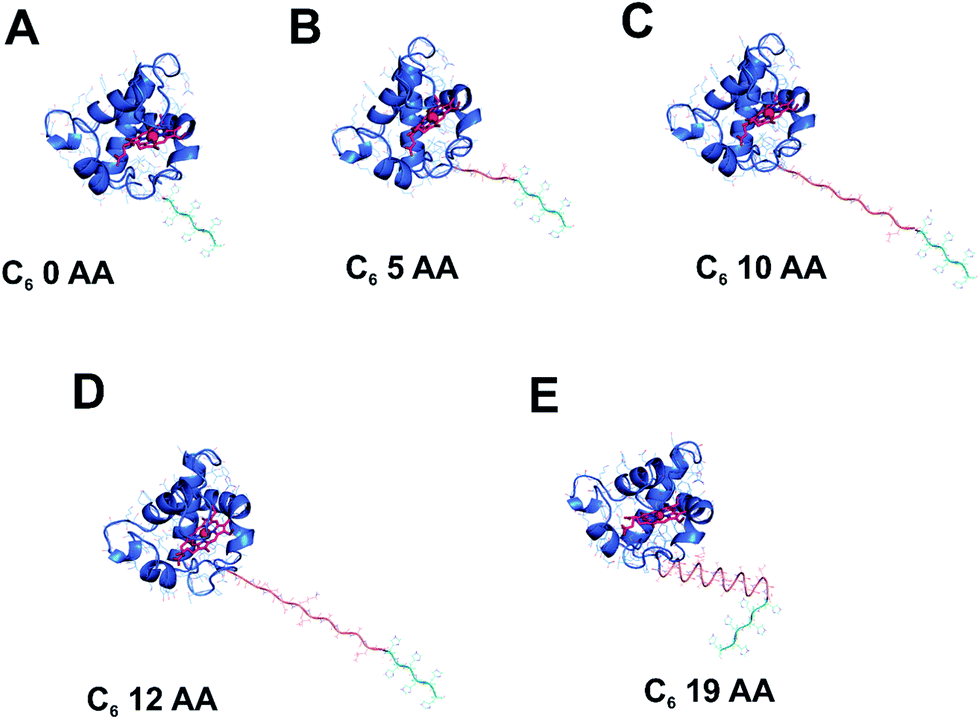 | ||
| Fig. 1 Modelling of the His6-tagged peptide linkers into the crystal structure of cyt c6 from Synechococcus sp. PCC 7002. The cyt c6 structure (PDB: 4EIC)34 is the highest resolution type I c-type cytochrome (a close homologue of cyt c553 from C. merolae) crystal structure available to date and was used to model in the five distinct peptide linkers used in this study. The haem group of cyt c6 with its central Fe atom is displayed in red. | ||
The following AA variant characteristics could be predicted based on the physico-chemical properties of the respective AA: (i), 0AA: most restrictive conformation allowing for no rotation; (ii), 5AA: a minor degree of rotation and movement; (iii), 10AA: a small degree of rotation or movement; (iv), 12AA: more rotation or movement, and (v), 19AA: L-shaped hinged linker, with a semi-α-helical structure predicted to allow for conformational flexibility of the immobilised cyt c553 protein. The 5AA linker peptide is composed mainly of small, polar amino acids allowing for limited structural flexibility, particularly at the more charged C-terminal end of the sequence. The 10AA linker peptide comprises mainly polar Gly/Ser repeats and a charged Glu residue allowing for more structural flexibility of the immobilised cyt c553 variant compared to the 5AA variant. The 12AA linker corresponds to the original linker peptide encoded within the multiple cloning site of the petJ gene expression vector that has been genetically engineered between the cyt c553 holoprotein and a C-terminal His6-tag. It comprises polar Gly/Ser residues, non-polar Val/Ala/Leu residues, and negatively charged Asp/Glu residues. Finally, the 19AA cyt variant was designed to introduce a semi-helical ‘hinged’ linker peptide to the immobilised cyt c553 holoprotein. For this variant, we defined the Phi/Psi angles of each residue to be equal to −62/−41, which corresponds to the average values found experimentally for α-helices which are non-geometrically ideal.38
2.2 Mapping of the Gibbs free energy of the cyt c553 variants and estimation of their structural flexibility
To obtain an insight into the thermodynamic properties of the distinct variants of cyt c553 upon their immobilisation on the electrode surface, we calculated a theoretical probability of acceptable conformation that each peptide linker can adopt under random Brownian motion. Firstly, we applied a Monte Carlo simulation to calculate the acceptance rate for each cyt c553 variant, corresponding to the ratio of a total number of energetically permissive moves divided by the total number of all the attempted moves. During the simulation, Phi and Psi dihedral angles of the linker were altered randomly from a uniform distribution whilst including the Si plane in the simulations (see Materials and Methods). The acceptance rate was used in this context as a measure of the thermodynamic ‘freedom of motion’ of each cyt c553 variant upon its attachment to the p-doped Si surface via a His6-tag. It is worth noting that the acceptance rate largely depends on the degree of flexibility of the different AA side chains present within the linker peptides,38 implying that longer linker peptides would have larger probabilities of adopting different three-dimensional conformations, compared to the shorter counterparts.Fig. 2 shows that the highest acceptance rate (0.6851 ± 0.04), corresponding to the most restricted free motion of the haem with respect to the Si surface, was calculated for the shortest (5AA) cyt c553 variant, whereas the lowest acceptance rate value (0.3868 ± 0.02), corresponding to the least restricted free motion of the haem with respect to the Si surface, was attributed to the cyt c553 variant containing a semi-α-helix (19AA) linker peptide within its C-terminal domain. The acceptance rate calculation was not applicable to the 0AA linker variant as no peptide was present between the C-terminus of the cyt c553 protein and the affinity His6-tag.
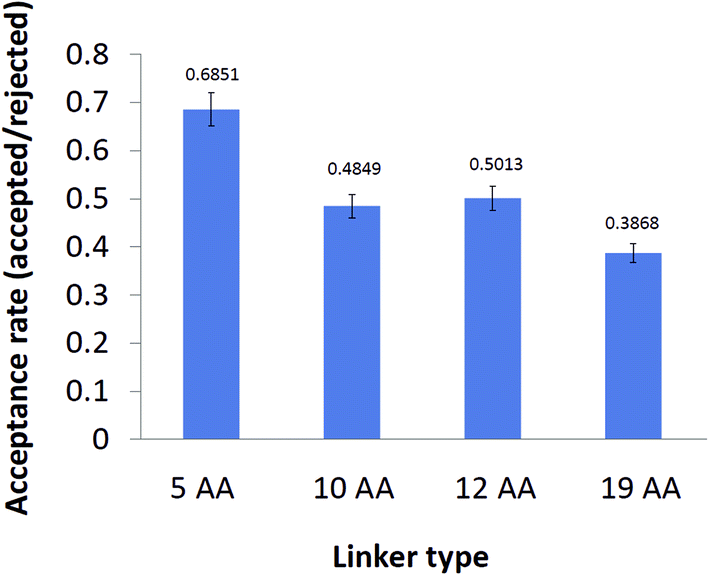 | ||
| Fig. 2 Acceptance rates of different cyt c553 variants after Monte Carlo simulation test. The acceptance rate was calculated as the ratio of the number of energetically permissive moves to the total number of moves using the standard Metropolis criterion.52 The acceptance rate calculation was excluded for the 0AA cyt variant as there is no peptide linker peptide between the two ab initio points of this simulation; namely the C-terminus of the holoprotein sequence and the first His residue of the His6-tag. Values of the acceptance rates are displayed above the error bars of each cyt c variant. | ||
We then calculated the 2D Gibbs free energy maps for each cyt c553 linker peptide variant as a function of the absolute distance of the haem group of cyt c (i.e., its central Fe atom) and the relative planar angle between the haem and a flat electrode surface (see Fig. 3). Fig. 4 shows that the presence of the longer peptide linkers within the cyt c structure evokes a significantly larger number of thermodynamically favourable conformations of the anchored cyt c (in terms of probability of energetically favourable alignment of the haem group to the electrode surface) after a given move, and consequently, a lower acceptance rate (see Fig. 2), as per Monte Carlo criteria. On the other hand, a short peptide linker is more likely to adopt a thermodynamically unfavourable conformation, since very few combinations of dihedral angles are possible within the whole linker length. Therefore, a lower probability of proximal alignment of the haem group to the Si surface exists (see Fig. 4).
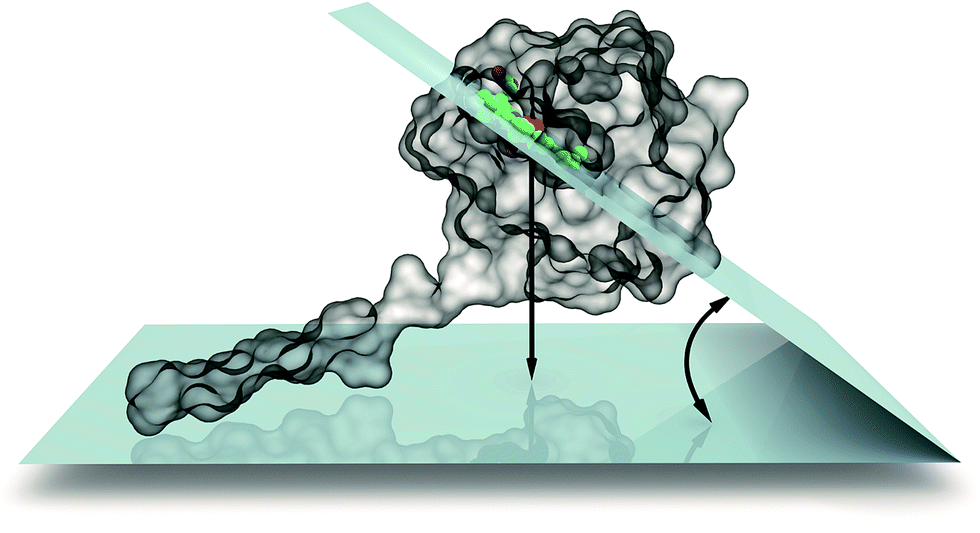 | ||
| Fig. 3 Definition of internal coordinate system used to describe haem–Si mutual orientation. We used two coordinates for this purpose: the dihedral angle between the haem group and silicon planes as well as the distance between the two planes. | ||
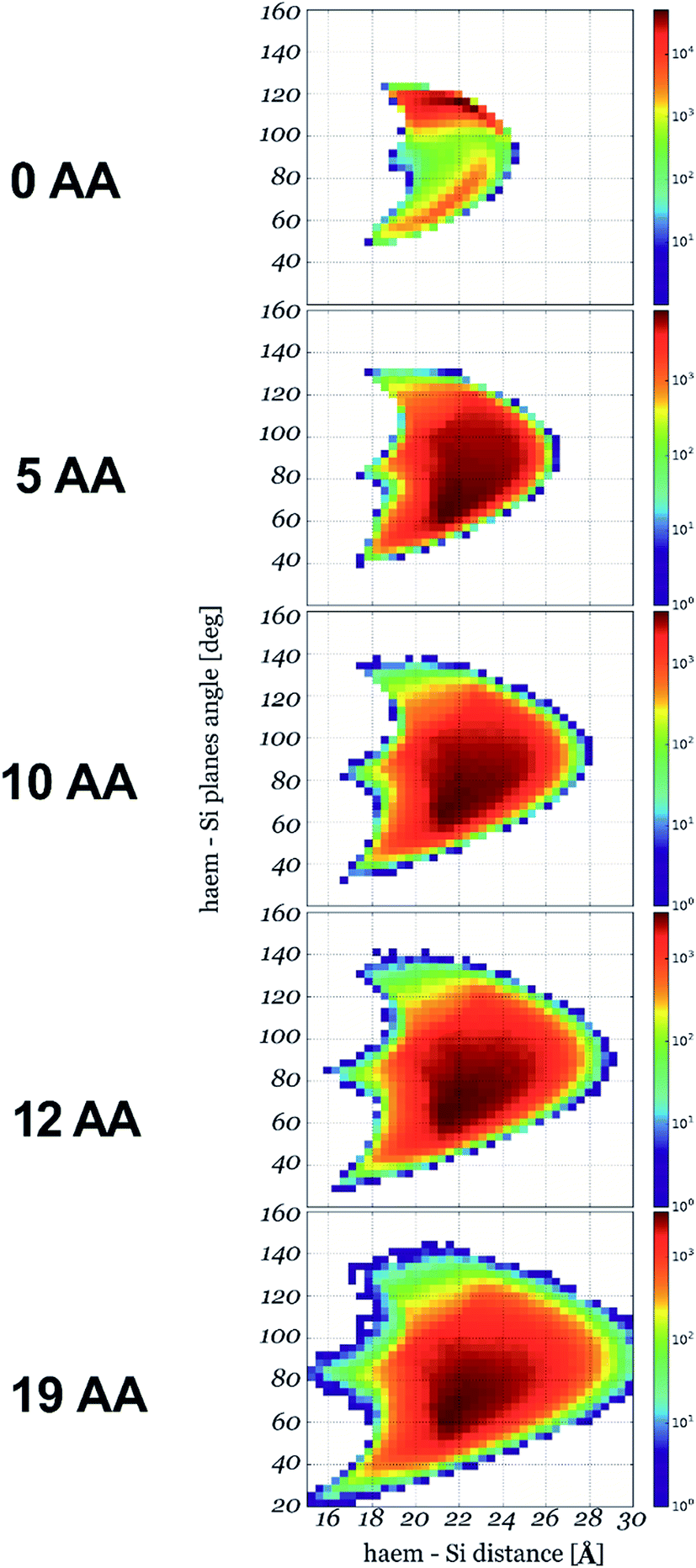 | ||
| Fig. 4 2D conformational flexibility maps for different cyt c553 variants. The maps show relative counts of energetically permissive conformations for a given orientation of haem plane described by its distance to the Si plane and the angle between the two planes: x-axis and y-axis, respectively (see Fig. 3 for the definition). The number of conformations in logarithmic scale is right proportional to the Gibbs free energy of a given state. | ||
To assess whether the calculated Gibbs free energy levels and conformational flexibility of the experimentally designed peptide linkers can be extended to much larger populations of the similar-length peptides, we conducted simulations on a high number of randomly de novo generated peptide linkers of 5AA, 7AA, 10AA, 12AA and 19AA in length (see ESI Fig. 1†). The aim of this in silico study was to investigate how far the cyt c553 protein can depart from the electrode for a given length of a peptide linker, independently of its actual AA sequence. To this end, 600 linker sequences were randomly generated for each given peptide length, with each AA residue equally probable. For this simulation, we employed the same set of randomly chosen movers and applied one of them at each iteration on a random residue of the linker. The de novo designed 7AA linker population provided a new class of peptide linkers that were omitted in the original experimental design.
The energy levels of experimentally and in silico designed linkers are similar, indicating energetically favourable properties of the experimental linkers compared to the whole random peptide dataset (see ESI Fig. 1†). Significantly, these simulations also revealed the existence of peptide linkers that exhibit more suitable levels of free energy, which potentially could be beneficial for the highly efficient DET between the semiconductor substrate and P700 reaction centre of PSI. This hypothesis will be verified in the future study.
2.3 Purification and biochemical characterisation of cytochrome c553 variants
We have cloned and heterologously overexpressed the transgenes encoding all the five variants of cyt c553 in E. coli using the tightly catabolically controlled co-expression system in conjunction with the constitutive expression of the haem maturation cassette to ensure synthesis of the fully redox-active cyt c holoproteins. All the five His6-tagged cyt c553 variants were purified to homogeneity using Ni–NTA immobilised metal affinity chromatography, as shown in Fig. 5. The predominant pools of each cyt c553 variant were obtained in the reduced state, with ∼20% of the total cyt yield obtained in the oxidised state (see Fig. 5B). The total yield of redox active cyt c553 variants was estimated in the range of 1–1.5 mg L−1. We confirmed the full redox activity of all the purified cyt c553 variants by measuring the redox difference absorption spectra, in which the disappearance of 553 nm and 521 nm haem-specific peaks was clearly observed upon chemical oxidation (see Fig. 5C and ESI Fig. 2†).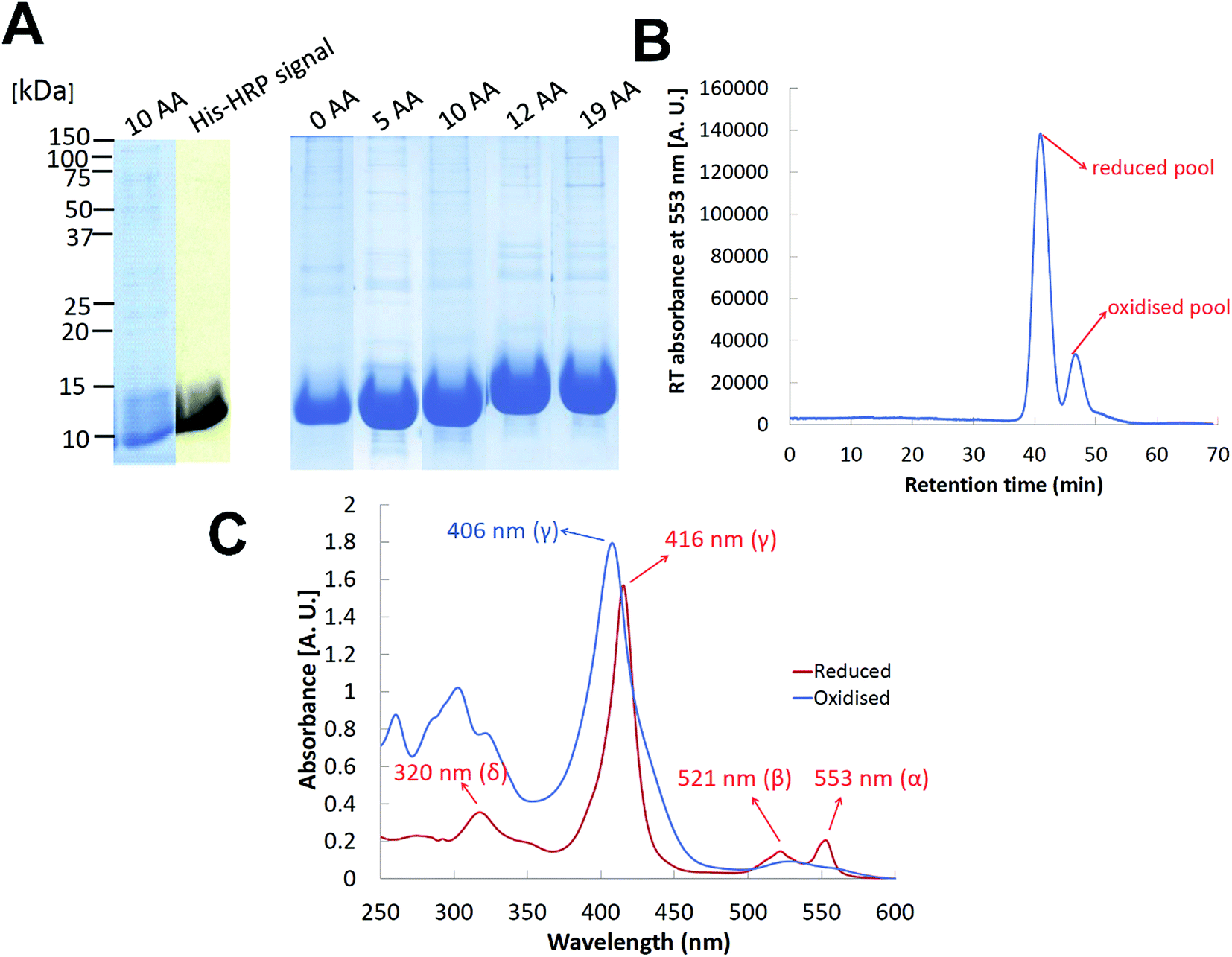 | ||
| Fig. 5 Purification and spectroscopic characterisation of cyt c553 variants. (A) SDS-PAGE protein profiles of overexpressed cyt c553 variants (30 μg of redox active cyt c553 per lane) in conjunction with a representative HisProbe detection of His6-tagged protein products (10AA cyt c553 variant). (B) A representative HPLC chromatogram of cyt c553 purification using Ni–NTA technology (C), a representative redox difference absorption spectrum of cyt c553 variant (10AA) showing maximally reduced and oxidised species. | ||
2.4 Construction and characterisation of the cyt c553/p-doped Si bioelectrodes
Five distinct bioelectrodes were constructed by immobilisation of the purified His6-tagged cyt c553 variants on a p-doped Si substrate. The electrode surface was chemically functionalised with a Ni2+–NTA self-assembled monolayer (SAM) to facilitate anchoring of cyt c553 on the electrode surface (see ESI Fig. 3†). We introduced the Ag and Au/Cr back and front contacts (200 and 50 nm, respectively) to perform J–V measurements, as schematically depicted in Fig. 6A. We have chosen p-doped Si as the electrode material as this semiconductor has been shown to serve as an excellent substrate for cyt c,39 due to the mid-point potential of this type of cyt (+210 mV) being close to the estimated Fermi level of the conduction band of p-doped Si (+500 mV vs. SHE),19,20 with the level of p-doping used in our study (see Fig. 6B for the energy band diagram). The thickness of a single layer of cyt c was estimated as 3 nm, taking account of the dimension of the close homologue of this protein, cyt c6, determined by X-ray crystallography (PDB: 4EIC).34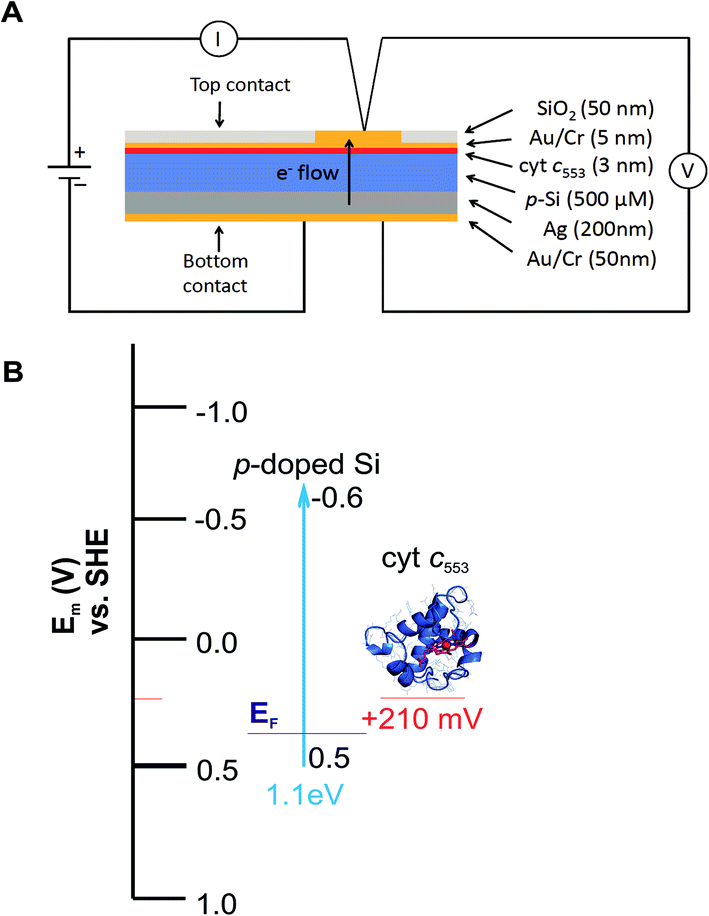 | ||
| Fig. 6 Construction of the cyt c553/Si bioelectrodes. (A) Diagrammatic representation of the all-solid state cyt c553/Si biodiodes. The thickness (nm or μm) of each layer, measured by sputtering detection, is indicated. The thickness of the Si wafer was 500 μM. (B) Energy band diagram of the cyt c553/Si bioelectrode. Indicated are valence (0.5 V vs. NHE) and conduction (−0.6 V vs. NHE) bands for the p-doped Si substrate, and cyt c553 redox mid potentials (Em) together with the estimated Fermi level (EF) for the used level of boron doping in Si. | ||
One of the major questions we set out to address in this study was to evaluate the effect of redox active cyt c553 layer on the inherent charge transfer within the p-doped Si substrate. To this end, we assessed the effect of the distinct cyt c553 variants on the electron diffusion and recombination processes at the surface of the biofunctionalised Si electrode by two independent approaches: J–V measurements in the dark (see Fig. 7) and electrochemically, by two-electrode all-solid-state linear sweep voltammetry (Fig. 8). All the biodevices displayed some diode-like behaviour. Notably, the dark saturation current (J0) of heavily p-doped Si, which is the recombination parameter within a given semiconductor in its solid-state configuration,40 was lower upon biofunctionalisation of the p-doped Si substrate with all the cyt c553 variants compared to the p-doped Si-SAM control (see Fig. 7, 8 and Table 2), albeit to a different degree depending on the peptide variant used. On one hand, the presence of short peptide linker cyt c553 variants on the Si surface resulted in the higher J0 values compared to variants with longer peptide linkers (see Table 2).
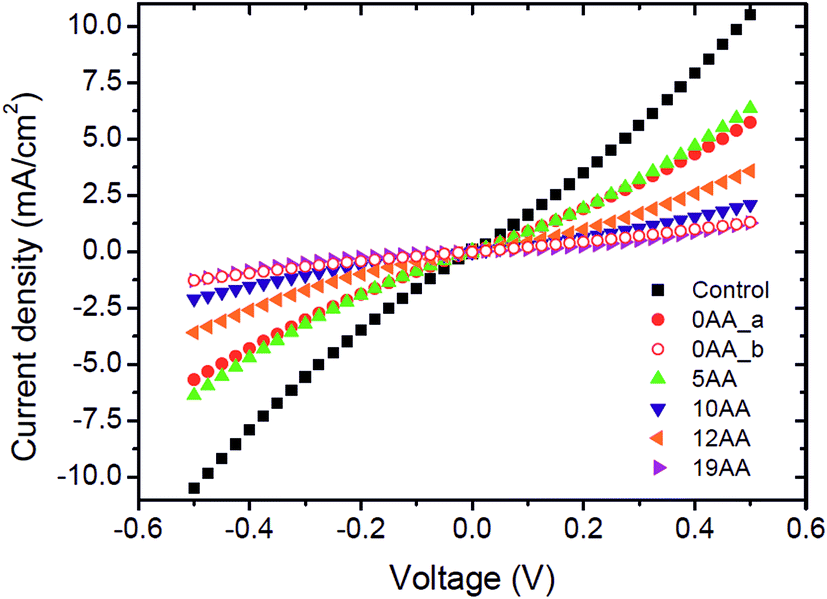 | ||
| Fig. 7 J–V characteristics of the cyt c553/Si bioelectrodes in the dark. The five types bioelectrodes constructed in this study display some biodiode characteristics, albeit at different kinetic rates of e− transfer. Short, rigid peptide cyt c variants evoke larger dark saturation currents (J0) compared to longer, more flexible peptide linker cyt c553 counterparts. The control is the p-doped Si with SAM only. The 0AA linker cyt c variant yielded two populations of bioelectrodes displaying either very low or high J0 values; we attribute this anomalous behaviour to the severe restriction of movement of this cyt c variant upon its anchoring onto the Si electrode surface. The J–V curves represent average values of 2–4 independent measurements. | ||
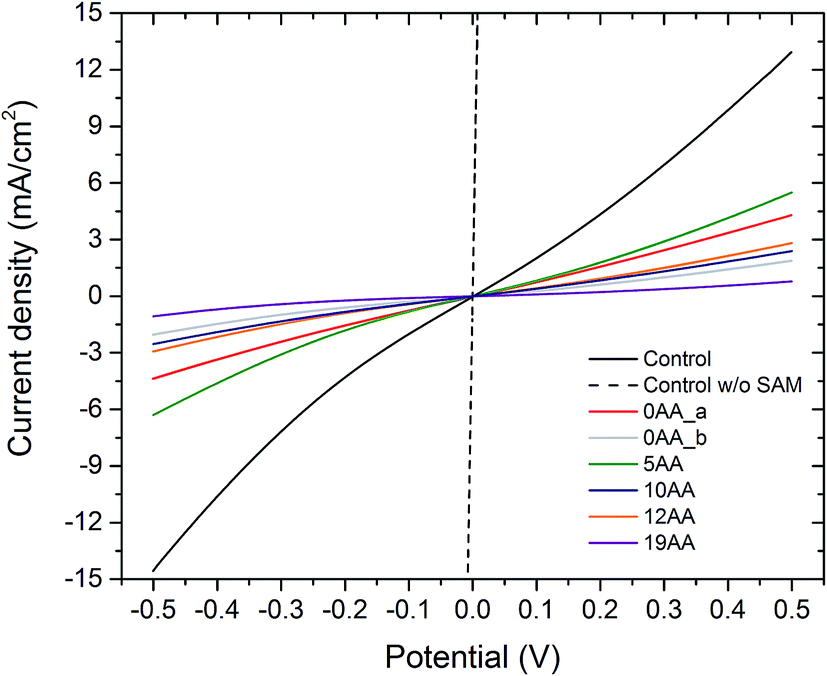 | ||
| Fig. 8 Linear sweep (two-electrode) voltammograms of the all-solid-state cyt c553/Si bioelectrodes. The cyclic voltammograms were obtained in a two-electrode measuring setup in the dark, at a scanning rate of 10 mV s−1. The potential was cycled from −500 mV to 500 mV. Note that the Ni–NTA SAM also contributes to the redox activity of the solid-state construct. The absolute value of the dark saturation current (mA) is inversely dependent on the length of the linker peptide, with the highest current for the 0AA linker variant and the lowest current for the 19AA cyt c553 variant electrode. | ||
| Bioelectrode type | J0e (mA cm−2) |
|---|---|
| a J0, dark saturation current (inherent recombination parameter) of each solid-state device. The J0 values were obtained from eqn (1) (assuming that at 300 K, kT/q = 25.8 mV) and were also estimated graphically at the y-intercept in ESI Fig. 4.b For a scheme depicting SAM synthesis refer to ESI Fig. 3.c Value was obtained for two 0AA linker variant electrodes which displayed similar J–V curves (see Fig. 7).d Value was obtained for two 0AA linker variant electrodes which displayed similar J–V curves (see Fig. 7).e Values were calculated for the six different bioelectrodes that were J–V characterised at 300 K. | |
| Control with SAMb | 1.2 × 10−1 |
| 0AAc | 4.5 × 10−2 |
| 0AAd | 2.8 × 10−3 |
| 5AA | 6.9 × 10−2 |
| 10AA | 2.3 × 10−2 |
| 12AA | 3.5 × 10−2 |
| 19AA | 7.5 × 10−3 |
Our Gibbs free energy modelling and calculation of movement acceptance rates (see Fig. 2 and 4) showed that the short linker peptide cyt c variants display limited structural flexibility upon their anchoring with the Si surface. It is therefore plausible that their redox active haem groups cannot properly align with the semiconductor surface to promote efficient reversible redox reactions, i.e., Fe3+/Fe2+ interconversion within the haem group. We identified two distinct populations of the electrodes biofunctionalised with the 0AA cyt c553 variant (see Table 2, Fig. 7 and 8), in line with identifying two distinct conformational populations of this variant (see Fig. 4). The existence of such distinct electrode populations for the 0AA variant can be due to the presence of favourable and unfavourable orientation of the haem group in terms of reversible redox reactions and minimisation of surface charge recombination.
On the other hand, the haem groups present in the longer linker peptide variants of cyt c553 are likely to be more capable of the favourable alignment with the semiconductor surface due to higher structural flexibility of the peptide linkers compared to their shorter counterparts (see Fig. 4). Consequently, of all the cyt c553/p-doped Si bioelectrodes analysed in this study, the device constructed with the 19AA peptide linker variant displays the lowest degree of p-doped Si surface charge recombination, as measured by the lowest dark J0 parameter (see Table 2), most likely due to the largest number of the thermodynamically favourable conformations of 19AA cyt c553 variant upon its immobilisation on the electrode surface, as well as the highest probability of the haem orientation favouring minimisation of the Si surface charge recombination.
The precise mechanism of electron transfer between the semiconductor electrode and the haem group of cyt c is still not fully understood. While some quantum mechanical calculations suggest that the haem group must be parallel to the electrode surface in order to promote the efficient DET,41,42 also described as “superexchange”,43 other studies suggest the possibility of electron transfer when the haem group is perpendicular to the surface of the electrode.44 Introduction of the 19AA and to a smaller degree, the 12AA linker peptides into the cyt c553 structure allows for a larger number of thermodynamically favourable conformations of immobilised cyt c with various types of the haem group alignment with respect to the plane of the electrode, as evidenced by both, our Gibbs free energy maps (see Fig. 4) and lower J0 values (see Table 2, Fig. 7 and 8). An alternative mechanistic explanation could be provided in the context of molecular orbital theory and electronic excitation of the haem group in the solid-state configuration. As short, rigid AA linkers yield more restricted conformations, there is less time for the haem group to relax electronically to the lower energy state (or energy well) characteristic for steady-state charge carrier hopping,45 as observed in solid-state electronic devices.46 This hypothesis would explain why the J0 parameter for the 5AA cyt c553 variant biodiode is much higher than those with longer (see Table 2), more flexible variants, whereby the haem group would be expected to relax longer, being in a further distance from the p-doped Si surface.
Although a number of studies have attempted to understand the intermolecular kinetics dominating the complex processes of electron transfer between the active site of the redox active protein and the semiconductor surface,47–50 to date only several reports have employed p-doped Si as a the semiconductor substrate together with cyt c protein either to investigate the level of conductance43,46,48,51 or the electrochemical properties of cyt c on p-doped Si.39 Nevertheless, none of these studies have addressed the electrochemical effects of cyt c on the inherent semiconductor solid-sate surface junction properties. Amdursky and colleagues46 showed that the specific electronic conductance to the haem group of bovine cyt c is directly related to the distance between the haem group and the electrode surface,43,46 in line with our observations of this study. However, the same authors focused mainly on defining the physico-chemical parameters governing the Fe–Si conductivity, rather than p-doped Si biopassivation by cyt c, nor did they display any charge transfer data from the p-doped Si control, in contrast to our study. Interestingly, although two of the aforementioned studies46,51 investigated the level of conductance rather than the overall effect of redox active proteins on the inherent charge transfer properties of the semiconductor substrate, their J–V curves show a striking similarity to our dark J–V data (Fig. 7 and 8), indicating that indeed, the cyt c553 may evoke similar redox processes at the surface of the semiconductor, promoting “superexchange” at the solid-state interface.43 Moreover, Amdursky and colleagues43 ascertain that distinctly high current density values (in the mA cm−2 range) may be attributed to properly linked cyt c holoprotein with a fully operational redox active haem in the vicinity of the p-Si surface, in line with our data. The same authors demonstrated the direct proportionality between haem group distance from the Si surface and an increased series resistance,46 similarly to the J–V data in the present study. Interestingly, such correlation between the level of passivation and the increased resistivity seems to be a general feature also in the case of chemically passivated semiconductor substrates, including Si.5
2.5 Construction and photovoltaic characterisation of the PSI/cyt c553/p-doped Si biophotoelectrodes
Our J–V and in silico data strongly suggest that the 19AA linker variant of cyt c553 is likely to be the best candidate for biopassivation of the p-doped Si substrate, as this variant significantly minimises the J0 parameter within the p-doped Si substrate, over and above the other cyt variants used in this study. We therefore hypothesised that this feature introduced with the specific immobilisation of the 19AA cyt c553 variant onto the p-doped Si substrate may lead to the overall improvement of the PSI-based biosolar cell's performance. Thus, the additional charge-separation event upon photoactivation of the PSI module on top of the cyt c553/p-doped Si electrode would promote the enhanced DET, resulting in the desired downhill energy transfer and enhanced power conversion efficiency.To verify this hypothesis, we constructed and characterised the photovoltaic properties of two types of nanodevices encompassing both 19AA cyt c553 variant and PSI biological redox active modules (see Fig. 9 for schematic depiction of the PSI-based biophotoelectrodes), and compared these characteristics with the control devices built exclusively with PSI photoactive layer on top of the p-doped Si substrate, and devoid of cyt c553. In order to assess the biophotovoltaic performance of the biophotoelectrodes, we determined the open-circuit potential (Voc) and the short-circuit current (Jsc) parameters by photochronoamperometry (see Fig. 10 and 11, respectively).
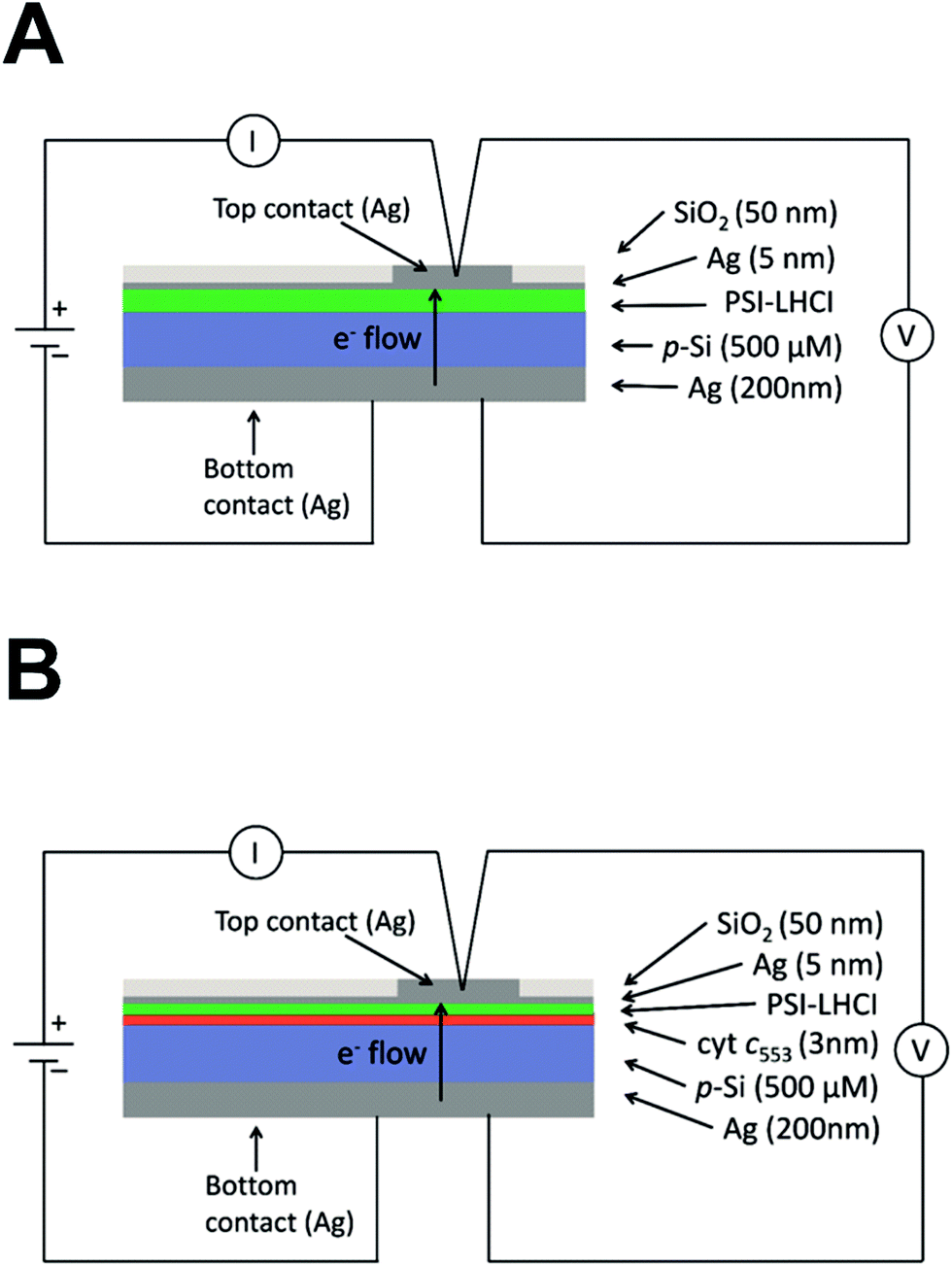 | ||
| Fig. 9 Diagrammatic representation of PSI-LHCI containing devices. (A) PSI-LHCI/p-doped Si nanodevice (third (III) generation biophotovoltaic device). The thickness of each layer is indicated on the right and was estimated by sputtering detection. The components are not to scale. (B) Diagrammatic representation of the PSI-LHCI/cyt c553/p-doped Si nanodevice (fourth (IV) generation biophotovoltaic device). The thickness of each layer indicated on the right was estimated by sputtering detection. The components are not to scale. All Si layers were functionalised and contained a Ni–NTA SAM, as depicted schematically in ESI Fig. 3.† | ||
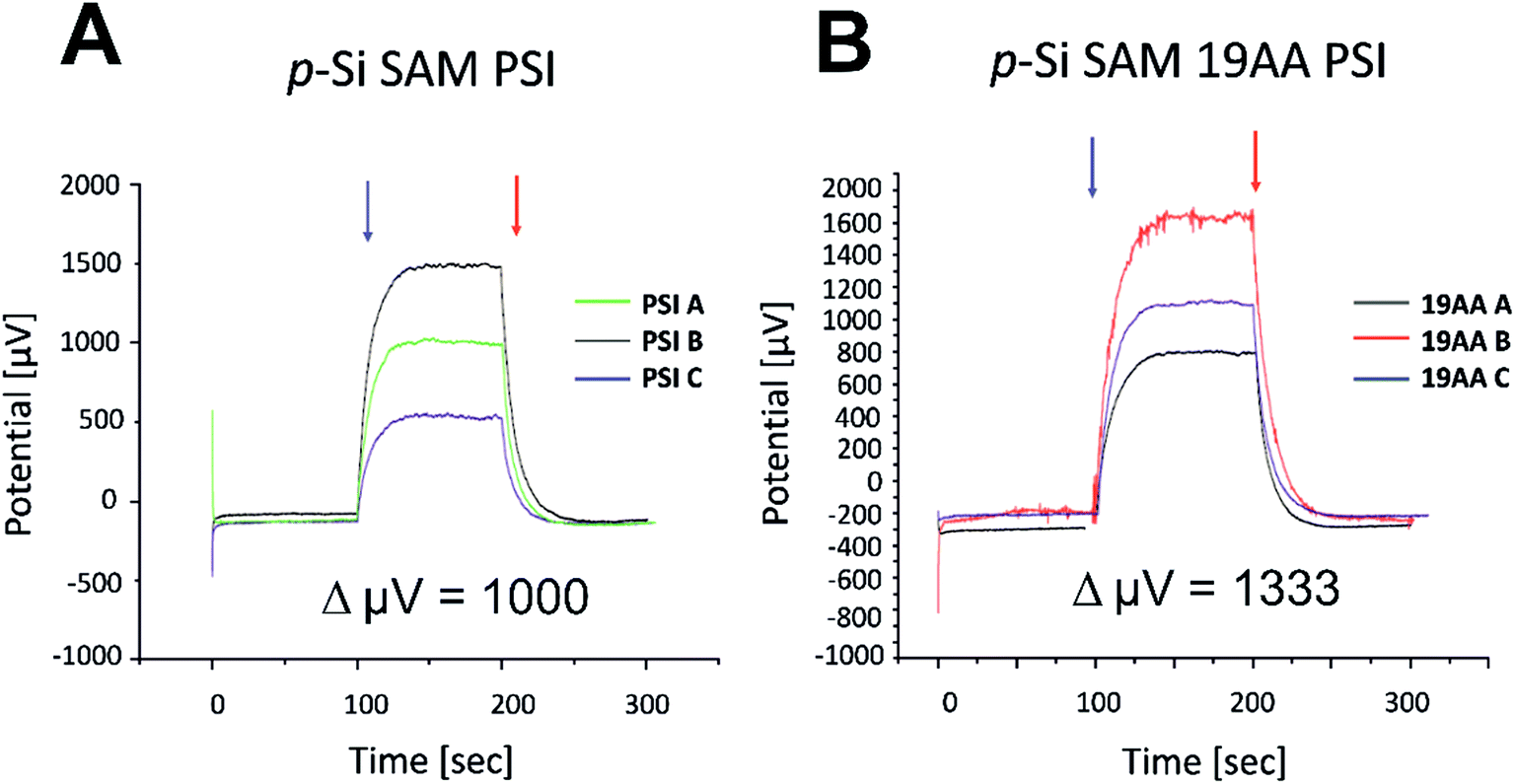 | ||
| Fig. 10 Photochronoamperometric determination of open-circuit potential (Voc) within PSI/p-doped Si (Panel A) and PSI/19AA cyt c553/p-doped Si biophotovoltaic devices (Panel B). After averaging the three independent devices, an increase in 333 μV is observed for the 19AA cyt variant-containing biophotoelectrode (Panel B) compared to the biophotoelectrode containing the PSI module alone (Panel A). The average Voc increment for the biophotoelectrode containing PSI-LHCI sample is 1000 μV (shown in Panel A), while the average Voc increment for the biophotoelectrode containing PSI-LHCI in addition to the 19AA linker variant is 1333 μV (shown in Panel B). Blue and red arrows symbolise light on and off, respectively. Potential (μV) values on the y-axis have been rescaled for the ease of ΔVoc estimation. | ||
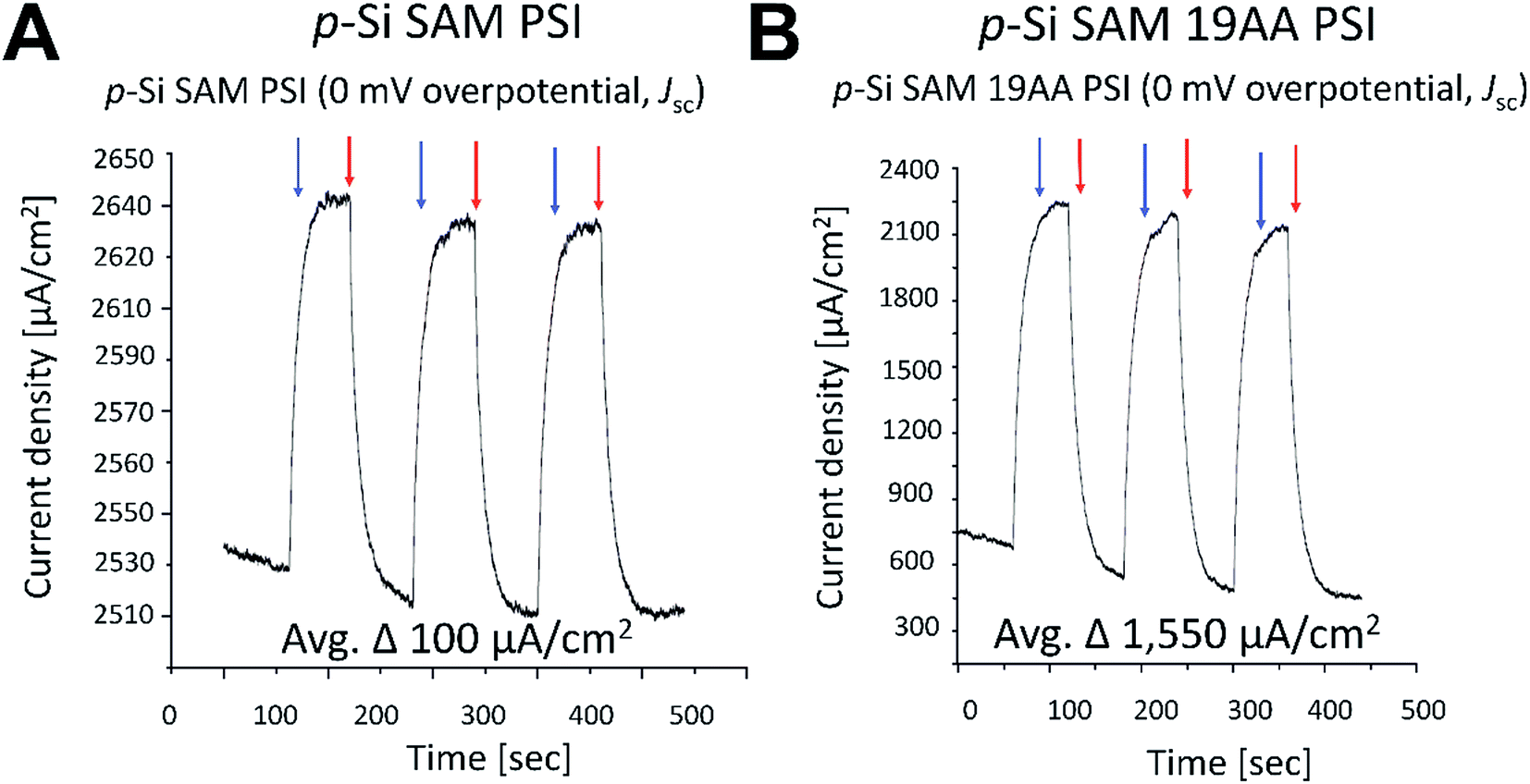 | ||
| Fig. 11 Photochronoamperometric determination of short circuit current (Jsc) within the best performing PSI/p-doped Si and PSI/19AA cyt c553/p-doped Si biophotovoltaic devices. No overpotential was applied to determine Jsc parameter. Blue and red arrows symbolise light on and off, respectively. Current density values on the y-axis have been rescaled for clarity. | ||
Incorporation of the 19AA linker variant between the p-Si substrate and the PSI layer resulted in a 333 μV increase of the Voc parameter compared to the device which contained PSI alone (see Fig. 10). In parallel, we observed a 15.5-fold enhancement of the Jsc parameter at the open circuit potential in conjunction with a 20-fold increase of external quantum efficiency when the 19AA cyt c553 variant was introduced between the p-doped Si substrate and the PSI photoactive layer (see Fig. 11 and Table 3). These results clearly confirm our hypothesis of cyt c553-mediated enhancement of the PSI-biophotoelectrode photovoltaic performance as a function of haem distance and orientation with respect to the electrode surface promoting minimisation of semiconductor surface charge recombination. Such an increment of Voc and Jsc parameters was observed only upon illumination, and both parameters remained unchanged in the dark. Thus, we confirmed that PSI operated as the photoactive module when built into the all-solid-state biophotoelectrodes.
| Device configuration | Jsc [μA cm−2] | Voc [V] | FF | η [%] | References |
|---|---|---|---|---|---|
| a Jsc; short circuit current, the current density when the voltage across the device is 0 V (see Fig. 11). Voc; open circuit voltage or open circuit potential (the photovoltage generated when no bias is present across the circuit) (see Fig. 10). FF; fill factor. η [%]; external power conversion efficiency of the biophotovoltaic device. The performance of the listed solid state biosolar cells was evaluated at standard white light illumination of 100 mW cm−2 (AM 1.5), as described in Materials and Methods.b Values were estimated for selected electrodes with the best performance. Significant variability was observed between devices due to fragile construction (brittle nature of the p-Si).c Calculation of fill factor and efficiency was performed with the open circuit voltage for the estimated values presented in the table assuming an ideality factor of 1 (n = 1) and a temperature of 300 K, as specified in Materials and Methods. | |||||
| p-Si/19AA/PSI | 1,550b | 0.001333b | 0.3c | 0.0006c | This work |
| p-Si/PSI | 100b | 0.001b | 0.3c | 0.00003c | This work |
| p-Si/PSI/ZnO/ITO | 127 | 0.214 | 0.28 | 0.0077 | 19 |
| PAni/PSI/TiO2/SnO2 | 72 | 0.299 | 0.42 | 0.0091 | 15 |
3. Concluding remarks
In summary, we provide the first evidence that incorporation of a redox-active robust biological component, the extremophilic cyt c553 protein, allows for passivation of the p-doped Si substrate by minimisation of the inherent dark saturation current (J0). By introducing the specific linker peptides within the cyt c553 structure, we achieved minimisation of surface charge recombination within the p-doped Si substrate by optimisation of the alignment of the haem group with respect to the semiconductor surface. We show that the cyt c553 variant characterised by the largest structural flexibility upon its specific immobilisation on the semiconductor surface allowed for a significantly improved biophotovoltaic performance of the all-solid-state PSI-based Si biophotoelectrode. Thus, our molecular approach has paved the way for developing of an alternative, cheap and facile route for significant reduction of the inherent minority charge recombination on the semiconductor surface. As such, it carries the great potential for future biotechnological applications, such as the development of viable ‘green’ solar-to-fuel devices.4. Materials and methods
4.1 Monte Carlo simulations
A Monte Carlo object was used to assess the probability of free motion of the various peptide linkers attached to cyt c. This object accepts or rejects a modified protein using standard Metropolis criterion. An acceptance rate is defined by PyRosetta® software and it corresponds to the number of accepted moves divided by the total number of moves attempted.52 The Monte Carlo test used a score function with the default Rosetta full-atom energy terms and weights (see http://www.pyrosetta.org). A set of the following movers, provided bys the standard PyRosetta® distribution, was employed for the simulation: (1) SmallMover: modifies the Phi or Psi angle of a randomly chosen residue, by a small random angle. The angles belonged to the allowable region of Ramachandran plot (Metropolis acceptance criterion based on rama score);52 (2) ShearMover: modifies the Phi and Psi angle of a randomly chosen residue, by a small random angle. It was also used under the Metropolis acceptance criterion;52 (3) SequenceMover: applies a sequence of several movers in succession. In our case, this mover contained the two previous movers. (4) RandomMover: applies a randomly chosen mover within a given list. For this simulation, we applied the SmallMover, the ShearMover, or both of them (SequenceMover); (5) RepeatMover: applies a mover n times, n being given by the user. The RandomMover simulation was repeated several times on each cyt c variant. The same set of randomly chosen movers were applied, one per each iteration, on randomly generated AA residues of the fixed-length linkers of 5AA, 7AA, 10AA, 12AA and 19AA in length. The standard full-atom scoring function was used without hydrogen bonds, solvation and electrostatics energy components. The process was repeated 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times and 600 linker sequences were randomly generated for every given AA length. The Si plane was introduced to the simulations as an infinite plane interacting only with hard repulsion i.e. infinite excluded volume energy for any atom crossing the plane. Such clashing conformations were rejected. For each of the linkers, the energy levels lower than 0.5% of the maximum energy value were extracted and, within this range, the maximum and minimum lengths in Å, as well as the maximum and minimum energies were stored and plotted in the graphs in ESI Fig. 1.† The same simulation was performed on the experimentally designed linkers.
000 times and 600 linker sequences were randomly generated for every given AA length. The Si plane was introduced to the simulations as an infinite plane interacting only with hard repulsion i.e. infinite excluded volume energy for any atom crossing the plane. Such clashing conformations were rejected. For each of the linkers, the energy levels lower than 0.5% of the maximum energy value were extracted and, within this range, the maximum and minimum lengths in Å, as well as the maximum and minimum energies were stored and plotted in the graphs in ESI Fig. 1.† The same simulation was performed on the experimentally designed linkers.
4.2 Bioinformatics calculations of the Gibbs free energy and modelling of solid-state cytochrome c-based bioelectrode
The Monte Carlo simulation protocol described above was used to characterise Gibbs free energy as a function of haem orientation relative to the electrode, as described by two internal coordinates. The distance between Fe atom and the flat electrode surface and the angle between haem plane and flat electrode surface (see Fig. 3) were used for this purpose. All the simulations were performed using the crystal structure of cyt c6 from Synechococcus sp. PCC 7002 (PDB: 4EIC)34 with an appropriate linker added. The geometry was calculated for each modelled conformation with the BioShell package.534.3 Cloning and expression of petJ gene in E. coli
Genomic DNA from C. merolae was used as a template for polymerase chain reaction to amplify the petJ gene (annotated CMV193C; http://merolae.biol.s.u-tokyo.ac.jp/db/) encoding the cyt c553 protein. The original verified sequence of petJ was cloned into pET28b-(+) vector (Invitrogen®) together with four distinct DNA sequences corresponding to the four distinct AA linkers (see Table 1 for AA sequences) between the petJ sequence and the C-terminal His6-tag. The entire constructs were excised with NcoI and PstI from pET28b-(+) vectors and subcloned into a pBAD/HisA expression vector (Invitrogen®) using the same restriction enzymes, then transformed into the E. coli strain DH5α. The correctness of the petJ::pBAD/HisA construct was confirmed by DNA sequencing. A specialised constitutively expressed vector comprising a multiple gene cassette encoding the cyt c type haem group maturation enzymes54 (a gift from Prof. A. Szczepaniak, Wrocław Univ., Poland), was co-transformed with the petJ::pBAD/HisA recombinant vector into the expression host strain of E. coli, TOP10. Expression cultures inoculated from the single recombinant colonies were supplemented with 30 μg mL−1 chloramphenicol, 100 μg mL−1 ampicillin, and grown overnight. The overnight saturated pre-cultures (1:100 volume ratio) were then grown to OD600 0.4. The cultures were then supplemented with 100 mg L−1 Fe2SO4 and 17 mg L−1 δ-aminolevulinic acid, and petJ gene expression was induced with 0.2% arabinose and allowed to proceed for 20 hours at 37 °C, with shaking at 160 rpm. The cells were harvested at 4000 × g for 15 min. at 15 °C and then chilled on ice for 20 min.
4.4 Cytochrome c553 purification
Recombinant E. coli cells expressing the petJ gene were resuspended in a buffer containing 20 mM imidazole, 100 mM standard phosphate buffer (PB) (pH 7.5), 25% glycerol (w/v), 30 μM PMSF and 20 μg of DNase I per 50 mL of suspension or a protease inhibitor cocktail (Roche®, 1 tablet per 50 mL of resuspension buffer). Cells were ruptured by vigorous agitation with 0.1 mm glass beads in a Bead Beater (BioSpec®) using 18 cycles of 45 s and interim 4 min. cooling-off periods. Cell lysate was separated from the beads by vacuum filtration. Cell debris was removed by ultracentrifugation for 45 min at 100000 × g at 4 °C. After centrifugation samples were filtered using 0.2 μm disposable filtering devices (Millipore®) and applied onto an HPLC preparative Ni–NTA GE Healthcare HisTrapTM® HPLC/FPLC column (1 mL bed volume) at a flow rate of 0.5 mL min−1 to ensure efficient binding of the His6-tagged cyt c553 variants. The column was then washed for 30 min at a flow rate of 0.5 mL min−1 with 50 mM imidazole and then the bound proteins were eluted with a linear gradient of 50–420 mM imidazole at a flow rate of 0.5 mL min−1. The column was further washed with 500 mM imidazole for 30 min to remove any residual proteins. Elution of cyt c553-containing fractions was monitored at 416 nm, 521 nm and 553 nm, corresponding to the absorption peaks of reduced cyt c553. Cyt-containing fractions were pooled and concentrated using VIVASPIN-2 devices (3000 MWCO, Sartorius-Intec®, Poland), then re-suspended in a buffer containing 100 mM PB (phosphate buffer, 100 mM Na2HPO4, 18 mM KH2PO4, pH 7.5) and 25% glycerol (w/v). The total protein concentration was determined using the Bradford assay55 using bovine serum albumin as a standard. Purity of the cyt c553 protein was verified on 16% Tris–Tricine SDS-PAGE gels using a standard protocol.56 The cyt c553 concentration was determined by redox difference absorption spectroscopy using extinction coefficient of 24.1 mM−1,57 as described below (refer to ESI Fig. 2† for exemplary redox difference spectra).
4.5 SDS-PAGE gel electrophoresis and His6-tag detection
SDS-PAGE was performed using a Tris–Tricine system.56 Protein bands of IMAC pooled fractions (normally 3–5 μg protein per lane or equal volume, see below) were resolved on a 16% polyacrylamide gel at a constant voltage of 30 V for 30 min. After 30 min., the voltage was increased to 200 V for 45–60 min. Proteins were visualised with Coomassie Brilliant Blue R-250 staining or electro-transferred onto a polyvinylidene difluoride membrane (Millipore®) for detection of His6-tagged proteins. Blots were probed with the HisProbe-HRP® conjugate (Thermo Fisher Scientific®, USA) and visualised by chemiluminescence, according to the manufacturer's protocol. Prior to SDS-PAGE, samples were resuspended in 5 × SDS-PAGE sample buffer (0.225 M Tris–HCl, pH 6.7, 50% glycerol, 5% SDS, 0.05% bromophenol blue, and 0.25 M DTT), vortexed vigorously and boiled at 95 °C for 8 min with continuous shaking. Samples were centrifuged at 14100 × g for 5 min and the supernatant was carefully loaded onto the SDS-PAGE gels. The volume was adjusted to 30 μg total protein per lane.
4.6 Redox difference absorption spectroscopy
UV-VIS spectra of all cyt c553 variants were recorded using a Shimadzu UV 1800 spectrophotometer at RT. The cyt c553 protein was titrated to the appropriate concentration (5–20 μg mL−1 cyt c553 in 100 mM standard phosphate buffer, pH 7.5) and the first RT absorbance spectrum was measured. To ensure the complete reduction of cyt, a few grains of sodium dithionite were added to the cuvette. After the grains were fully dissolved, another spectrum was measured, and if necessary more grains of sodium dithionite were added (grain by grain) until the fully reduced state of cyt was achieved. Subsequently, a grain of FeCN was added to oxidise the cyt c553 protein. A spectrum was measured, in which flattening of the 521 nm and 553 nm peaks was observed. A grain of FeCN was added repeatedly until cyt was fully oxidised displaying a shift in the blue region from 416 nm to 406 nm. The cyt c protein was subsequently re-reduced with addition of approximately two times the amount of sodium dithionite upon which the reappearance of the 416, 521, 553 nm peaks occurred. Delta A553 (difference between maximally reduced and maximally oxidised species) was used to calculate the exact concentration of redox active cyt c553 using the molar extinction coefficient of 24.1 mM−1.57 For all the cyt c553 preparations, the absolute amounts of protein determined via the Bradford assay and by redox difference absorption spectroscopy were comparable. Refer to ESI Fig. 2† for exemplary redox difference spectra of the 4 cyt c553 variants used in this study excluding the 10AA linker variant spectra which is displayed in Fig. 5C.4.7 Construction of the cyt c553 bioelectrodes and dark current characterisation
Heavily p-doped Si wafers were obtained from Department of Silicon Technology in the Institute of Electronic Materials Technology (ITME, Warsaw, Poland). Silicon wafers had a thickness of 500 ± 25 μm and a resistivity of 0.001–0.005 Ω cm equivalent to a boron doping density of 2.13 × 1019–1.3 × 1020 cm−3. The wafers were pre-treated by submerging in SWIPE® for 24 hours. After removal of the detergent, the wafers were treated with hot, concentrated sulfuric acid (96%), a solution of ammonia in hydrogen peroxide and water (H2O:H2O2:NH4OH/1:1:0.05), and finally in diluted hydrofluoric acid in water (HF:H2O/1:10). Subsequently a 200 nm Ag layer was deposited through chemical evaporation on the back side of the wafers. Rapid Thermal Processing (RTP) was performed to improve metal/semiconductor contact. Subsequently, the wafers were again treated with diluted hydrofluoric acid (as above) and then functionalised with Ni–NTA according to a protocol from58 (See ESI Fig. 3†). After functionalisation, cyt c553 variants were deposited using vacuum-assisted spin coating to generate a thin layer of cyt chelated to the Ni–NTA moiety. For this, a solution of 500 μL of 125 μM cyt c553 variant was employed for deposition via vacuum-assisted spin coating. After deposition cyt c553 was initially dried with a nitrogen stream and then left for further drying in nitrogen cabinet for 24 h. A transparent 5 nm layer of an Au–Cr alloy was then deposited to ensure the proper metal/cyt c553 electrical contact. Several contact points were grown by chemical evaporation of this Au–Cr alloy employing laser-cut masks and the rest of the surface was sealed by a transparent 50 nm SiO2 layer. Four to six biodiodes were cut into the precise square shapes and measured to determine exact area (3.24 cm2) of the biodiodes for each single variant electrode. An extra 50 nm layer of the Au–Cr alloy was chemically deposited on top of the bottom Ag contact to avoid any unwanted electrochemical Ag oxidation/reaction upon further manipulation for all-solid-state (mediatorless) electrochemical experiments.
4.8 J–V measurements of cyt c553/p-doped Si bioelectrodes
All the J–V data was acquired using a custom-made setup comprising a PC with an interface card (Texas Instruments®), an AGILENT 34401A digital multimeter, the AGILENT 6634B system DC power supply and a home-made interface card (Department of Optoelectronics, ITME). Data was processed using ITME-customised LAB VIEW 8.6 software. Data was averaged using four independently prepared bioelectrodes using the selected area of 3.24 cm2 which was employed as a standard area for all the electrodes constructed in this study. The entire protocol was repeated twice to ensure reproducibility of the J–V results. The J–V values were averaged for three independent bioelectrodes using the data from three most similar J–V curves from a total set of four measurements. All the J0 values were calculated from eqn (1) below, and were also determined from the J–V curves presented on a semi-logarithmic scale, where the J0 value corresponds to the y-intercept (see ESI Fig. 4†).40| J = J0(e[qV/nkT]−1) | (1) |
4.9 All-solid-state two-electrode cyclic voltammetry of cyt c553/p-doped Si bioelectrodes
The CVs were obtained in a two-electrode configuration at a scan rate of 10 mV s−1 using an SP-300 Bio-Logic Potentiostat. The potential was cycled from −500 mV to 500 mV. The bioelectrodes were connected by gold-plated Kelvin clamp. The top contact of the bioelectrode was connected as a working electrode and the counter and reference electrodes were connected to the bottom contact.4.10 Construction of PSI/p-doped Si and PSI/cyt c553/p-doped Si devices
The p-doped Si wafers were prepared essentially as described in Section 4.7, with or without chemical modification, where appropriate. After biofunctionalisation with cyt c553, the Si wafers were dried in a semi-anaerobic nitrogen box for 24 h. A 500 μL suspension of the C. merolae PSI-LHCI complex at 1 mg mL−1 Chla was overlaid on the pre-treated Si wafers and the protocol described in Section 4.7 was followed for the vacuum-assisted spin coating of the protein sample, with the exception that instead of a 5 nm of Au layer, a 5 nm of Ag was employed. For a flow-chart depicting preparation of all-solid-state PSI/cyt c553/p-doped Si biophotovoltaic devices refer to ESI Fig. 5.†4.11 Photochronoamperometry
All photoelectrochemical measurements on PSI-based devices were performed with a CH Instruments CHI660a electrochemical workstation (CH instruments, Inc.®). The working electrode clip was attached to the Ag bottom contact layer, and the counter and reference electrodes were clipped on the top Ag layer contact as depicted in Fig. 9. The light source employed was an Industrial Solar Cell Testing Light Source (150/300 W) (Solar Light®) and the intensity output was 100 mW cm−2 for all the photocurrent measurements, reassessed independently with a quantum metre (HD 2302.01) (DeltaOhm®) to a value of 2200 μmol photons per m2 per s. Samples were positioned in the same distance from the light source for all the measurements, with frequent verification of constant light intensity. Photochronoamperometric data was collected with no bias between the reference and working electrodes for measurement of photo-driven open-circuit voltage changes. Short circuit current was measured when the voltage across the p–n-junction was 0 V.The following equations were employed for determining fill factor (FF) and external quantum efficiency (η) of the PSI-containing biophotoelectrodes (see Table 3):
| FF = (Voc − ln(Voc + 0.72))/Voc + 1 | (2) |
| η = (Voc × Jsc × FF)/Pin × 100% | (3) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JDJO and JK are grateful for funding from the National Science Centre of Poland (grants no. UMO-2013/11/N/NZ1/02390 and UMO-2014/15/B/NZ1/00975) and from the Polish National Centre for Research and Development (grant no. DZP/POLTUR-1/50/2016, agreement no. 5/POLTUR-1/2016). We gratefully acknowledge invaluable help from Dr Eng. Grzegorz Bubak (Solar Fuels Laboratory, CeNT UW, Poland) with J–V measurements, laboratory and software training of JDJO and useful discussion on the manuscript. We also thank A. Dawid (Laboratory of Theory of Biopolymers, Faculty of Chemistry, University of Warsaw, Poland) for the preparation of Fig. 3. We thank Prof. Andrzej Szczepaniak (Wrocław University, Poland) for the pEC86 vector comprising a gene expression cassette for cyt c type haem maturation. We are grateful to Prof. Jan Augustyński and Olga Krysiak (CeNT UW, Poland) for access to their CV equipment. T. Krupnik (Faculty of Biology, Univ. of Warsaw) is acknowledged for his assistance with preliminary cloning and expression studies of cyt c553. Prof. Dariusz Bartosik (Faculty of Biology, Univ. of Warsaw) is gratefully acknowledged for providing the pBAD/HisA vector and host E. coli strain TOP10, and for his advice during the early cyt c553 expression studies.References
- N. Armaroli and V. Balzani, Chem.–Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- G. Kavlak, J. McNerney, R. L. Jaffe and J. E. Trancik, Energy Environ. Sci., 2015, 8, 1651–1659 Search PubMed.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2015, 23, 1–9 Search PubMed.
- K. Yoshikawa, H. Kawasaki, W. Yoshida, T. Irie, K. Konishi, K. Nakano, T. Uto, D. Adachi, M. Kanematsu, H. Uzu and K. Yamamoto, Nat. Energy, 2017, 2, 17032 CrossRef CAS.
- J. Schmidt, F. Werner, B. Veith, D. Zielke, S. Steingrube, P. P. Altermatt, S. Gatz, T. Dullweber and R. Brendel, Energy Procedia, 2012, 15, 30–39 CrossRef.
- J. Tian, R. Gao, Q. Zhang, S. Zhang, Y. Li, J. Lan, X. Qu and G. Cao, J. Phys. Chem. C, 2012, 116, 18655–18662 CAS.
- J. Tian, Q. Zhang, E. Uchaker, R. Gao, X. Qu, S. Zhang and G. Cao, Energy Environ. Sci., 2013, 6, 3542 CAS.
- H. Savin, P. Repo, G. von Gastrow, P. Ortega, E. Calle, M. Garín and R. Alcubilla, Nat. Nanotechnol., 2015, 10, 1–6 CrossRef PubMed.
- S. W. Hogewoning, E. Wientjes, P. Douwstra, G. Trouwborst, W. van Ieperen, R. Croce and J. Harbinson, Plant Cell, 2012, 24, 1921–1935 CrossRef CAS PubMed.
- T. Kothe, S. Poeller, F. Zhao, P. Fortgang, M. Roegner, W. Schuhmann and N. Plumere, Chem.–Eur. J., 2014, 20, 11029–11034 CrossRef CAS PubMed.
- M. Kamran, J. D. Delgado, V. Friebe, T. J. Aartsma and R. N. Frese, Biomacromolecules, 2014, 15, 2833–2838 CrossRef CAS PubMed.
- J. Ihssen, A. Braun, G. Faccio, K. Gajda-Schrantz and L. Thony-Meyer, Curr. Protein Pept. Sci., 2014, 15, 374–384 CrossRef CAS PubMed.
- O. Yehezkeli, R. Tel-Vered, D. Michaeli, I. Willner and R. Nechushtai, Photosynth. Res., 2014, 120, 71–85 CrossRef CAS PubMed.
- A. Mershin, K. Matsumoto, L. Kaiser, D. Yu, M. Vaughn, M. K. Nazeeruddin, B. D. Bruce, M. Graetzel and S. Zhang, Sci. Rep., 2012, 2, 234 CrossRef PubMed.
- E. A. Gizzie, J. Scott Niezgoda, M. T. Robinson, A. G. Harris, G. Kane Jennings, S. J. Rosenthal and D. E. Cliffel, Energy Environ. Sci., 2015, 8, 3572–3576 CAS.
- P. I. Gordiichuk, G. J. A. H. Wetzelaer, D. Rimmerman, A. Gruszka, J. W. De Vries, M. Saller, D. A. Gautier, S. Catarci, D. Pesce, S. Richter, P. W. M. Blom and A. Herrmann, Adv. Mater., 2014, 26, 4863–4869 CrossRef CAS PubMed.
- K. Ocakoglu, T. Krupnik, B. Van Den Bosch, E. Harputlu, M. P. Gullo, J. D. J. Olmos, S. Yildirimcan, R. K. Gupta, F. Yakuphanoglu, A. Barbieri, J. N. H. Reek and J. Kargul, Adv. Funct. Mater., 2014, 24, 7467–7477 CrossRef CAS.
- V. B. Shah, W. R. Henson, T. S. Chadha, G. Lakin, H. Liu, R. E. Blankenship and P. Biswas, Langmuir, 2015, 31, 1675–1682 CrossRef CAS PubMed.
- J. C. Beam, G. LeBlanc, E. A. Gizzie, B. L. Ivanov, D. R. Needell, M. J. Shearer, G. K. Jennings, C. M. Lukehart and D. E. Cliffel, Langmuir, 2015, 31, 10002–10007 CrossRef CAS PubMed.
- G. Leblanc, G. Chen, E. A. Gizzie, G. K. Jennings and D. E. Cliffel, Adv. Mater., 2012, 24, 5959–5962 CrossRef CAS PubMed.
- J. Kargul, J. D. Janna Olmos and T. Krupnik, J. Plant Physiol., 2012, 169, 1639–1653 CrossRef CAS PubMed.
- H. Krassen, A. Schwarze, B. Friedrich, K. Ataka, O. Lenz and J. Heberle, ACS Nano, 2009, 3, 4055–4061 CrossRef CAS PubMed.
- J. D. Janna Olmos and J. Kargul, Acta Soc. Bot. Pol., 2014, 83, 423–440 CrossRef.
- J. D. Janna Olmos and J. Kargul, Int. J. Biochem. Cell Biol., 2015, 66, 37–44 CrossRef CAS PubMed.
- P. N. Ciesielski, F. M. Hijazi, A. M. Scott, C. J. Faulkner, L. Beard, K. Emmett, S. J. Rosenthal, D. Cliffel and G. Kane Jennings, Bioresour. Technol., 2010, 101, 3047–3053 CrossRef CAS PubMed.
- P. N. Ciesielski, D. E. Cliffel and G. K. Jennings, J. Phys. Chem. A, 2011, 115, 3326–3334 CrossRef CAS PubMed.
- F. Zhao, F. Conzuelo, V. Hartmann, H. Li, M. M. Nowaczyk, N. Plumeré, M. Rögner and W. Schuhmann, J. Phys. Chem. B, 2015, 119, 13726–13731 CrossRef CAS PubMed.
- A. Efrati, C.-H. Lu, D. Michaeli, R. Nechushtai, S. Alsaoub, W. Schuhmann and I. Willner, Nat. Energy, 2016, 1, 15021 CrossRef CAS.
- M. Hippler, J. Reichert, M. Sutter, E. Zak, L. Altschmied, U. Schröer, R. G. Herrmann and W. Haehnel, EMBO J., 1996, 15, 6374–6384 CAS.
- M. Szalkowski, J. D. Janna Olmos, D. Buczyńska, S. Maćkowski, D. Kowalska and J. Kargul, Nanoscale, 2017, 9, 10475–10486 RSC.
- K. R. Stieger, S. C. Feifel, H. Lokstein and F. Lisdat, Phys. Chem. Chem. Phys., 2014, 16, 15667–15674 RSC.
- V. M. Friebe, J. D. Delgado, D. J. K. Swainsbury, J. M. Gruber, A. Chanaewa, R. Van Grondelle, E. Von Hauff, D. Millo, M. R. Jones and R. N. Frese, Adv. Funct. Mater., 2016, 26, 285–292 CrossRef CAS.
- K. R. Stieger, S. C. Feifel, H. Lokstein, M. Hejazi, A. Zouni and F. Lisdat, J. Mater. Chem. A, 2016, 4, 17009–17017 CAS.
- W. Bialek, S. Krzywda, M. Jaskolski and A. Szczepaniak, FEBS J., 2009, 276, 4426–4436 CrossRef CAS PubMed.
- T. Krupnik, E. Kotabová, L. S. Van Bezouwen, R. Mazur, M. Garstka, P. J. Nixon, J. Barber, R. Kaňa, E. J. Boekema and J. Kargul, J. Biol. Chem., 2013, 288, 23529–23542 CrossRef CAS PubMed.
- I. J. Iwuchukwu, M. Vaughn, N. Myers, H. O'Neill, P. Frymier and B. D. Bruce, Nat. Nanotechnol., 2010, 5, 73–79 CrossRef CAS PubMed.
- S. Yamada, S.-Y. Park, H. Shimizu, Y. Koshizuka, K. Kadokura, T. Satoh, K. Suruga, M. Ogawa, Y. Isogai, T. Nishio, Y. Shiro and T. Oku, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 1577–1582 CrossRef CAS.
- D. J. Barlow and J. M. Thornton, J. Mol. Biol., 1988, 201, 601–619 CrossRef CAS PubMed.
- N. S. Lewis and M. S. Wrighton, Science, 1981, 211, 944–947 CAS.
- A. Cuevas, Energy Procedia, 2014, 55, 53–62 CrossRef.
- G.-X. Wang, W.-J. Bao, M. Wang and X.-H. Xia, Chem. Commun., 2012, 48, 10859–10861 RSC.
- C. Peng, J. Liu, Y. Xie and J. Zhou, Phys. Chem. Chem. Phys., 2016, 1–4 Search PubMed.
- N. Amdursky, I. Pecht, M. Sheves and D. Cahen, J. Am. Chem. Soc., 2013, 135, 6300–6306 CrossRef CAS PubMed.
- J. Zhou, J. Zheng and S. Jiang, J. Phys. Chem. B, 2004, 108, 17418–17424 CrossRef CAS.
- D. Segal, A. Nitzan, W. B. Davis, M. R. Wasielewski and M. A. Ratner, J. Phys. Chem. B, 2000, 104, 3817–3829 CrossRef CAS.
- N. Amdursky, D. Ferber, C. A. Bortolotti, D. a. Dolgikh, R. V. Chertkova, I. Pecht, M. Sheves and D. Cahen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5556–5561 CrossRef CAS PubMed.
- E. Topoglidis, A. E. G. Cass, G. Gilardi, S. Sadeghi, N. Beaumont and J. R. Durrant, Anal. Chem., 1998, 70, 5111–5113 CrossRef CAS PubMed.
- H. Tajima, S. Ikeda, M. Matsuda, N. Hanasaki, J. W. Oh and H. Akiyama, Solid State Commun., 2003, 126, 579–581 CrossRef CAS.
- T. Lee, S. U. Kim, J. Min and J. W. Choi, Adv. Mater., 2010, 22, 510–514 CrossRef CAS PubMed.
- T. Liu, J. Zhong, X. Gan, C. Fan, G. Li and N. Matsuda, ChemPhysChem, 2003, 4, 1364–1366 CrossRef CAS PubMed.
- M. I. Schukfeh, L. Sepunaru, P. Behr, W. Li, I. Pecht, M. Sheves, D. Cahen and M. Tornow, Nanotechnology, 2016, 27, 115302 CrossRef PubMed.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS.
- D. Gront and A. Kolinski, Bioinformatics, 2008, 24, 584–585 CrossRef CAS PubMed.
- F. Fischer, P. Künzler, D. Ritz, H. Hennecke, F. Fischer, P. Ku and M. Institut, J. Bacteriol., 1995, 177, 4321–4326 CrossRef.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- H. Schagger, Nat. Protoc., 2006, 1, 16–22 CrossRef PubMed.
- A. Díaz, F. Navarro, M. Hervás, J. A. Navarro, S. Chávez, F. J. Florencio and M. A. De la Rosa, FEBS Lett., 1994, 347, 173–177 CrossRef.
- P. Maury, M. Escalante, M. Péter, D. N. Reinhoudt, V. Subramaniam and J. Huskens, Small, 2007, 3, 1584–1592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10895h |
| This journal is © The Royal Society of Chemistry 2017 |