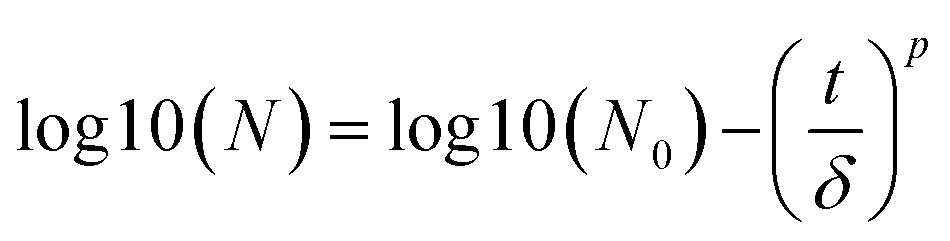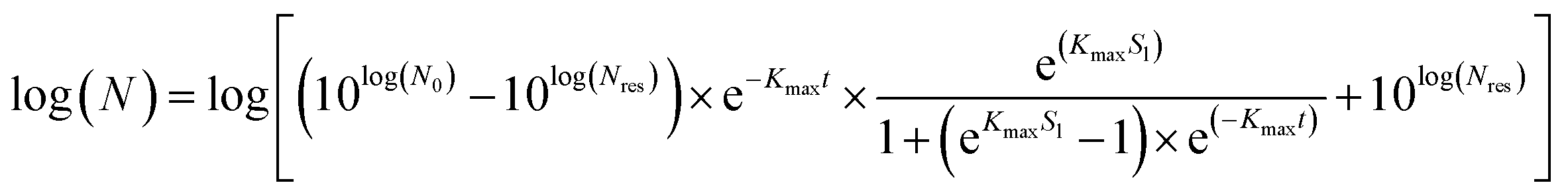DOI:
10.1039/C7RA10605J
(Paper)
RSC Adv., 2017,
7, 51822-51830
Size-controlled synthesis of Cu2O nanoparticles: size effect on antibacterial activity and application as a photocatalyst for highly efficient H2O2 evolution†
Received
25th September 2017
, Accepted 23rd October 2017
First published on
7th November 2017
Abstract
We propose a green approach for the size-controlled synthesis of Cu2O nanoparticles (NPs) with simplified chemical deposition technology by adjusting the concentration of the precursor copper source. The morphologies, phase compositions and structures were recorded for the obtained Cu2O NPs with sizes of 10, 50, 100 and 200 nm. In the dark, all of the Cu2O NPs showed good antibacterial activity towards Escherichia coli K-12 (E. coli). A size-dependent antibacterial effect was also observed as a result where decreasing the size of the Cu2O NPs led to increasing the antibacterial activity. The smallest Cu2O NPs (Cu2O-10) with the minimum inhibitory concentration (≤1 µg mL−1) achieved the highest antibacterial efficiency, with the inactivation of 7-log E. coli in 80 min with a concentration of 5 µg mL−1 in the dark. Under visible light (VL), medium concentrations (10, 20 and 50 µg mL−1) of Cu2O-10 showed enhanced antibacterial activity compared to that in the dark. For low (5 µg mL−1) or high concentrations (200 µg mL−1) of Cu2O-10, the antibacterial activity under VL was almost the same as that in the dark. The mechanisms of antibacterial activity are proposed and discussed in detail. “Contact killing” and photogenerated h+ together with reactive oxygen species (such as ˙OH and H2O2) were found to be responsible for bacterial inactivation in the dark and under irradiation, respectively. Cu2O-10 proved to be a good photocatalyst, capable of the highly efficient evolution of H2O2, whose stable equilibrium concentration could be as high as 150 µM (using 20 µg mL−1 of Cu2O-10). Study on the membrane-separated system shows that the concentration of diffusing H2O2 can be as high as 50 µM, contributing to a complete inactivation of 7-log E. coli in 7 h. For comparison, the inactivation kinetics of the Cu2O NPs and H2O2 were calculated, and could be fitted into the Weibull and shoulder-linear-tail models, respectively.
1. Introduction
Synthesis of cuprous oxide (Cu2O) nanostructures is becoming increasingly attractive due to their relatively easy preparation, low cost and wide range of potential applications. In particular, shape-controlled synthesis of Cu2O nanostructures has been extensively studied because precise control of shapes offers an opportunity for discovering potentially exciting and unique shape-dependent properties.1 As a result, a wide variety of Cu2O nanostructures with controlled shapes such as rods,2 wires,3 spheres,4 different polyhedrals5–8 and hollow structures9–11 have been successfully synthesized, and some shape-dependent properties have been widely investigated as well.12–14 However, a size-controlled synthesis of Cu2O nanoparticles (NPs) has so far been absent and remains a challenging task owing to the intrinsic difficulties in controlling the sizes of NPs while keeping their shapes uniform and unchanged. Thus, the size-controlled synthesis of Cu2O NPs is highly interesting not only for further development of synthetic strategies, but also for the examination of their size-dependent properties.
Cu2O NPs, which are a good antibacterial, are widely used as an antiseptic and germicide in daily life15 and are a promising antibacterial for medical treatment16 and the removal of pathogens from aquatic environments.17 In recent years, the engineering of micro- and nanostructures of Cu2O has tended to attract particular interest owing to their distinctive and outstanding antibacterial activity. Microstructured Cu2O was found to have morphology-dependent antibacterial activity and specificity against Gram-positive and Gram-negative bacterial strains.17–20 Nanostructured Cu2O was studied and demonstrated higher antibacterial activity than its high valence counterpart (CuO)21,22 and even most metal oxide NPs such as TiO2,21 ZnO,21,23 WO321 and NiO.23 However, there has still been a lack of studies on the relationship between the antibacterial activity and particle size of Cu2O NPs. Thus, in this study, we initiate an investigation on the effect of size on the antibacterial activity of Cu2O NPs towards Escherichia coli K-12 (E. coli) in the dark.
Cu2O NPs are also an efficient visible-light-driven photocatalyst with a high absorption coefficient and theoretical solar conversion efficiency. However, studies on pristine Cu2O NPs only center on either the photocatalytic degradation of organics10,24,25 or H2 production,26–28 and their application in photocatalytic water disinfection has not been reported till now. Moreover, the excellent photocatalytic degradation activity of organics suggests that Cu2O NPs might have high photoactive antibacterial effects, inspiring us to further investigate their properties and functions related to photoactive antibacterial effects under visible light (VL).
In this study, we developed an effective protocol for the size-controlled preparation of Cu2O NPs by improving a previous chemical deposition method29 at ambient temperature without a high energy demand, complicated apparatus, or any organic compounds or surfactants. In particular, we paid much attention to the inactivation kinetics of H2O2 towards E. coli because H2O2 is widely involved in Cu2O29–31 and Cu2O-based32 photocatalytic systems but its role is still unclear for photocatalytic disinfection. Moreover, for comparison, the rates of inactivation of E. coli in different conditions were calculated and fitted according to the inactivation kinetics of models proposed by Geeraerd et al. (2000).33 We expect that this study can offer a significant reference for the selection of Cu2O in real applications.
2. Experimental section
2.1. Preparation and characterization of Cu2O NPs
A stock colorless 500 mL solution was prepared by adding 400 mL of sodium thiosulfate (Na2S2O3) to 100 mL of copper sulfate (CuSO4·5H2O) solution. The concentrations of CuSO4·5H2O were adjusted to 0.05, 0.1, 0.6, 1, 1.2 and 2 M. The concentrations of Na2S2O3 were adjusted correspondingly and the molar ratio of CuSO4·5H2O and Na2S2O3 was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The same volume (500 mL) of 5 M NaOH solution was placed in another beaker. The mixture was poured into the above NaOH solution with vigorous stirring. Cu2O powder was obtained by centrifugation at 12000 rpm, washed with deionized water and dried under vacuum at 60 °C. The phase purity of the Cu2O powder was characterized by X-ray powder diffraction (XRD) using an X-ray diffractometer (Y-2000) with Cu Kα radiation (λ = 1.5418 Å). A scan efficiency of 0.1° S−1 was applied to record the powder patterns in the range of 10° ≤ 2θ ≤ 80°. The Brunauer–Emmett–Teller (BET) surface areas of the samples were determined by a high-speed automated area and pore size analyzer (NOVA 2000e). Scanning electron microscopy (SEM) images were obtained on a JEOL SM-6700F microscope operating at 5 kV. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were carried out with a JEOL JEM-100CXII.
:4. The same volume (500 mL) of 5 M NaOH solution was placed in another beaker. The mixture was poured into the above NaOH solution with vigorous stirring. Cu2O powder was obtained by centrifugation at 12000 rpm, washed with deionized water and dried under vacuum at 60 °C. The phase purity of the Cu2O powder was characterized by X-ray powder diffraction (XRD) using an X-ray diffractometer (Y-2000) with Cu Kα radiation (λ = 1.5418 Å). A scan efficiency of 0.1° S−1 was applied to record the powder patterns in the range of 10° ≤ 2θ ≤ 80°. The Brunauer–Emmett–Teller (BET) surface areas of the samples were determined by a high-speed automated area and pore size analyzer (NOVA 2000e). Scanning electron microscopy (SEM) images were obtained on a JEOL SM-6700F microscope operating at 5 kV. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were carried out with a JEOL JEM-100CXII.
2.2. Bacterial inactivation by Cu2O NPs in the dark and under VL irradiation
The inactivation of E. coli was conducted using a 300 W xenon lamp (Beijing Perfect Light Co. Ltd., Beijing) with a UV cutoff filter (λ < 400 nm) as the light source (Fig. S1†). The VL intensity was measured by a light meter (LI-COR, USA) and adjusted to 100 mW cm−2. The bacteria strain E. coli was received from the American Type Culture Collection and chosen for this study. One pure bacterial colony grown on the nutrient agar plate was inoculated in a 2.5% nutrient broth solution at 37 °C with 200 rpm agitation for 17 h. The culture was then washed twice with sterilized 0.9% NaCl solution (saline) by centrifugation at 5000 rpm for 5 min using a Hermle Z323 centrifuge (Hermle Labortechnik, Germany). The bacterial cells and Cu2O NPs were suspended in saline in a partly-filled beaker and the cell density was finally adjusted to about 2 × 107 colony forming unit (cfu) per mL or 2 × 105 cfu mL−1 by dilution with an appropriate volume of saline to conduct different antibacterial experiments. The reaction temperature was maintained at 25 °C. At different time intervals, aliquots of the sample were collected and serially diluted with saline. 0.1 mL of the diluted sample was then immediately spread on nutrient agar (Lancashire, UK) plates and incubated at 37 °C for 24 h to determine the number of viable cells (in cfu). Light controls (light alone without Cu2O NPs) and dark controls (without Cu2O NPs and light) were also carried out for each set of experiments. All of the experimental controls and treatments were performed in triplicates.
To compare the rate of inactivation of E. coli, the inactivation kinetics in different conditions were calculated using the GInaFit software according to the microbial survival models proposed by Geeraerd et al.33 Two models were used in this study named the Weibull and shoulder-linear, which can be referred to as eqn (1) and (2), respectively.
| | 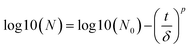 | (1) |
where
N0 and
N are the initial and surviving number of cells (log, in cfu), respectively,
t = reaction time [min] and
δ [time unit] is a scale parameter.
| | 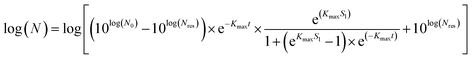 | (2) |
where
Kmax = specific inactivation rate [min
−1],
Sl = shoulder length [min],
Nres = residue population density [cfu mL
−1],
N0 and
N are the initial and surviving number of cells (log, in cfu), respectively, and
t = reaction time [min].
2.3. Fluorescence spectroscopy
The membrane-separated (or partition) system is shown in Fig. S2.† The E. coli in no-Cu2O cell with different irradiation times was collected and stained with the dyes of the LIVE/DEAD BacLight bacterial viability kit (L7012, Molecular Probes, Inc. Eugene, OR) following the protocol recommended by the manufacture. After being incubated at 25 °C for 15 min in the dark, the samples were transferred to a coverslip and examined with a fluorescence microscope (Nikon ECLIPSE 80i, Japan) equipped with an NUV-2A filter block consisting of an Ex 400–600 (Nikon, Japan) excitation filter and a Spot-K slider CCD camera (Diagnostic instruments. Inc. Sterling Heights, MI).
2.4. Determination of the concentrations of copper ion
The concentration of cuprous ion (Cu+) was measured by colorimetric analysis with bathocuproine disulfonic acid, which selectively chelates Cu+. The total concentration of the copper ion was also measured by adding ascorbic acid to reduce all of the cupric ions (Cu2+) to Cu+.34 The corresponding absorbance spectra (457 nm) were recorded using a UV/Vis spectrophotometer (Blue Star A, US).
2.5. Detection of H2O2, ˙O2− and ˙OH
H2O2 was analyzed photometrically by the peroxidase-catalyzed oxidation product of N,N-diethyl-p-phenylenediamine,23,24 which was measured using a UV/Vis spectrophotometer (Blue Star A, US) at 551 nm.31 Superoxide radical (˙O2−) was measured by 2,3-bis(2-methoxy-4-nitro-5-sulfophehyl)-2H-tet-razolium-5-carboxanilide (XTT),35–37 which can be reduced by ˙O2− to form XTT-formazan. The formazan has an absorption spectrum (measured by a UV/Vis spectrophotometer (Blue Star A, US)) with a peak at 470 nm that can be used to quantify the relative amount of ˙O2−. Terephthalic acid (TA) is a well-known probe to detect ˙OH due to its high reactivity with ˙OH. The reaction between TA and ˙OH results in the formation of 2-hydroxy-terephthalic acid38 which can be detected using an HPLC-FLD system.39 The steady state concentration of the ˙OH can be calculated using the following eqn (3) and (4):40| | | ˙OH + Terephthalic acid (TA) → 2-hydroxyterephthalic acid (2HTA) | (3) |
| | 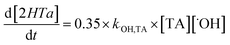 | (4) |
where k = 3.3 × 109 M−1 s−1.
3. Results and discussions
3.1. Synthesis and characterization of Cu2O NPs
Cu2O can be produced by a chemical deposition method29 described in eqn (5)–(7):| | | 2Cu2+ + 4S2O32− = 2[CuS2O3]− + [S4O6]2− | (5) |
| | | [CuS2O3]− ↔ Cu+ + S2O32− | (6) |
| | | 2Cu+ + OH− → Cu2O + H+ | (7) |
In a typical procedure, NaOH, CuSO4·5H2O and Na2S2O3 were introduced as source materials, in which CuSO4·5H2O and Na2S2O3 were used as the precursors of the Cu2O and reducing agent, respectively. The formation of Cu2O can be expressed as the reaction of free Cu+ ions with OH− ions (eqn (7)). In this work, this deposition technique was improved and an effective protocol was developed for size-controlled preparation of the Cu2O NPs. In the improved deposition method, a relatively high concentration of NaOH (5 M) was used to suppress the agglomeration of Cu2O NPs due to the generation of strongly electrostatic repulsion amongst the Cu2O NPs. Different sizes of Cu2O NPs were successfully prepared by simply adjusting the concentration of Cu(II) in eqn (5). Here, the concentrations of CuSO4·5H2O as the precursor of Cu2O were adjusted ranging from 0.05 to 2 M. Fig. 1A–D show the representative SEM images of the Cu2O NPs prepared with concentrations of 0.1, 0.6, 1 and 1.2 M, respectively. The obtained Cu2O NPs were uniform in size and their size distributions are shown in Fig. S3.† The corresponding sizes are around 10, 50, 100 and 200 nm, and so they were named Cu2O-10, Cu2O-50, Cu2O-100 and Cu2O-200, respectively. Increasing the concentration of precursor significantly increased the size of the Cu2O NPs (Fig. 1A–D). However, further increasing the concentration of the precursor did not contribute to an increased size of the Cu2O NPs. This is due to excessive copper precursor counteracting the function of NaOH for suppression of the agglomeration of the Cu2O NPs, resulting in the agglomeration of Cu2O NPs (Fig. 1D). A much more serious agglomeration of Cu2O NPs was observed when using 2 M of the copper precursor (Fig. S4†) due to interconnection of these Cu2O NPs to form irregular structures. This result was expected and is similar to that of our previous report.29 On the other hand, an excessively low concentration of precursor failed to achieve smaller Cu2O NPs (less than 10 nm). The SEM image of the Cu2O NPs (not shown here) prepared with 0.05 M of precursor is similar to that with 0.1 M of precursor. The results suggest that 10 nm is close to the size of the crystal nucleus for the formation of Cu2O NPs in the current synthesis conditions.
 |
| | Fig. 1 SEM images of Cu2O NPs prepared with (A) 0.1, (B) 0.6, (C) 1 and (D) 1.2 M of Cu(II) ion solution. The concentration of NaOH solution was fixed at 5 M. | |
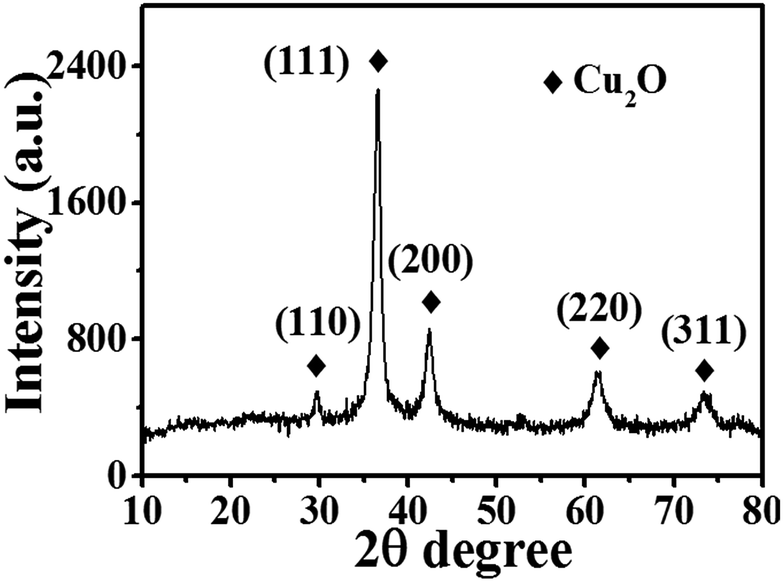
Fig. 2 shows the XRD pattern of Cu2O-10. Five peaks at 2θ = 29.78°, 36.56°, 42.39°, 62.51°, and 73.46° can be indexed to the (110), (111), (200), (220) and (311) planes of the Cu2O cubic phase, which is very close to the values in the JCPDS – International Centre for Diffraction Data (PDF, Powder Diffraction File, no. 05-0667, 1996). No other peaks suggesting impurities, or significant changes in the peak locations of the XRD spectra were detected indicating that pure Cu2O powders were obtained under the current synthetic conditions. The high intensity of the (111) diffraction peak suggests that the obtained crystals are mainly dominated by (111) facets. The XRD spectra of the other Cu2O samples were as good as that of Cu2O-10 (data not shown here), indicating that variation of the Cu(II) ion concentration does not change the phase composition of the Cu2O NPs. The average size of the nanocrystals for these samples was estimated using the Debye–Scherrer formula from the full width at half maximum (FWHM) of the diffraction peaks. According to the FWHM of the diffraction peaks of the (111) plane of Cu2O, the average size of the Cu2O crystals was determined to be about 7.1 nm.
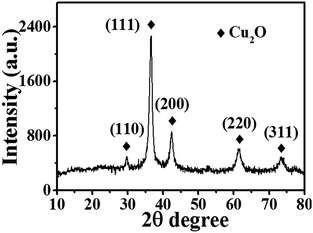 |
| | Fig. 2 XRD pattern of Cu2O-10 NPs. | |
TEM images of Cu2O-10, Cu2O-50, Cu2O-100 and Cu2O-200 are shown in Fig. 3A–D, and display almost the same sizes as the corresponding SEM data. HRTEM was also carried out to further investigate the crystal structure of the Cu2O NPs. Fig. 3E is a typical HRTEM image of Cu2O-10. The Fourier transform pattern [Fig. 3E, inset (I)] corresponding to the red square region with two-dimensional lattice fringes indicates that Cu2O-10 was composed of small single crystals with sizes of 5–10 nm. Fig. 3F shows the bright field of the HRTEM image for an individual nanosphere of Cu2O-200. The non-uniform contrast of the HRTEM image suggests that the large nanosphere is also assembled with small crystals with sizes of about 5–10 nm (Fig. 3F inset). The results suggest that the sizes of the obtained Cu2O NPs are different, but they are all composed of small single crystals with sizes of 5–10 nm, which is in good agreement with the size data (7.1 nm) obtained from the XRD analysis. In addition, the circle corresponding to the (111) plane is much brighter than the others in the selected area electron diffraction pattern (Fig. 3E, inset (II)), indicating that the formation of the Cu2O NPs is highly orientated to the (111) plane and is also in agreement with the data of the XRD analysis.
 |
| | Fig. 3 TEM images of (A) Cu2O-10, (B) Cu2O-50, (C) Cu2O-100 and (D) Cu2O-200, and HRTEM images of (E) Cu2O-10 and (F) Cu2O-200. Insets (I) and (II) are the Fourier transform pattern corresponding to the red square region and the selected-area electron diffraction pattern corresponding to the whole of image E, respectively. | |
3.2. Comparison of bacterial inactivation by Cu2O NPs with different sizes in dark
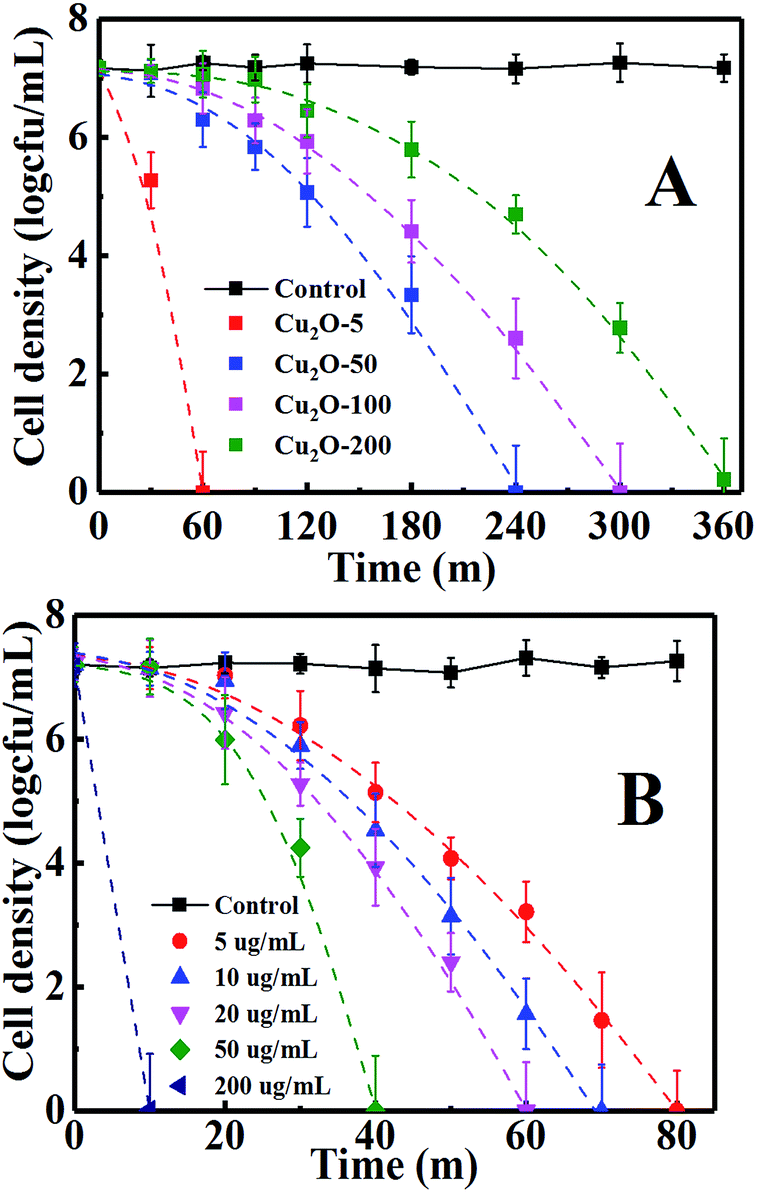
The antibacterial activity and inactivation kinetics of E. coli in the dark by the obtained Cu2O NPs are shown in Fig. 4A. Control experiments confirmed that E. coli was not inactivated only in the dark control. A cell loss of about 2 × 107 was achieved for 20 µg mL−1 of Cu2O-10, Cu2O-50, Cu2O-100 and Cu2O-200 within 60, 240, 300 and 360 min, respectively, indicating that an increase in the size of the Cu2O NPs resulted in a decrease in the antibacterial activity. They are characterized by the convex curves (dashed lines of Fig. 4A) of the Weibull model whose parameters are given in Table S1(a).† The inactivation kinetics of E. coli by the Cu2O NPs in the dark are in good agreement with those of the thermal inactivation of microbial cells which can also be described by the Weibull model.41,42 The variation of the scale parameter δ (time) indicates a negative correlation between the inactivation effects and the sizes of the Cu2O NPs. The dimensionless parameter p denotes the shape of the fitted curves. The dashed lines of Fig. 4A are visualized as convex curves (p > 1, Table S1(a)†), indicating that the remaining cells became increasingly damaged.41
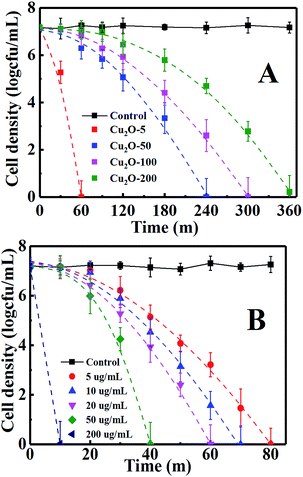 |
| | Fig. 4 Inactivation of E. coli by (A) 20 µg mL−1 of Cu2O-10, Cu2O-50, Cu2O-100 and Cu2O-200, respectively, in the dark and by (B) different amounts of Cu2O-10. The log inactivation was plotted versus the time with dashed lines. | |
3.3. Comparison of bacterial inactivation by Cu2O-10 with different concentrations in the dark
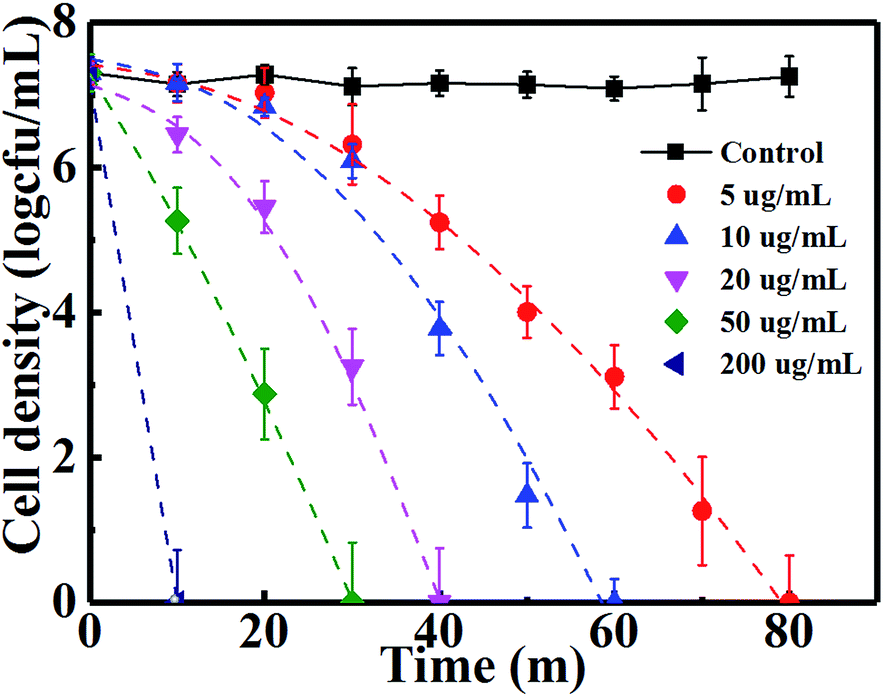
The antibacterial effects were further studied for different concentrations of Cu2O-10 NPs, whose antibacterial activity and inactivation kinetics are recorded in Fig. 4B. Complete inactivation of 7-log E. coli by 5, 10, 20, 50, and 200 µg mL−1 of Cu2O-10 was observed within 80, 70, 60, 40 and 10 min, respectively. We also determined the minimum inhibitory concentration (MIC) for Cu2O-10 to be ≤1 µg mL−1, which is much lower than the 6.25,18 100,21 and 1270 (ref. 43) µg mL−1 reported for the MICs of E. coli in cited references. The inactivation kinetics (dashed lines of Fig. 4B) of E. coli were also fitted with the Weibull model (Table S1(b)†). The scale parameters p > 1 and δ decreased with an increase in the Cu2O content (Table S1(b)†) indicating that the remaining cells became increasingly damaged41 and that a high Cu2O content resulted in a high inactivation efficiency.
3.4. Photocatalytic inactivation activity
Fig. 5 shows the photocatalytic inactivation of E. coli by Cu2O-10 with different concentrations. Control experiments indicated that E. coli was not inactivated only under the light control. Under VL the antibacterial activity of Cu2O-10 with a medium concentration (10, 20 and 50 µg mL−1) was substantially improved, indicating that Cu2O-10 can be driven by VL and enjoys photocatalytic antibacterial activity. However, there was no significant difference in antibacterial activity between the VL irradiation and dark conditions for both low (5 µg mL−1) and high (200 µg mL−1) concentrations of Cu2O-10. These results suggest that the inactivation of E. coli is dominated by its own antibacterial activity and its photocatalytic antibacterial activity is negligibly small when a low or high concentration of Cu2O-10 is used. The inactivation kinetics of Cu2O-10 with different concentrations under VL can all be fitted into the convex curves of the Weibull model (Table S2†). The very slight variation of the parameters p and δ (see and compare the rows highlighted with red in Tables S1(a) and S2†) further suggests that the antibacterial activity of Cu2O was dominated by the toxicity of Cu2O.
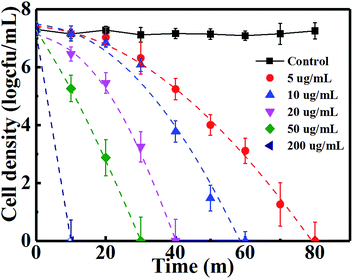 |
| | Fig. 5 Inactivation of E. coli by different amounts of Cu2O-10 under VL. | |
3.5. Mechanisms of inactivation of E. coli by Cu2O NPs
3.5.1 Mechanisms of inactivation of E. coli by Cu2O NPs in the dark.
The origin of the antibacterial activities of Cu2O in the dark was first investigated by Sunada et al.34 who proposed that contact with the surface of Cu2O caused denaturation or degradation of bacteria, resulting in their inactivation. This so-called “contact killing” was widely adopted by many researchers as a mechanism for the antibacterial activities of Cu2O.22,34,44 Meghana et al.45 further proved that the cell envelope of bacteria initially suffered severe damage from Cu2O, which added strong support to the contact killing mechanism. Here, we agree with the contact killing mechanism according to the results of the antibacterial testing of Cu2O-10 under aerobic and anaerobic conditions. As we know, reactive oxygen species (ROS) can be generated under aerobic conditions, but cannot exist in anaerobic conditions. Fig. S5† shows that Cu2O-10 displayed the same inactivation efficiency in aerobic conditions (pink line) as in anaerobic conditions (blue line), suggesting that E. coli was inactivated by contact killing due to exclusion of the effect of ROS on the inactivation of the bacteria. In this case, the more contact between the Cu2O-10 and bacterial cells, the higher the antibacterial activity was. Thus, it is expected that Cu2O NPs with larger BET surface areas contribute a higher antibacterial activity. We determined the BET surface areas of Cu2O-10, Cu2O-50, Cu2O-100 and Cu2O-200 to be 26.24, 6.467, 4.421 and 3.228 m2 g−1, respectively, which can demonstrate well that decreasing the size of the Cu2O NPs results in increasing antibacterial activity. Similar size-dependent antibacterial activity was reported by B. Ajitha et al. who observed that the smallest size of Ag NPs displayed the highest antibacterial activity.46
3.5.2 Mechanisms of the inactivation of E. coli by Cu2O NPs under VL.
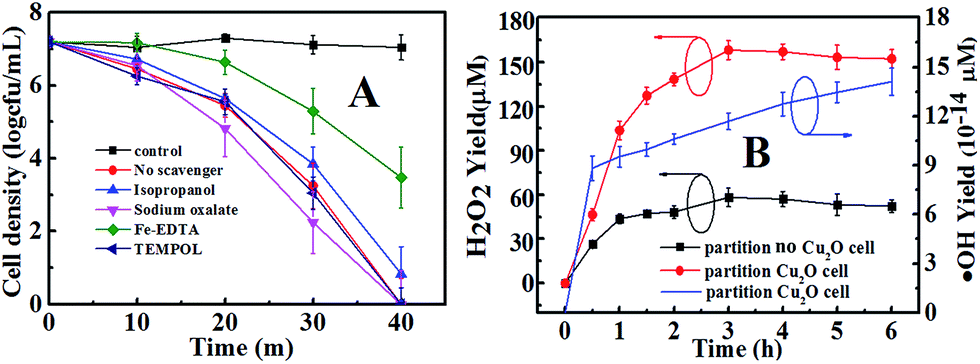
Under VL, Cu2O acts as a photocatalyst. Photocatalysis is known to produce several ROS (˙OH, H2O2, ˙O2−, and h+) that are potentially involved in the inactivation of bacterial cells.47 The roles of ROS for the photocatalytic inactivation of E. coli by Cu2O-10 are clarified according to the scavenging study and testing results for ROS shown in Fig. 6. Scavengers are widely used to determine the bactericidal contribution of each reactive species, including sodium oxalate for h+, isopropanol for ˙OH, TEMPOL for ˙O2− and Fe(II)–EDTA for H2O2.48–50 Before conducting the experiments, the applied concentration of each scavenger was optimized to ensure its maximum scavenging effect without causing any inactivation to the bacterial cell.48–50 As discussed above, 20 µg mL−1 of Cu2O-10 can inactivate 7-log of E. coli within 40 min. In this case, no scavenger was involved. With the addition of Fe(II)–EDTA (H2O2 scavenger) or isopropanol (˙OH scavenger), the inactivation of E. coli was partly prohibited, suggesting that both H2O2 and ˙OH are primary ROS associated with the bacterial inactivation process. H2O2 and ˙OH are generated through either an oxidative process or a reductive process. In an oxidative process, a hole can abstract electron from water and/or hydroxyl ions to produce ˙OH and H2O2 (eqn (8) and (9)).
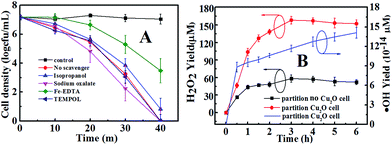 |
| | Fig. 6 (A) Photocatalytic inactivation of E. coli by Cu2O-10 under VL irradiation with different scavengers (50 mmol L−1 sodium oxalate, 0.5 mmol L−1 isopropanol, 0.1 mmol L−1 Fe(II)–EDTA, and 2 mmol L−1 TEMPOL), and (B) H2O2 and ˙OH yields in a partition system. The concentration of Cu2O-10 was 20 µg mL−1. | |
However, the hole from the valence band of Cu2O is produced at about +0.6 V potential, and no overpotential is available for the oxidation of water (about +0.57 V, eqn (9)).28,51 It also cannot oxidize H2O and OH− to form H2O2 (+1.763 V) and ˙OH (about +2.8 V), respectively. Thus, the origin of H2O2 and ˙OH should be the electron from the conduction band of Cu2O by a possible reductive process (eqn (10)–(13)).29
| | | O2 + 2H2O + 2e− → H2O2 + 2OH− | (11) |
| | | ˙O2− + H2O2 → ˙OH + OH− + O2 | (12) |
| | | H2O2 + e− → ˙OH + OH− | (13) |
It was observed that the addition of TEMPOL (˙O2− scavenger) did not significantly prohibit the inactivation of E. coli, suggesting that ˙O2− was not involved in the bacterial inactivation. From Fig. S6,† one can see that no ˙O2− signal was detected due to the absence of an absorption peak (470 nm), further confirming that ˙O2− was not produced in the current conditions. In this case, the electrons followed the reductive process of eqn (11) and (13), through which H2O2 and ˙OH were generated.
As mentioned above, h+ is not involved in an oxidative process for the evolution of ROS. However, whether h+ can directly inactivate E. coli or not needs to be clarified. Thus, sodium oxalate was used as a scavenger to determine the bactericidal contribution of h+. Interestingly, the inactivation process was enhanced a bit (Fig. 6A) with the addition of sodium oxalate. This can be explained by the removal of h+ by sodium oxalate enhancing the h+–e− separation efficiency, thus kinetically favoring the remaining e− to form more ROS to inactivate cells in the system.52 To eliminate the influence of e− from the conduction band of Cu2O, argon (Ar) was aerated to remove the dissolved O2 in solution and hence prohibit the formation of ROS, leaving only the function of h+ from the valence band. Control experiments with Ar in the dark and under VL (black and red curves in Fig. S5†) confirm that E. coli was not inactivated. In this case, the addition of sodium oxalate gave rise to the decrease in the inactivation efficiency (cyan and purple curves in Fig. S5†). This result indicates that when removing only the h+ inactivation is inhibited, proving that h+ on its own can directly inactivate the bacterial cells. Moreover, comparing the blue and green curves in Fig. S5,† one can conclude that the h+ can directly kill the bacterial cells since Cu2O-10 can more significantly inactivate E. coli under anaerobic conditions (green curve) than aerobic conditions (blue curve). Concerning holes produced at about +0.6 V, the holes can directly inactivate E. coli by electrochemically oxidizing co-enzyme A (CoA) in the cell wall to dimeric CoA, resulting in the death of E. coli.53
3.6. The role of H2O2
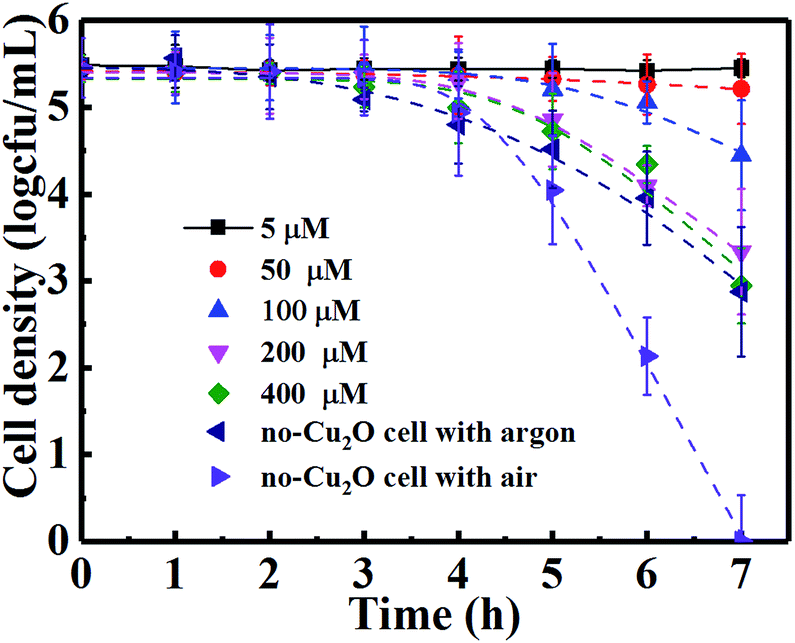
The leading bactericidal effect of H2O2 rather than ˙OH was expected because H2O2 production is thermodynamically favored by directly reducing O2 to H2O2 (eqn (11)) while ˙OH is produced through a second-order reaction (eqn (13)). This can be partly supported by the scavenging study data in Fig. 6. The addition of isopropanol inhibited the bacterial inactivation efficiency (over 6-log reduction) a little while the corresponding efficiency was greatly inhibited (only 1-log reduction) upon the addition of Fe(II)–EDTA, indicating that H2O2 is much more deeply involved in the disinfection than ˙OH. To explore the role of H2O2, we conducted a photocatalytic inactivation of E. coli using a partition system (Fig. S2†) and quantitatively assessed the H2O2 yield. The partition system was used to block direct contact between the bacterial cell and the Cu2O, and excluded the function of toxicity of the Cu2O, ˙OH and h+. Only H2O2 can diffuse across the membrane due to the long life of H2O2,31 which is convenient for independent investigations on the inactivation of E. coli by H2O2. Fig. 6B shows that the H2O2 yields in both cells of the partition system increased at first and became stable over a prolonged time, due to the decomposition of H2O2 in parallel with its production.54 The equilibrium concentrations of H2O2 in the Cu2O cell and no-Cu2O cell of the partition system were estimated to be 150 and 50 µM, respectively, which are much higher than the corresponding data (7.28 and 2.56 µM) from our previous Cu2O film partition system.31 The results indicate that Cu2O-10 is a photocatalyst capable of highly-efficient H2O2 evolution. This ability of Cu2O-10 for highly efficient H2O2 evolution is probably due to its high BET surface area and crystal structure with a high oriented (111) plane exhibiting higher photocatalytic activity than the other faces.55,56 We observed that the 5-log E. coli was completely inactivated in the no-Cu2O cell of the partition system within 7 h (Fig. 7). The inactivation efficacy of H2O2 in the no-Cu2O cell was also confirmed by the BacLight kit fluorescent microscopic method (Fig. S7†). To investigate the dependence of the concentration of H2O2 on the antibacterial effect, we recorded the data of the inactivation of E. coli by direct addition of different concentrations of H2O2 (50–800 µM). A direct addition of H2O2 (50 µM) did not have significant antibacterial effect (Fig. 7). Note that the total amount of H2O2 was much higher than the actual measured value of 50 µM as H2O2 was generated continuously and dynamically consumed in situ in the system. Thus, the actual amount of H2O2 available to interact with the bacterial cell was much higher than the amount detected.46 We observed that the equilibrium concentration of H2O2 was as high as 50 µM in the no-Cu2O cell contributing to a similar inactivation efficiency to H2O2 (800 µM) (not shown in Fig. 7) by direct addition. The inactivation kinetics (dashed lines of Fig. 7) of E. coli by H2O2 in different conditions can be fitted into the shoulder-linear-tail model (eqn (2)). The rate of inactivation (Kmax) increased and the shoulder length (Sl) decreased when the applied H2O2 concentration increased (Table S3†). Notably, the Kmax of inactivation by 800 µM H2O2 (7.46 h−1) is comparable to that of 50 µM H2O2 (7.49 h−1) under VL.
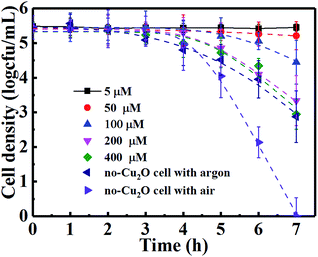 |
| | Fig. 7 Inactivation of E. coli by different amounts of H2O2 and in a no-Cu2O cell of the partition system under VL irradiation. | |
3.7. The role of copper ions
Copper ion leakage from Cu2O may contribute to the inactivation of E. coli because it is toxic at high concentrations31,57 and underlying Fenton-like reactions (eqn (14) and (15)) produce ROS to inactivate E. coli.57,58| | | Cu(I) + O2 → Cu(II) + ˙O2− | (14) |
| | | Cu(I) + ˙O2− + 2H+ → Cu(II) + H2O2 | (15) |
Fig. S8† shows the copper ion content, including the Cu+ and Cu2+ leakage from a 20 µg mL−1 Cu2O-10 suspension under irradiation and in the dark as a function of time. The copper ion leakage from Cu2O-10 increased quickly in the first 2 h and slowly later, but with a low total leakage rate. After 7 h of VL irradiation, 0.34 mg L−1 of total copper ion (including 0.26 mg L−1 Cu2+ and 0.08 mg L−1 Cu+) and 0.29 mg L−1 total copper ion (including 0.22 mg L−1 Cu2+ and 0.07 mg L−1 Cu+) were present under VL and in the dark, respectively. The low concentration of copper ion leakage from Cu2O-10 did not contribute to the antibacterial activity.31 In addition, since ˙O2− and ˙OH were not detected, the copper-catalyzed Haber–Weiss reaction was probably too weak to be observed under the current experimental conditions. In all, the copper ion leakage from Cu2O-10 did not contribute to the inactivation of E. coli.
4. Conclusions
In this study, a simplified and green approach for the size-controlled synthesis of Cu2O nanoparticles (NPs) was achieved by chemical deposition technology. The obtained Cu2O NPs presented size-dependent antibacterial activity, i.e. decreasing the size of the Cu2O NPs resulted in increasing the antibacterial activity. The smallest size of Cu2O-10 had a relatively low C of ≤1 µg mL−1 and enjoyed the highest antibacterial activity which could completely inactivate 7-log E. coli in 80 min with a concentration of 5 µg mL−1 in the dark. Under VL, the medium concentrations (10, 20 and 50 µg mL−1) of Cu2O-10 showed enhanced antibacterial activity compared with that in the dark, whereas low and high concentrations (5 and 200 µg mL−1) of Cu2O-10 presented almost the same antibacterial activity as that in dark. “Contact killing” and ROS (˙OH, H2O2 h+) were found to be responsible for bacterial inactivation in the dark and under irradiation, respectively. Cu2O-10 was proved to be effective for evolution of H2O2. A stable equilibrium concentration of H2O2 (as high as 150 µM) could be achieved by using 20 µg mL−1 Cu2O-10 under VL. The inactivation kinetics of the Cu2O NPs in different conditions and that of H2O2 were calculated and they were fitted into Weibull and shoulder-linear-tail models, respectively.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Science Foundation of Hubei Province (2016CFB511), the Hubei Provincial Department of Education (D20152702), and the National Natural Science Foundation of China (11547018, 11647122, and 61705064). Dr Chen appreciates the support of the Project of the Open Fund of Hubei Key Laboratory of Ferro & Piezoelectric Materials and Devices (201605), the Outstanding Young and Middle-aged Innovation Team of Hubei (T201521) and the Major Project of Hubei Polytechnic University (16xjz03A).
References
- A. S. Zoolfakar, R. A. Rani, A. J. Morfa, A. P. O’mullane and K. Kalantar-Zadeh, J. Mater. Chem. C, 2014, 2, 5247 RSC.
- W. Chen, W. Zhang, L. Chen, L. Zeng and M. Wei, J. Alloys Compd., 2017, 723, 172 CrossRef CAS.
- F. Wu, S. Banerjee, H. Li, Y. Myung and P. Banerjee, Langmuir, 2016, 32, 4485 CrossRef CAS PubMed.
- M. Pang and H. C. Zeng, Langmuir, 2010, 26, 5963 CrossRef CAS PubMed.
- D. Nunes, A. Pimentel, P. Barquinha, P. A. Carvalho, E. Fortunato and R. Martins, J. Mater. Chem. C, 2014, 2, 6097 RSC.
- K. X. Yao, X. M. Yin, T. H. Wang and H. C. Zeng, J. Am. Chem. Soc., 2010, 132, 6131 CrossRef CAS PubMed.
- A. Mishra and D. Pradhan, Cryst. Growth Des., 2016, 16, 3688 CAS.
- M. Leng, M. Liu, Y. Zhang, Z. Wang, C. Yu, X. Yang, H. Zhang and C. Wang, J. Am. Chem. Soc., 2010, 132, 17084 CrossRef CAS PubMed.
- Q. Wang, Y. Jia, M. Wang, W. Qi, Y. Pang, X. Cui, W. Ji and J. Yi, J. Phys. Chem. C, 2015, 119, 22066 CAS.
- H. Xu, W. Wang and L. Zhou, Cryst. Growth Des., 2008, 8, 3486 CAS.
- C. H. Kuo and M. H. Huang, J. Am. Chem. Soc., 2008, 130, 12815 CrossRef CAS PubMed.
- C. Tan, S. Hsu, W. Ke, L. Chen and H. Huang, Nano Lett., 2015, 15, 2155 CrossRef CAS PubMed.
- Y. Xu, H. Wang, Y. Yu, L. Tian, W. Zhao and B. Zhang, J. Phys. Chem. C, 2011, 115, 15288 CAS.
- W. Ke, C. Hsia, Y. Chen and H. Huang, Small, 2016, 12, 3530 CrossRef CAS PubMed.
- L. Kiaune and N. Singhasemanon, Rev. Environ. Contam. Toxicol., 2011, 213, 1 CAS.
- R. P. Allaker and K. Memarzadeh, Int. J. Antimicrob. Agents, 2014, 43, 95 CrossRef CAS PubMed.
- W. Fan, X. Wang, M. Cui, D. Zhang, Y. Zhang, T. Yu and L. Guo, Environ. Sci. Technol., 2012, 46, 10255 CrossRef CAS PubMed.
- H. Pang, F. Gao and Q. Lu, Chem. Commun., 2009, 1076 RSC.
- J. Ren, W. Wang, S. Sun, L. Zhang, L. Wang and J. Chang, Ind. Eng. Chem. Res., 2011, 50, 10366 CrossRef CAS.
- K. Giannousi, G. Sarafidis, S. Mourdikoudis, A. Pantazaki and C. Dendrinou-Samara, Inorg. Chem., 2014, 53, 9657 CrossRef CAS PubMed.
- M. A. Vargas-Reus, K. Memarzadeh, J. Huang, G. G. Ren and R. P. Allaker, Int. J. Antimicrob. Agents, 2012, 40, 135 CrossRef CAS PubMed.
- M. Hans, A. Erbe, S. Mathews, Y. Chen, M. Solioz and F. Mucklich, Langmuir, 2013, 29, 16160 CrossRef CAS PubMed.
- J. Hrenovic, J. Milenkovic, N. Daneu, R. M. Kepcija and N. Rajic, Chemosphere, 2012, 88, 1103 CrossRef CAS PubMed.
- C. H. Kuo, Y. C. Yang, S. Gwo and M. H. Huang, J. Am. Chem. Soc., 2011, 133, 1052 CrossRef CAS PubMed.
- A. Singhal, M. R. Pai, R. Rao, K. T. Pillai, I. Lieberwirth and A. K. Tyagi, Eur. J. Inorg. Chem., 2013, 2013, 2640 CrossRef CAS.
- S. Kakuta and T. Abe, Electrochem. Solid-State Lett., 2009, 12, P1 CrossRef CAS.
- M. K. I. Senevirathna, P. K. D. D. P. Pitigala and K. Tennakone, J. Photochem. Photobiol., A, 2005, 171, 257 CrossRef CAS.
- P. E. De Jongh, D. Vanmaekelbergh and J. J. Kelly, J. Electrochem. Soc., 2000, 147, 486 CrossRef CAS.
- L. Xiong, H. Yu, G. Yang, M. Qiu, J. Chen and Y. Yu, Thin Solid Films, 2010, 518, 6738 CrossRef CAS.
- L. Huang, F. Peng, H. Yu and H. Wang, Solid State Sci., 2009, 11, 129 CrossRef CAS.
- L. Xiong, T. W. Ng, Y. Yu, D. Xia, H. Y. Yip, G. Li, T. An, H. Zhao and P. K. Wong, Electrochim. Acta, 2015, 153, 583 CrossRef CAS.
- Y. Zhang, L. Ma, J. Li and Y. Yu, Environ. Sci. Technol., 2007, 41, 6264 CrossRef CAS PubMed.
- A. H. Geeraerd, C. H. Herremans and J. F. Van Impe, Int. J. Food Microbiol., 2000, 59, 185 CrossRef CAS PubMed.
- K. Sunada, M. Minoshima and K. Hashimoto, J. Hazard. Mater., 2012, 235–236, 265 CrossRef CAS PubMed.
- M. W. Sutherland and B. A. Learmonth, Free Radical Res., 1997, 27, 283 CrossRef CAS PubMed.
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164 CrossRef CAS PubMed.
- L. Brunet, D. Y. Lyon, E. M. Hotze, P. J. J. Alvarez and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4355 CrossRef CAS PubMed.
- K. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, J. Photochem. Photobiol., A, 2000, 134, 139 CrossRef CAS.
- H. Xu, W. J. Cooper, J. Jung and W. Song, Water Res., 2011, 45, 632 CrossRef CAS PubMed.
- D. Zhang, S. Yan and W. Song, Environ. Sci. Technol., 2014, 48, 12645 CrossRef CAS PubMed.
- M. a. J. S. Van Boekel, Int. J. Food Microbiol., 2002, 74, 139 CrossRef PubMed.
- M. Peleg and M. B. Cole, Crit. Rev. Food Sci. Nutr., 1998, 38, 353 CrossRef CAS PubMed.
- M. Turalija, P. Merschak, B. Redl, U. Griesser, H. Duelli and T. Bechtold, J. Mater. Chem. B, 2015, 3, 5886 RSC.
- G. S. Theja, R. C. Lowrence, V. Ravi, S. Nagarajan and S. P. Anthony, CrystEngComm, 2014, 16, 9866 RSC.
- S. Meghana, P. Kabra, S. Chakraborty and N. Padmavathy, RSC Adv., 2015, 5, 12293 RSC.
- B. Ajitha, Y. Reddy, P. Reddy, H. Jeon and C. Ahn, RSC Adv., 2016, 6, 36171 RSC.
- W. Wang, L. Zhang, T. An, G. Li, H. Yip and P. Wong, Appl. Catal., B, 2011, 108–109, 108 CrossRef CAS.
- Y. Chen, A. Lu, Y. Li, L. Zhang, H. Y. Yip, H. Zhao, T. An and P. K. Wong, Environ. Sci. Technol., 2011, 45, 5689 CrossRef CAS PubMed.
- W. Wang, Y. Yu, T. An, G. Li, H. Y. Yip, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2012, 46, 4599 CrossRef CAS PubMed.
- L. S. Zhang, K. H. Wong, D. Q. Zhang, C. Hu, J. C. Yu, C. Y. Chan and P. K. Wong, Environ. Sci. Technol., 2009, 43, 7883 CrossRef CAS PubMed.
- P. E. De Jongh, D. Vanmaekelbergh and J. J. Kelly, Chem. Commun., 1999, 1069 RSC.
- D. Xia, Z. Shen, G. Huang, W. Wang, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2015, 49, 6264 CrossRef CAS PubMed.
- T. Matsunaga, Y. Namba and T. Nakajima, Bioelectrochem. Bioenerg., 1984, 13, 393 CrossRef.
- F. Shiraishi and C. Kawanishi, J. Phys. Chem. A, 2004, 108, 10491 CrossRef CAS.
- L. Feng, C. Zhang, G. Gao and D. Cui, Nanoscale Res. Lett., 2012, 7, 1 CrossRef PubMed.
- H. Xu, W. Wang and W. Zhu, J. Phys. Chem. B, 2006, 110, 13829 CrossRef CAS PubMed.
- H. J. Park, T. T. Nguyen, J. Yoon and C. Lee, Environ. Sci. Technol., 2012, 46, 11299 CrossRef CAS PubMed.
- N. Perez-Almeida, M. Gonzalez-Davila, J. M. Santana-Casiano, A. G. Gonzalez and M. Suarez De Tangil, Environ. Sci. Technol., 2013, 47, 1239 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10605j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Huaqing
Yu
*a,
Changjiang
Nie
*a,
Huaqing
Yu
*a,
Changjiang
Nie