
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-active ionic liquid electrolyte with multi energy storage mechanism for high energy density supercapacitor†
Duck-Jae Youa,
Zhenxing Yina,
Yong-keon Ahnb,
Seong-Hun Leea,
Jeeyoung Yoo*a and
Youn Sang Kim *ac
*ac
aProgram in Nano Science and Technology, Graduate School of Convergence Science and Technology, Seoul National University, Seoul 151-744, Republic of Korea. E-mail: younskim@snu.ac.kr; jyoo78@snu.ac.kr
bGraduate School of Energy and Environment, Seoul National University of Science and Technology, Seoul 01811, Republic of Korea
cAdvanced Institutes of Convergence Technology, 864-1 Iui-dong, Yeongtong-gu, Suwon-si, Gyeonggi-do 443-270, Republic of Korea
First published on 8th December 2017
Abstract
A bimodal redox-active ionic liquid electrolyte for supercapacitors with high energy density was demonstrated. The suggested bimodal electrolyte, which consists of 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMITFSI) and 1-ethyl-3-methylimidazolium halide (EMI-X, X = Br, I) as a redox active couple, shows the three types of energy storage mechanism: a classical EDL capacitance; a pseudo-capacitance from the redox reaction of halide species, such as bromide and iodide; and an EDL capacitance strongly enhanced by ion size effects. When EMITFSI is mixed with small ions, the thickness of the ionic layer becomes thinner and even more ions are packed into the electrode due to the decrement of excluded-volume effects and the increment of electrostatic interactions. The supercapacitor containing a mixture of EMITFSI and EMI-I showed a considerably high performance with 175.6 W h kg−1 and 4994.5 W kg−1 at 1 A g−1 and excellent cycling stability up to 5000 cycles.
A new industry using energy storage devices is emerging. Various types of energy storage devices are striving to enter the industrial market.1–4 In particular, in the case of electric vehicles (EVs), lithium ion batteries (LIBs), fuel cells (FCs) and supercapacitors (SCs) are in a competitive relationship.5–7 LIBs are the most promising candidates because of their high energy density and technical maturity. However, there is a problem with safety.8 Also, the unstable supply and high price of lithium and cobalt are critical issues.9 Meanwhile, FCs have difficulties in commercialization due to the disadvantage of obtaining hydrogen from petrochemistry and the resistance to establishing hydrogen stations.10 Recently, as an alternative to these situations, SCs were strongly proposed. Because the large power density of SCs is suitable for an intermittent power supply, up to now, SCs have been applied in an electric bus using regenerative brakes and an electric train operated by pulse power.11,12 If SCs store high energy density without sacrificing power density, SCs can be realized as good alternative energy storage systems replacing LIBs and FCs in various EVs.
There are two ways to increase the energy density of SCs. One is the enhancement of capacitance, and the other is the increment of operating voltages. Because energy density is proportional to capacitance and operating voltage, at the initial stage, many researchers focused on widening the electrode area for enhancing electric double layer (EDL) capacitance. The various types of activated carbon (AC) are commercialized as the result of these efforts.13 Carbon nano-materials like carbon nano tubes (CNTs) and graphene are highlighted as large capacitance materials.14–18 Additionally, the materials for a redox active electrode like metal oxide and conductive polymer are proposed to enlarge total capacitance with pseudo-capacitance.19,20 However, the redox active electrode has some problems such as lower electrical conductivity than carbonaceous, a limitation of adopting the electrolyte with an aqueous system, and relatively short life time by a partially irreversible chemical reaction.21,22 Another approach to redox active materials is the use of advanced electrolyte containing redox active couples. Representative redox couples are alkali metal halides, vanadium complexes, copper salts, hydroquinone, methylene blue, indigo carmine, p-phenylenediamine, m-phenylenediamine, lignosulfonates, and sulfonated polyaniline in aqueous medium.21,23–28 Another effort for increasing energy density is widening the operating voltages. In the early stages, classical salt and solvent systems are suggested. Quaternary ammonium salts and acetonitrile are representative electrolytes of SCs. Although these electrolytes show high ionic conductivity, organic solvent is highly flammable and thermally unstable. SCs including these electrolytes must be operated under 80 °C.28 To solve these issues, ionic liquids (ILs), which have a good thermal stability induced by negligible vapor pressure, are investigated.29–31 More importantly, ILs are very effective for widening the operating voltage due to their wide electrochemical stability window over 3 V.21,30–33 Among these various ILs, ILs containing imidazolium cations are broadly studied due to their high ionic conductivities and relatively low viscosities. Especially, 1-ethyl-3-methyl imidazolium tetrafluoroborate (EMIBF4) and 1-ethyl-3-methyl imidazolium bis(trifluoromethanesulfonyl)imide (EMITFSI) exist as a liquid state at ambient conditions. Therefore they can be applied as an electrolyte in SCs without any additional solvent.30,34 Conventional electrolyte consists of salts and solvent. Solvent is an inert medium for transporting ions from salts. Consequently, most parts of an electrode pore are filled with inert solvent, and the energy density of the SC is strongly reduced. This problem can be solved by adopting the liquid state of ILs such as EMIBF4 and EMITFSI. For these reasons, ILs are very attractive electrolyte material for high energy density. Despite these advantages, ILs still have room for improvement in energy density as an electrolyte of SCs. Most of ILs, which have been studied as an electrolyte for SCs, store charges with only EDL capacitance. Generally, a capacitance from EDL is much smaller than pseudo-capacitance for redox-active materials.21 This means that if the ILs have pseudo-capacitive behaviour, they can improve the energy density. Some researchers applied ILs to a pseudo-electrode such as MnO2 with organic solvent. However, clear redox behaviour was not observed even though specific capacitance was increased.35–37 The second approach is the adoption of ILs containing a redox active couple. Redox active electrolyte using 1-ethyl-3-methylimidazolium iodide (EMI-I) into EMIBF4 was reported.38–41 However, in most cases, the redox active couple was iodide, and EMI-I was added by weight ratio. As EDL capacitance is proportional to the number of ions (charges) and a weight ratio is not appropriate for representing the number of ions in EMI-I/EMIBF4 mixture. For this reason, research on ILs containing redox species should be more accurate with a molar ratio.
In this study, we proposed a binary ILs electrolyte system containing EMITFSI and 1-ethyl-3-methyl imidazolium halide (EMI-X) for enhancing energy densities of SCs. We selected a mole ratio to equalize the number of ions. Additionally, bromide anion is also used to compare the redox activity between bromide and iodide. The proposed electrolyte includes three types of energy storage mechanism. The first role of the suggested electrolyte system is classical EDL capacitance of ILs. The second is pseudo-capacitance from redox reaction of EMI-X. The third is a suggested concept in which different ion size improves the EDL capacitance due to increasing the packing density of the EDL by bimodal effect from different anion size of TFSI− and X−. TFSI anions were selected to obtain dramatic size distribution with halide because this TFSI− is about three times larger than X−. Although some researches have analysed the effect on cations of different sizes, they mostly discuss the potential of zero charge at the interface and AC characteristics were evaluated to exclude the influence on the electrochemical reaction induced by DC energy.42–45 The SCs adopting 0.12 mole fraction of EMI-I show the highest specific capacity of 144.4 F g−1 and this value of capacitance is increased by 33.5% in comparison to bare EMITFSI. In addition, its energy density is 175.6 W h kg−1 at 1 A g−1, when the power density is 4994.5 W kg−1. Obtained energy density value is more than 10 times higher when comparing the recently reported results at similar power density.46,47
The storage mechanism of redox-active electrolyte on the positive electrode is shown in Fig. 1. Consequently, the redox reaction of halide ion and different size of anion could help to increase the capacitance of SC. EMI-Br, EMI-I and EMITFSI were synthesized for the bimodal redox active electrolyte. The reaction mechanism, is described in Fig. S1† and the structural analysis of prepared ILs, were performed with 600 MHz proton nuclear magnetic resonance (1H NMR), Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) and Ion Chromatography (IC). EMI-X were synthesized by quaternarization of 1-methyl imidazole and ethyl halide.48 EMITFSI were prepared with anion exchange reaction between EMIT-Br and LiTFSI.49 Cation (EMI+) structure was confirmed by 1H NMR. CD3OD and D2O were used as the standard solvent for EMITFSI and EMI-X, respectively, and chemical shifts for H2O for each 1H NMR solvent were observed around 4.8 ppm. Chemical shift, coupling constant, and resonance multiplicities of prepared ionic liquids are summarized in Fig. S2 and Table S1.† Halides were confirmed by IC and bromide and iodide that were detected at the amount of 41.45% and 57.1%, respectively. These values are in good agreement with the theoretical content of halide (bromide: 41.82% and iodide: 57.31%). TFSI anion was analysed with ATR-FTIR Fig. S3.† The absorption peaks located in the 1171 cm−1 assign to S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric stretching are weakly coupled with S–N asymmetric stretching. Bands at 1128 and 1046 cm−1 correspond to C–F stretching and C–N stretching.50 Other peaks are summarized in Table S2.†
O symmetric stretching are weakly coupled with S–N asymmetric stretching. Bands at 1128 and 1046 cm−1 correspond to C–F stretching and C–N stretching.50 Other peaks are summarized in Table S2.†
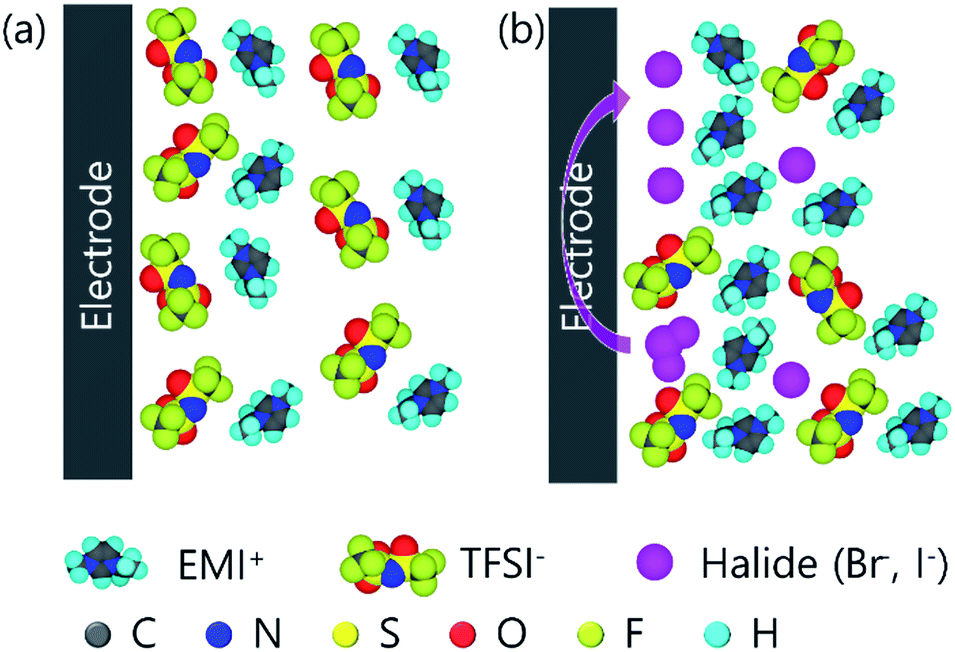 | ||
| Fig. 1 EDL model of (a) EMITFS and (b) mixture of EMITFSI and EMI-halide. | ||
The Redox active electrolytes were prepared with EMITFSI and EMI-X at different mole ratio. Mole fraction of EMI-X and their abbreviations are organized in Table 1. Each EMI-X were dissolved in EMITFSI to maximum solubility.
| Abbreviation | Halide type | Halide mole ratio | EMITFSI mole ratio | Halide weight % [%] |
|---|---|---|---|---|
| EMIT-Br0.04 | Bromide | 0.04 | 0.96 | 1.99 |
| EMIT-Br0.08 | Bromide | 0.08 | 0.92 | 4.07 |
| EMIT-Br0.12 | Bromide | 0.12 | 0.88 | 6.24 |
| EMIT-I0.04 | Iodide | 0.04 | 0.96 | 2.47 |
| EMIT-I0.08 | Iodide | 0.08 | 0.92 | 5.02 |
| EMIT-I0.12 | Iodide | 0.12 | 0.88 | 7.66 |
Fig. 2(a) and (b) show thermal stability of prepared redox active electrolyte. Thermal decomposition temperatures were analysed from the cross-point of the tangent line to the decrement starting portion and baseline (initial flat section) in the thermogravimetric analysis (TGA) graph by extrapolation. Every prepared electrolyte is thermally stable over 300 °C. The Fig. 2(b) display the pyrolysis temperature of prepared electrolytes. The prepared electrolyte (EMI-X) thermally decomposed through two step. Each thermal decomposition temperatures were denoted at Table S3.† The first thermal decomposition was occurred from EMI-X because the thermal decomposition temperature of EMI-X (X = Br or I) is lower than that of EMITFSI. After the first decomposition, the tendency of residual mass fractions was consistent with the weight ratio of EMI-X described in Table 1.
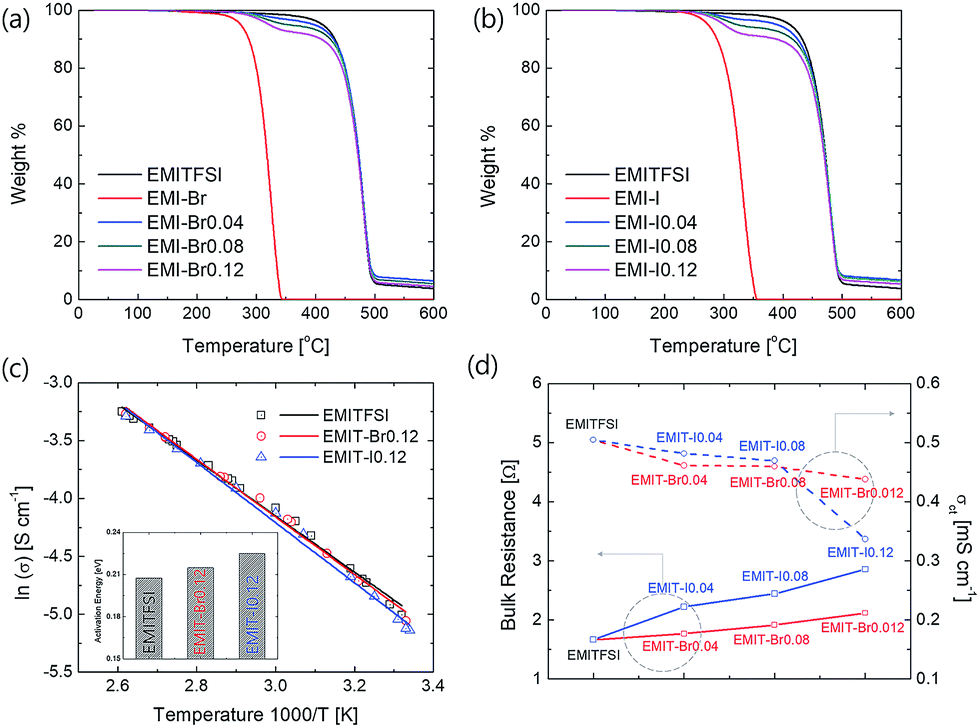 | ||
| Fig. 2 TGA profile of (a) EMIT-Br and (b) EMIT-I, (c) ionic conductivities of EMITFSI, EMIT-Br0.12 and EMIT-I0.12 (inset: activation energy) and (d) bulk resistance and charge transfer conductivities of EMITFSI, EMIT-Br0.12 and EMIT-I0.12. | ||
In order to verify the electrical properties of redox active electrolyte, ionic conductivity of the electrolytes was measured using a conductivity meter (cell constant, κ = 1). Fig. 2(c) shows the Arrhenius plots for the conductivity of redox active electrolyte with various temperature. EMIT-Br0.12 and EMIT-I0.12 were chosen representing EMI-X added electrolyte. The ionic conductivity of EMI-X added electrolyte was slightly changed and the ionic conductivities of EMITFSI, EMIT-Br0.12, and EMIT-I0.12 were 6.7 mS cm−1, 6.36 mS cm−1, and 5.86 mS cm−1, respectively, at room temperature. At a high temperature over 100 °C, the ionic conductivity of EMITFSI, EMIT-Br0.12, and EMIT-I0.12 were 33.8 mS cm−1, 33.7 mS cm−1, and 33.0 mS cm−1, respectively. The linearly of slopes from Fig. 2(c) shows that the conductivity of SCs with redox-active electrolyte has Arrhenius behaviour. The activation energy values were calculated from Fig. 2(c) using the Arrhenius equation
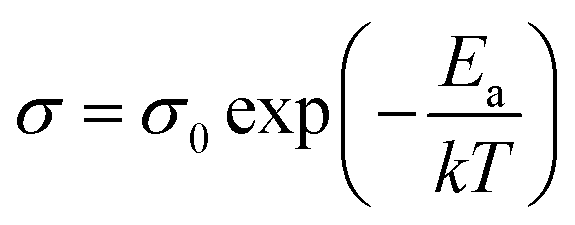 | (1) |
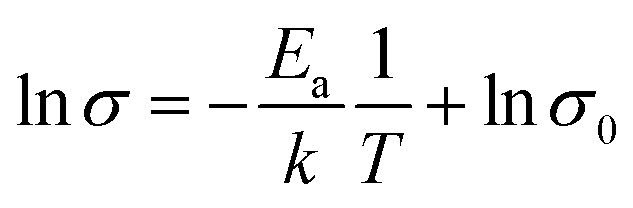 | (2) |
In order to investigate electrochemical stability of prepared electrolyte, Linear Sweep Voltammetry (LSV) was performed with a stainless steel (SUS)//ionic liquid electrolyte//SUS cell from 0 V to 10 V with a scan rate of 10 mV s−1. The results of the LSV tests with proposed electrolytes are displayed in Fig. S5(a).† As shown in Fig. S5(a),† every prepared electrolyte was stable up to 3.5 volts. The redox peaks were observed at 3.8 V, and 4.7 V for EMI-Br and EMI-I, respectively. For EMI-Br0.04, EMI-Br0.08 and EMI-012, the electrochemical degradation started at about 5.5 V. In the case of EMI-I added electrolyte, the electrochemical decomposition started at about 5.0 V to 6.0 V. The electrochemical degradation voltage slightly increased as the content of redox species increased. This is related to the ionic bonding between cations and anions. Ionic bonding strength was increased in order of I− > Br− > TFSI−. As this reason, iodide added electrolyte was the most electrochemically stable.
Additionally, faradic fraction was calculated from the anodic charge Qa and the cathodic charge Qc of cyclo-voltammogram with scan rate 5 mV S−1 to verify the actual cell voltage limitation.32 Tested cells were manufactured using a symmetrical porous carbon electrode.
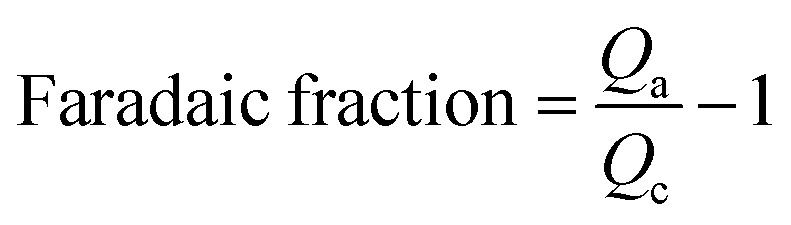 | (3) |
Fig. 3(a) is a plot of the faradaic fraction against the potential. The pure EMITFSI was stable up to 3.7 V. Moreover, EMIT-Br0.12, and EMIT-I0.12 were stable at 3.5 V. These results of LSV and faradaic fraction were based on setting the operating voltage of SCs with proposed electrolytes to 3.5 V in this study.
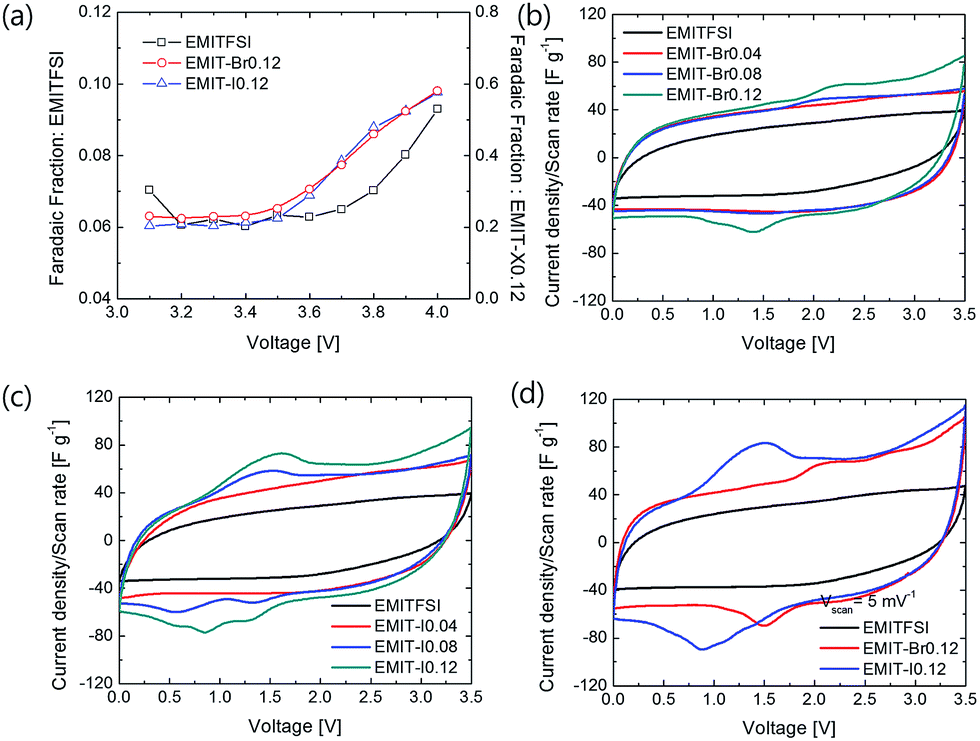 | ||
| Fig. 3 (a) Faradaic fraction of EMITFSI, EMIT-Br0.12 and EMIT-I0.12, cyclo-voltammogram of (b) EMI-Br and (c) EMI-I at 10 mV s−1, and (d) EMITFSI, EMIT-X0.12 at 5 mV s−1. | ||
Fig. 3(b–d) shows the cyclo-voltammogram of EMITFSI and EMI-X added electrolytes.
The SCs adopting EMI-X added electrolyte show not only the quasi-rectangular curve of EDL but also that the redox peaks appear clearly in the cyclo-voltammogram. These redox peaks were generated because of halide redox reaction. The redox mechanism is denoted below as eqn (4).
| 3X− ↔ X3− + 2e− | (4) |
Redox peaks of EMIT-I and EMIT-Br were compared in Fig. 3(d). The anodic peak means oxidation and it is observed at 1.4 V for iodide and 2.1 V for bromide. Cathodic peak means reduction and it appeared at 0.9 V for triiodide and 1.5 V for tribromide. As shown in Fig. 3(b) and (c), peak intensities were increased with increment of EMI-X contents. This redox reaction could contribute to enhance the capacitance.51 In the case of iodide, redox reaction occurred more actively than bromide as described in Fig. 3(d). Bromide is a stronger acid than iodide in terms of Lewis acid. Thus, the ionic bonding strength between bromine and EMI+ is stronger than that of iodide.52,53 In general, in organic solvent, bromide is easily solvated and shows higher reactivity. However, the behavior of bromide in ionic liquids is different. This is because the solvent surrounding the bromide in the solvated form of the bromide is an EMI cation. EMI+ prevents the redox reaction of bromide. This type of solvated bromide is less reactive than solvated iodide and is the same as the represented protic solvent medium, aqueous system.21 Therefore, the redox peak of iodide is stronger than bromide. At the same time, the quasi-rectangular curve of EDL also becomes larger than SCs by applying neat EMITFSI. The larger quasi-rectangular curve of EMI-X added EMITFSI is caused by the size difference of anion as mentioned in Fig. 1 because the moles of cations are equal regardless of the change in halide anion mole fraction. The increments of the quasi-rectangular curve of CV are caused by the addition of smaller ions to electrolytes. The size of ion in the electrolyte is an important factor for the capacitance of EDLC.54–56 The capacitance of the EDL capacitor using EMITFSI electrolyte is increased by the addition of a smaller anion.57 When the EMITFSI is mixed with a small anion due to the decrement of excluded-volume effect and increased electrostatic interactions, the thickness of the ionic layer becomes thinner and more counterion of EMI+ packed in the positive electrode.57 Thus, capacitance of the EDL capacitor are increased by enhancing the counterion density in the EDL structure. The size of ions in the proposed electrolyte is 0.5 nm, 0.6 nm, 0.196 nm, and 0.219 nm for EMI, TFSI, bromide, and iodide,57 respectively. Thus, the use of electrolytes composed of various sizes of anion increases for the specific capacitance. In conclusion, the enhancement of capacitance in our study is caused by the redox reaction of halide ions and ion size effect. The specific capacitance was measured by cycle voltammetry (CV) with the operating voltage of 3.5 V at the scan rate of 5 mV s−1 and they are summarized in Fig. S6.† As estimated by the area of Fig. 3(c) and (d), gravimetric capacitances are increased by the addition of EMI-X to EMITFSI electrolyte. Gravimetric capacitances of EMITFS, EMIT-Br0.12 and EMIT-I0.12 are 110.0 F g−1, 171.8 F g−1 and 200.6 F g−1 at 5 mV s−1, respectively, when maximum mole ratio of 0.12 were applied. In Fig. S5(b),† the specific capacitance of SCs using pure EMITFSI electrolyte depended on the various scan rates and the quasi-rectangular CV curves are obtained from 1 mV s−1 to 100 mV s−1. The gravimetric capacitance for pure EMITFSI was changed from 148.7 F g−1 at 1 mV s−1 to 32.5 F g−1 at 100 mV s−1.
The Fig. S5(c) and (d)† show that the gravimetric capacitances of SC with mixture of ionic liquids with maximum mole ratio. The values of specific capacitance of SCs using EMIT-I electrolyte, which measured at the scan rate of 10 mV s−1, are 138.1 F g−1, 166.6 F g−1 and 185.9 F g−1 at a mole fraction of 0.04, 0.08, and 0.12, respectively. The specific capacitance of proposed SCs using EMIT-Br electrolyte was increased from 148.5 F g−1 to 160.1 F g−1 depending on the mole ratio. Large capacitances of SCs adopting EMIT-I are explained with redox activity and anion size effect.
The Galvanostatic charge–discharge (GCD) test was performed to evaluate the energy density of SCs adopting EMTFSI/EMI-X0.12, which is able to achieve the highest gravimetric capacitance in cyclo-voltammogram and neat EMITFSI. Specific capacitances (Csp), energy densities (E) and power densities (P), were calculated from the GCD profile on the gravimetric basis of half-cell,
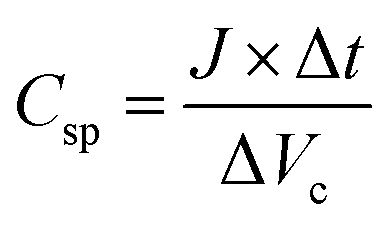 | (5) |
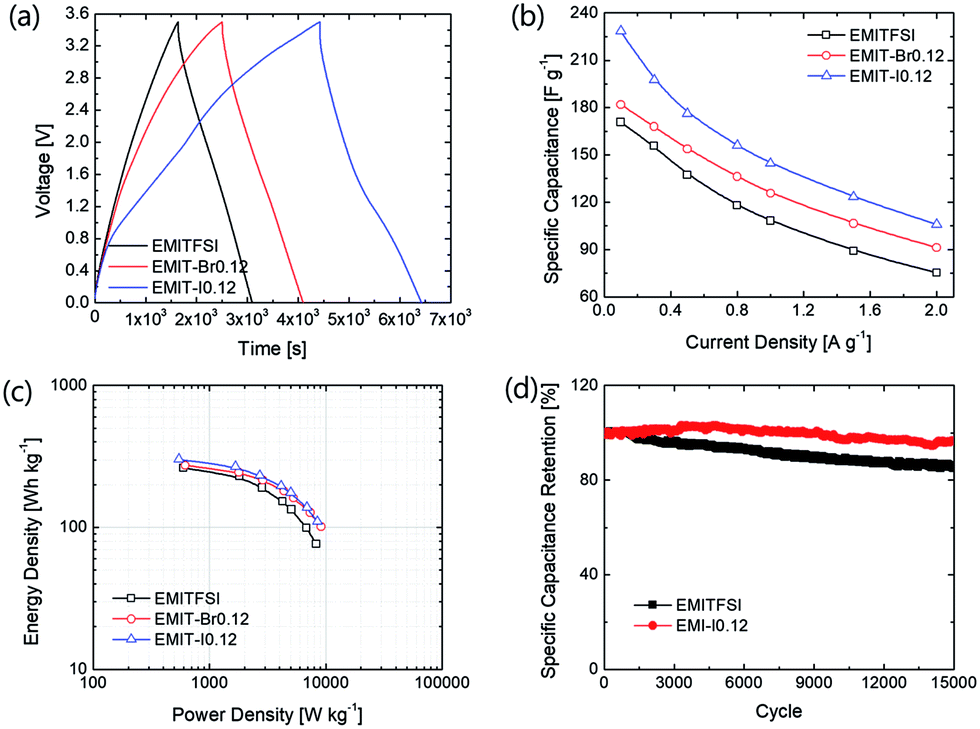 | ||
| Fig. 4 (a) The Galvanostatic charge–discharge (GCD) of SCs adopting EMITFSI, EMI-Br0.12 and EMIT-I0.12, (b) specific capacitance of EMITFSI, EMIT-Br0.12 and EMIT-I0.12 depending on current densities, (c) Ragone plot of EMITFSI, EMIT-Br0.12 and EMIT-I0.12 and (d) cycle-ability of EMITFSI and EMIT-I0.12. | ||
These fading rates relate with cycle-ability of SCs. The cycling tests were performed to evaluate the reliability of prepared SCs at 1 A g−1 (25C-rate). As shown in Fig. 4(d), the SCs containing EMIT-I0.12 show excellent cycle durability. The cycle performance was conducted after aging to stabilize cell and remove residual moisture in cell.
After stabilization, SCs applying EMIT-I0.12 maintained specific capacitance evenly. On the contrary, specific capacitance of EMITFSI is gradually faded like a conventional EDL capacitor. They also maintained a high coulombic efficiency over 90% during cycling. In the case of EMIT-Br0.12, failure was observed after 200 cycles. EMIT-Br0.12 was electrochemically degraded and the SCs did not charged to 3.5 V because of simultaneous electrolysis during 3.5 V. The feature of EMIT-Br0.12 after 200 cycles was demonstrated in Fig. S6(h)† as compared with EMIT-I0.12. We estimated that the black part of Fig. S6(h)† is a product of electrolyzed EMIT-Br0.12. The Ragone plots of SCs with a proposed electrolyte are displayed in Fig. 4(c). The specific energy density and power density were calculated using galvanostatic charge–discharge within the operating voltage range from 0 V to 3.5 V by eqn (6) and (7).
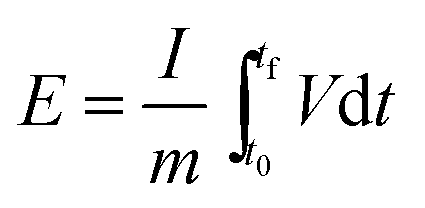 | (6) |
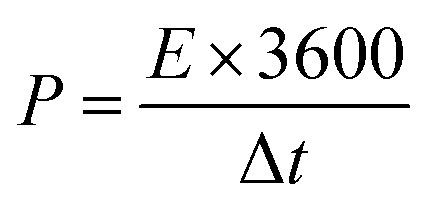 | (7) |
The t0, and tf are initial time and end time for the discharge, respectively. The I is current. The using of EMIT-I0.12 is led to enhance the energy density and power density in comparison with the EDL capacitor using pure EMITFSI. The proposed bimodal SCs using EMIT-I0.12 show the maximum energy density of 175.6 W h kg−1, and the power density of 4994.5 W kg−1 at 1 A g−1. The energy densities are 161.2 W h kg−1, and 133.8 W h kg−1 for EMIT-Br0.12, and EMITFSI, respectively. The power densities are 5267.0 W kg−1, and 5080.3 W kg−1 for EMIT-Br 0.12 and EMITFSI, respectively. These results show significant increment in energy density of about 31% in comparison with neat EMITFSI by the addition of EMI-I. These results mean that the proposed bimodal energy storage mechanism is a very effective method to enhance the performance of SCs. We summarize a table to be compared with other researchers' result in Table S4.† Taking all of the above results into consideration, the proposed bimodal electrolytes give high-performance for SC, and are applicable to practical devices, which required high energy density and stability.
Conclusions
In summary, we demonstrated a bimodal redox-active ionic liquid electrolyte for high energy density SCs. The suggested bimodal electrolyte system is a new type electrolyte, which shows high energy density due to the redox reaction of halide species. Furthermore, EDL capacitance was increased by size distribution of anions. The proposed electrolytes consist of EMITFSI and EMI-X (X = Br, I) as redox active couples. When EIMT-I0.12 is applied, the SC showed the best performance. The EMIT-I0.12 electrolytes remarkably improved the energy density of SCs without sacrificing power density (175.6 W h kg−1, 4994.5 W kg−1 at 1 A g−1). Additionally, the cycle durability of SCs was excellent up to 5000 cycles despite adopting redox active material (EMI-I). Therefore, since the proposed bimodal redox-active ionic liquid electrolytes for SC give a novel approach for developing high energy density SCs, this study provides good inspiration to researchers in the field of energy storage and conversion devices.Experimental details
The EMI-X (X = Br, I) was prepared by quaternarization of 1-methyl imidazole with ethyl halide at a mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1. The ethyl halides (iodoethane and bromoethane) were used as quaternarization agents. First, the 1-methyl imidazole and ethyl halide were mixed and stirred without solvents for 4 hours. After reaction, the solution changed to a solid phase and then unreacted ethyl halide were removed under vacuum at 100 °C for 6 hours. EMITFSI was synthesized by an anion exchange reaction of EMI-Br and lithium bis(trifluoromethylsulfonyl)imide (LiTFSI). EMI-Br and LiTFSI were mixed in deionized water at 60 °C for 3 hours at molar ratio 1:1.03 (EMI-Br:LiTFSI). The EMITFSI was washed using water to remove residual LiTFSI and then the product was dried under vacuum at 120 °C for 24 hours. The synthesized EMI-X was dissolved in EMITFSI, and then the mixed solution was stirred in a glove box filled with argon gas. The molar ratio of EMI-X varied from 0.04 to 0.12. The chemical structure of synthesized electrolytes is investigated by 600 MHz proton nuclear magnetic resonance (1H NMR), Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR), and ion chromatography (IC). The thermal stability of synthesized ionic liquid electrolyte was investigated by thermogravimetric analysis (TGA) at the heating rate of 5 °C min−1 under nitrogen condition. The electrochemical properties of bimodal redox-active electrolyte were investigated by Electrochemical Impedance Spectroscopy (EIS) (VSP, Bio-Logic SAS) from 0.1 Hz to 1 MHz. The ion conductivity of synthesized ionic liquid electrolytes was evaluated by a conductivity meter (EC-400L, Istek) in a glove box filled with argon gas.
:1.1. The ethyl halides (iodoethane and bromoethane) were used as quaternarization agents. First, the 1-methyl imidazole and ethyl halide were mixed and stirred without solvents for 4 hours. After reaction, the solution changed to a solid phase and then unreacted ethyl halide were removed under vacuum at 100 °C for 6 hours. EMITFSI was synthesized by an anion exchange reaction of EMI-Br and lithium bis(trifluoromethylsulfonyl)imide (LiTFSI). EMI-Br and LiTFSI were mixed in deionized water at 60 °C for 3 hours at molar ratio 1:1.03 (EMI-Br:LiTFSI). The EMITFSI was washed using water to remove residual LiTFSI and then the product was dried under vacuum at 120 °C for 24 hours. The synthesized EMI-X was dissolved in EMITFSI, and then the mixed solution was stirred in a glove box filled with argon gas. The molar ratio of EMI-X varied from 0.04 to 0.12. The chemical structure of synthesized electrolytes is investigated by 600 MHz proton nuclear magnetic resonance (1H NMR), Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR), and ion chromatography (IC). The thermal stability of synthesized ionic liquid electrolyte was investigated by thermogravimetric analysis (TGA) at the heating rate of 5 °C min−1 under nitrogen condition. The electrochemical properties of bimodal redox-active electrolyte were investigated by Electrochemical Impedance Spectroscopy (EIS) (VSP, Bio-Logic SAS) from 0.1 Hz to 1 MHz. The ion conductivity of synthesized ionic liquid electrolytes was evaluated by a conductivity meter (EC-400L, Istek) in a glove box filled with argon gas.
To characterize the electrical performance of the supercapacitor, the symmetrical cells with bimodal redox-active electrolyte were fabricated with porous carbon electrode in a 2032 cell. The porous carbon has a large surface area of 2131 m2 g−1 Fig. S7.† The porous carbon electrodes were prepared by the casting method with Meyer rod on 15 ìm etched aluminum foil (KJCC). The slurry consisted of porous carbon as an active material, carbon black as a conductive agent (super P, Timcal), styrene-butadiene rubber (SBR, JSR Corporation) as a binder, and carboxy methyl cellulose (CMC, Dai-Ichi Kogyo Seiyaku) as the viscosity agent in the mass ratio of 80:10:5:5. The planetary centrifugal mixer (AR 100, Thinky mixer) was used to disperse the slurry. The AC electrode, synthesized ionic liquid electrolyte, and aramid fiber separator (Mitsubishi paper mill) were assembled using in a glove box filled with the argon gas. The linear sweep voltammetry (LSV) was confirmed from 0 V to 10 V and the cyclic voltammetry were investigated to observe pseudo-capacitive behavior by potentiostat from 0 V to 3.5 V (VSP, Bio-Logic SAS). The galvanostatic charge–discharge tests of a symmetrical cell were performed to determine specific capacitance, specific energy density, specific power density, and cycle-ability using a battery cycler system (WBCS3000, Wonathech Co., LTD). The performance of supercapacitors were measured after the aging process. The aging conditions is to charge and discharge 10 times at a current density of 0.1 A g−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP, Ministry of Science, ICT & Future Planning) (No. 2016R1C1B2013145) and (No. 2016M3A7B4910458).Notes and references
- F. Zhang, T. F. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. S. Chen, Energy Environ. Sci., 2013, 6, 1623–1632 CAS.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef CAS.
- I. Hadjipaschalis, A. Poullikkas and V. Efthimiou, Renewable Sustainable Energy Rev., 2009, 13, 1513–1522 CrossRef.
- T. M. I. Mahlia, T. J. Saktisahdan, A. Jannifar, M. H. Hasan and H. S. C. Matseelar, Renewable Sustainable Energy Rev., 2014, 33, 532–545 CrossRef.
- S. F. Tie and C. W. Tan, Renewable Sustainable Energy Rev., 2013, 20, 82–102 CrossRef.
- M. A. Hannan, F. A. Azidin and A. Mohamed, Renewable Sustainable Energy Rev., 2014, 29, 135–150 CrossRef.
- M. A. Hannan, M. M. Hoque, A. Mohamed and A. Ayob, Renewable Sustainable Energy Rev., 2017, 69, 771–789 CrossRef.
- S. Abada, G. Marlair, A. Lecocq, M. Petit, V. Sauvant-Moynot and F. Huet, J. Power Sources, 2016, 306, 178–192 CrossRef CAS.
- L. Ghadbeigi, J. K. Harada, B. R. Lettiere and T. D. Sparks, Energy Environ. Sci., 2015, 8, 1640–1650 CAS.
- I. A. Richardson, J. T. Fisher, P. E. Frome, B. O. Smith, S. Guo, S. Chanda, M. S. McFeely, A. M. Miller and J. W. Leachman, Int. J. Hydrogen Energy, 2015, 40, 8122–8127 CrossRef CAS.
- Z. Zhang, X. Zhang, W. Chen, Y. Rasim, W. Salman, H. Pan, Y. Yuan and C. Wang, Appl. Energy, 2016, 178, 177–188 CrossRef.
- Z. Zou, J. Cao, B. Cao and W. Chen, ISA Trans., 2015, 55, 234–240 CrossRef PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- V. V. N. Obreja, Phys. E, 2008, 40, 2596–2605 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS.
- Q. Chen, Y. Zhao, X. Huang, N. Chen and L. Qu, J. Mater. Chem. A, 2015, 3, 6761–6766 CAS.
- K. S. Ryu, K. M. Kim, N.-G. Park, Y. J. Park and S. H. Chang, J. Power Sources, 2002, 103, 305–309 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- S. T. Senthilkumar, R. K. Selvan and J. S. Melo, J. Mater. Chem. A, 2013, 1, 12386–12394 CAS.
- I. Shown, A. Ganguly, L.-C. Chen and K.-H. Chen, Energy Sci. Eng., 2015, 3, 2–26 CrossRef CAS.
- E. Frackowiak, M. Meller, J. Menzel, D. Gastol and K. Fic, Faraday Discuss., 2014, 172, 179–198 RSC.
- D. Gastol, J. Walkowiak, K. Fic and E. Frackowiak, J. Power Sources, 2016, 326, 587–594 CrossRef CAS.
- X. Wang, R. S. Chandrabose, S.-E. Chun, T. Zhang, B. Evanko, Z. Jian, S. W. Boettcher, G. D. Stucky and X. Ji, ACS Appl. Mater. Interfaces, 2015, 7, 19978–19985 CAS.
- J. Lee, S. Choudhury, D. Weingarth, D. Kim and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 23676–23687 CAS.
- S. E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- D.-J. You, Z. Yin, Y.-k. Ahn, S. Cho, H. Kim, D. Shin, J. Yoo and Y. S. Kim, J. Ind. Eng. Chem., 2017, 52, 1–6 CrossRef CAS.
- M. P. S. Mousavi, B. E. Wilson, S. Kashefolgheta, E. L. Anderson, S. He, P. Bühlmann and A. Stein, ACS Appl. Mater. Interfaces, 2016, 8, 3396–3406 CAS.
- C. Arbizzani, M. Biso, D. Cericola, M. Lazzari, F. Soavi and M. Mastragostino, J. Power Sources, 2008, 185, 1575–1579 CrossRef CAS.
- Y.-k. Ahn, B. Kim, J. Ko, D.-J. You, Z. Yin, H. Kim, D. Shin, S. Cho, J. Yoo and Y. S. Kim, J. Mater. Chem. A, 2016, 4, 4386–4391 CAS.
- A. Balducci, R. Dugas, P. L. Taberna, P. Simon, D. Plée, M. Mastragostino and S. Passerini, J. Power Sources, 2007, 165, 922–927 CrossRef CAS.
- H. Kurig, M. Vestli, K. Tõnurist, A. Jänes and E. Lust, J. Electrochem. Soc., 2012, 159, A944–A951 CrossRef CAS.
- X. Zhang, D. Zhao, Y. Zhao, P. Tang, Y. Shen, C. Xu, H. Li and Y. Xiao, J. Mater. Chem. A, 2013, 1, 3706–3712 CAS.
- H.-Q. Wang, Z.-S. Li, Y.-G. Huang, Q.-Y. Li and X.-Y. Wang, J. Mater. Chem., 2010, 20, 3883–3889 RSC.
- B. Shen, X. Zhang, R. Guo, J. Lang, J. Chen and X. Yan, J. Mater. Chem. A, 2016, 4, 8180–8189 CAS.
- T. Tooming, T. Thomberg, L. Siinor, K. Tõnurist, A. Jänes and E. Lust, J. Electrochem. Soc., 2014, 161, A222–A227 CrossRef CAS.
- E. Lust, L. Siinor, H. Kurig, T. Romann, V. Ivaništšev, C. Siimenson, T. Thomberg, J. Kruusma, K. Lust, P. Pikma, E. Anderson, V. Grozovski, R. Palm, L. Läll, A. Ruzanov, T. Tooming, O. Oll and A. Jänes, ECS Trans., 2016, 75, 161–170 CrossRef.
- C. Siimenson, L. Siinor, K. Lust and E. Lust, ECS Electrochem. Lett., 2015, 4, H62–H65 CrossRef CAS.
- C. Siimenson, M. Lembinen, O. Oll, L. Läll, M. Tarkanovskaja, V. Ivaništšev, L. Siinor, T. Thomberg, K. Lust and E. Lust, J. Electrochem. Soc., 2016, 163, H723–H730 CrossRef CAS.
- V. Lockett, R. Sedev, J. Ralston, M. Horne and T. Rodopoulos, J. Phys. Chem. C, 2008, 112, 7486–7495 CAS.
- M. T. Alam, M. M. Islam, T. Okajima and T. Ohsaka, J. Phys. Chem. C, 2008, 112, 16600–16608 CAS.
- M. T. Alam, J. Masud, M. M. Islam, T. Okajima and T. Ohsaka, J. Phys. Chem. C, 2011, 115, 19797–19804 CAS.
- V. Lockett, M. Horne, R. Sedev, T. Rodopoulos and J. Ralston, Phys. Chem. Chem. Phys., 2010, 12, 12499–12512 RSC.
- D. Zhou, H. Wang, N. Mao, Y. Chen, Y. Zhou, T. Yin, H. Xie, W. Liu, S. Chen and X. Wang, Microporous Mesoporous Mater., 2017, 241, 202–209 CrossRef CAS.
- G. Sun, K. Li, L. Xie, J. Wang and Y. Li, Microporous Mesoporous Mater., 2012, 151, 282–286 CrossRef CAS.
- A. Stark, B. L. MacLean and R. D. Singer, J. Chem. Soc., Dalton Trans., 1999, 63–66, 10.1039/A806708B.
- C. Zhu, H. Cheng and Y. Yang, J. Electrochem. Soc., 2008, 155, A569–A575 CrossRef CAS.
- J. Li, J. Tang, J. Yuan, K. Zhang, Q. Shao, Y. Sun and L.-C. Qin, Electrochim. Acta, 2016, 197, 84–91 CrossRef CAS.
- S. Yamazaki, T. Ito, M. Yamagata and M. Ishikawa, Electrochim. Acta, 2012, 86, 294–297 CrossRef CAS.
- K. M. Abraham, J. Electrochem. Soc., 2015, 162, A3021–A3031 CrossRef CAS.
- I. V. Nelson and R. T. Iwamoto, J. Electroanal. Chem., 1959, 1964(7), 218–221 Search PubMed.
- G. Feng, R. Qiao, J. Huang, S. Dai, B. G. Sumpter and V. Meunier, Phys. Chem. Chem. Phys., 2011, 13, 1152–1161 RSC.
- J. Andrews and S. Das, RSC Adv., 2015, 5, 46873–46880 RSC.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- C. Lian, K. Liu, K. L. Van Aken, Y. Gogotsi, D. J. Wesolowski, H. L. Liu, D. E. Jiang and J. Z. Wu, ACS Energy Lett., 2016, 1, 21–26 CrossRef CAS.
- L. G. Bettini, M. Galluzzi, A. Podestà, P. Milani and P. Piseri, Carbon, 2013, 59, 212–220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic mechanism of ionic liquids, H NMR, IR, AC Impedance spectroscopy, galvanostatic charge discharge profile. See DOI: 10.1039/c7ra10772b |
| This journal is © The Royal Society of Chemistry 2017 |