
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual redox-responsive PEG–PPS–cRGD self-crosslinked nanocapsules for targeted chemotherapy of squamous cell carcinoma†
Jianjun Zhang *a,
Yunxia Lib,
Jiexin Wanga,
Shengpei Qia,
Xiaoqing Songa,
Cheng Taoa,
Yuan Lea,
Ning Wen*b and
Jianfeng Chena
*a,
Yunxia Lib,
Jiexin Wanga,
Shengpei Qia,
Xiaoqing Songa,
Cheng Taoa,
Yuan Lea,
Ning Wen*b and
Jianfeng Chena
aCollege of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: zhangjj@mail.buct.edu.cn
bDepartment of the Prosthodontics, The General Hospital of Chinese PLA, Beijing 100853, P. R. China. E-mail: wenningchn@163.com
First published on 21st November 2017
Abstract
Polymer nanogels/nanocapsules with encapsulation stability, stimuli responsiveness and tumor targeting have emerged as one of the most remarkable carriers for anticancer drug delivery. In this work, we design a multifunctional, four-armed, branched copolymer, PEG–PPS–cRGD, and use it to develop a dual redox-responsive and αvβ3 integrin-targeting nanocapsule via a simple and straightforward self-crosslinking strategy through the disulfide exchange reaction between the polymer arms. The dissolution rate studies illustrate that the PEG–PPS–cRGD nanocapsules had robust drug release profiles under both oxidation and reduction conditions. The in vitro and in vivo investigations demonstrate that the nanocapsules exhibited precise tumor targeting, outstanding antitumor effect and excellent biological safety in the treatment of squamous cell carcinoma. Therefore, this work provides a promising drug delivery platform for cancer therapy and other applications.
Introduction
Over the past several decades, unprecedented numbers of studies have focused on the development of high-performance drug nanocarriers for cancer chemotherapy.1–4 Polymer nanocarriers, as a promising candidate, have attracted enormous attention. To date, various polymer nanocarriers have been explored, including amphiphilic copolymer micelles/polymersomes,5–7 polyester-based polymer nanoparticles,8–10 and polymer nanogels/nanocapsules.11–13 The polymer micelles may be able to carry the drugs but with poor stability, leading to serious side effects due to the premature leakage of the drugs during in vivo circulation.14,15 Alternatively, the polyester-based polymer nanoparticles, including polylactide (PLA), poly(ε-caprolactone) (PCL), and poly(lactide-co-glycolide) (PLGA), have high drug loading capacity and outstanding encapsulation efficiency, but the therapeutic applications of these polymer nanoparticles are hampered by the slow drug release rate and poor dispersibility in the aqueous phase due to the slow biodegradation rate and high hydrophobic properties of such a polymer main backbone.16,17 Recently, polymer nanogels/nanocapsules have emerged as one of the most remarkable drug delivery carriers to overcome the defects of polymer micelles and polyester-based polymer nanoparticles. However, the preparation of polymer nanogels was usually complicated because a series of monomers, crosslinkers and initiators must be introduced into the synthetic process, which are difficult to purify and may lead to serious toxicity and side effects in further treatment.18,19A variety of stimuli-responsive factors, such as acidity, light, magnetism, temperature, enzyme, reduction, and reactive oxygen species (ROS) have been utilized to design and fabricate smart drug delivery systems to improve the efficiency of the drugs in cancer therapy.20 Owing to the high level of reducing agents of glutathione (GSH) in intracellular fluids of cancer cells, the reduction-responsive nanocarriers containing disulfide bonds, which can be triggered to burst release the payload drugs by collapsing the original structures by dividing the disulfide bonds under the reduction environment, have been extensively explored for designing drug nanocarries.21–23 Otherwise, ROS is another important stimuli-responsive factor for anticancer drug delivery because that many tapes of cancer cells generate higher levels of ROS that can be used as a trigger to release the loading drugs by oxidating and degrading ROS-responsive carriers.24–26 The sulfur(II)-containing polymers such as poly(propylene sulfide) (PPS) have been used for fabricating ROS-responsive drug delivery systems due to it's hydrophobicity can be transformed into hydrophilicity upon oxidation conditions.27
Recently, star/branched polymers based on multi-arm PEG have been exploited for biomedical applications such as drug delivery and tissue engineering.28–30 The drug nanocarriers modified with PEG corona have multiple advantages including low toxicity, aqueous stability as well as long blood circulation.
In contrast with linear PEG, the multi-arm PEG of similar mass possess better water solubility and faster resolubilization rate due to high PEG densities, that makes these PEG more valuable for applying in drug delivery systems.31
The other main challenge in the applications of drug nanocarries for anticancer drug delivery is the low accumulation in tumor tissues.32 To date, there were so many research strategies focus on developing efficient drug nanocarries to overcome this fundamental limitation, such as fabricating worm-like nanoparticles or micelles,33,34 applying magnetic nanomaterials,35,36 and modifying targeting molecules.37,38 As we know, integrin plays a critical role in many types of the tumors,39,40 and therefore the peptide cyclic(Arg-Gly-Asp-D-Phe-Lys) (cRGD), which effectively bonds to tumor cells that overexpress αvβ3 integrins was often used for targeting anticancer drug delivery to promote tumor accumulation of the drugs.41
Herein, we use four-arm PEG as a water-soluble polymer backbone with PPS as a hydrophobic component and pyridine dithione as the end-capping agent to design a PEG–PPS amphiphilic branched copolymer terminated with protected disulfide bonds. The end-capping pyridine dithione groups were partly replaced to modify the targeting peptide thiol-cyclic(Arg-Gly-Asp-D-Phe-Lys) (cRGD–SH), and thereby synthesize PEG–PPS–cRGD copolymers through a disulfide exchange reaction. This PEG–PPS–cRGD branched copolymers can self-assemble into micelles via the physical association of the hydrophobic PPS blocks in water and can further self-crosslink to form nanocapsules via disulfide exchange reaction of the pyridine dithione groups under alkaline conditions.42 Furthermore, by using these multifunctional PEG–PPS–cRGD nanocapsules, we developed a smart, dual redox-responsive, drug-delivery system for the targeted chemotherapy of squamous cell carcinoma.
Our synthesis strategy is described in Fig. 1. First, doxorubicin (DOX) was chosen as model drug to encapsulate into PEG–PPS–cRGD by solvent–antisolvent method to form DOX-loaded PEG–PPS–cRGD nanocapsules, which self-crosslinked by disulfide bonds with initiation of triethylamine (TEA). Subsequently, DOX-loaded PEG–PPS–cRGD nanocapsules can effectively bond with αvβ3 integrins, which are overexpressed on the surface of squamous cells (SCC-15 cell-line), and further enter cells via the αvβ3-mediated endocytosis pathway.41 After the nanocapsules accumulate in tumor tissues and enter into the endocytosis, the high level of ROS can oxidate diethyl sulfide to swell the nanocapsules for releasing the loading DOX; further, the disulfide bonds in the nanocapsules will be cut off in GSH-rich cytoplasm to promote a second phase of loading drug release.43 Finally, the free DOX will diffuse into cell nucleus and intercalate on DNA to induce cell death. Moreover, in vitro studies demonstrated that the nanocapsules exhibit significant cytotoxicity against SCC-15 cells. The in vivo investigations proved that the nanocapsules can effectively target the tumor site in a xenograft model of squamous cell carcinoma in nude mice and strongly inhibit the tumor growth with high biological safety.
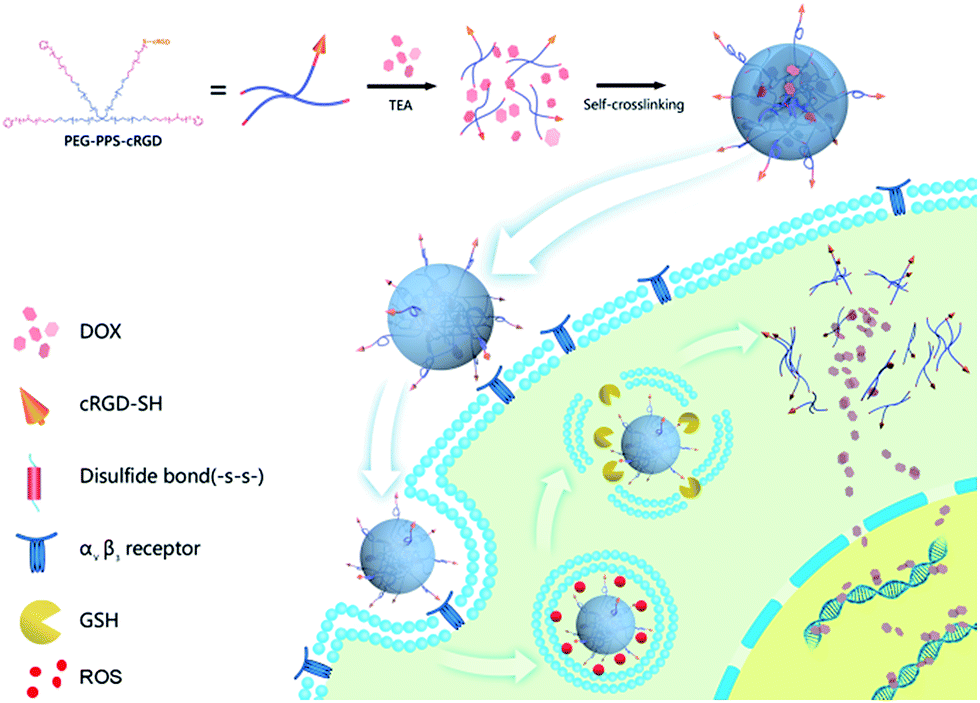 | ||
| Fig. 1 Schematic illustration showing the preparation and cellular uptake of DOX-loaded PEG–PPS–cRGD nanocapsules. I, DOX-loaded PEG–PPS–cRGD nanocapsules were prepared by a solvent–antisolvent process; II, these DOX-loaded PEG–PPS–cRGD nanocapsules can target cells via the αvβ3-integrin receptor; III, the nanocapsules undergo further uptake into cells by receptor-mediated endocytosis pathway; IV, the nanocapsules disassemble to release DOX cargo due to the oxidation and reduction conditions; V, released DOX enters into nucleus and intercalates on DNA to induce cell death. | ||
Experimental section
Materials
Sodium hydride (NaH), allyl bromide, thioacetic acid, 2,2′-azobisisobutyronitrile, sodium methoxide (NaOMe), propylene sulfide, and 2,2′-dithiodipyridine were purchased from Sigma-Aldrich. Four-arm poly(ethylene glycol) (PEG) (MW: 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Creative PEGWorks. Cyclic(Arg-Gly-Asp-D-Phe-Lys)-thioglycolic acid (cRGD–SH) and cyclic(Arg-Gly-Asp-D-Phe-Lys) (cRGD) were synthesized by Scilight Biotechnology, LLC. All other chemicals were purchased from Fisher Scientific unless otherwise noted.
000) was purchased from Creative PEGWorks. Cyclic(Arg-Gly-Asp-D-Phe-Lys)-thioglycolic acid (cRGD–SH) and cyclic(Arg-Gly-Asp-D-Phe-Lys) (cRGD) were synthesized by Scilight Biotechnology, LLC. All other chemicals were purchased from Fisher Scientific unless otherwise noted.
Instruments
Proton nuclear magnetic resonance (1H NMR) spectra were recorded at room temperature with a Bruker spectrometer (AV600) using CDCl3 as solvent. The particle size of the nanocapsules was determined using a Malvern ZetaSizer Nano instrument (ZS90). The morphology of the samples was examined using transmission electron microscopy (TEM, Tecnai 20). The drug-loading capacity and dissolution rate of DOX-loaded nanocapsules were calculated based on the absorbance value using a UV-visible spectrophotometer (Varian Cary 50).Synthesis of PEG–PPS and PEG–PPS–cRGD copolymers
Four-arm PEG–PPS amphiphilic branched copolymer was synthesized by three steps according to a previous work (Fig. S1†).42 Briefly, four-arm PEG (20 kDa) was dissolved in anhydrous THF and further reacted with allyl bromide by adding NaH to generate the alcoholate to synthesize PEG–allyl ether. Second, PEG–allyl ether was reacted with thioacetic acid via the initiation of AIBN in anhydrous degassed toluene to prepare PEG–thioacetate. Finally, the PEG–PPS copolymer was synthesized by open-ring polymerization. PEG–thioacetate was dissolved in degassed THF, and then NaOMe, propylene sulfide (PPS), and dipyridine dithione were sequentially added into reaction system. After the reaction, a PEG–PPS copolymer terminated with a capping group of pyridine dithione by disulfide bond was formed. All intermediate and final products were characterized by 1H NMR, and the data are shown in Fig. S2–S4.†PEG–PPS–cRGD copolymer was synthesized using cRGD–SH to partially substitute the capping group of PEG–PPS through a disulfide exchange reaction. PEG–PPS was dissolved in ultrapure water at a concentration of 0.5 wt%, and PBS buffer (150 mM, pH 7.4) was used to adjust the pH of the PEG–PPS solution to 7.4. Then, cRGD–SH was added into the PEG–PPS solution under vigorous stirring for 120 min at room temperature. The modification rate of cRGD on PEG–PPS was calculated by the absorbance change of reaction solution at wavelength of 342 nm (Fig. S5†).42
Preparation of DOX-loaded nanocapsules
Three milligrams of DOX·HCl and 2.5 μL of triethylamine (TEA) were dissolved in 0.5 mL of DMSO and mixed for 15 min under stirring to obtain the DOX base. Further, 5 mL of PEG–PPS or PEG–PPS–cRGD aqueous solution (0.5 wt%) was slowly dropped into the DOX solution. After adding PEG–PPS or PEG–PPS–cRGD, the reaction solution was stirred at 45 °C for another 2 h. Finally, the product was dialyzed against ultrapure water by dialysis bag (MWCO: 3500 Da) to remove unloaded DOX and DMSO. The DOX-loaded nanocapsules were then freeze dried.The stability test of DOX-loaded nanocapsules in both PBS and DMEM medium was studied by DLS. The DOX-loaded nanocapsules were dispersed in PBS (pH 7.4, 150 mM) or complete DMEM medium (DMEM + 10% serum + antibiotics) as final concentration of 0.2 mg mL−1, and the detection period was 48 h.
The drug-loading capacity (DLC) of the nanocapsules was determined using a UV-visible spectrophotometer with an absorption wavelength at 485 nm and calculated according to the formula: DLC (wt%) = (weight of loaded drug/weight of drug − loaded nanoparticles) × 100%.
In vitro DOX release from the nanocapsules
Each 1 mg of DOX-loaded PEG–PPS or PEG–PPS–cRGD nanocapsules was dispersed in 1 mL of PBS buffer and transferred into a dialysis bag with MWCO of 3500 Da. Then, this dialysis bag was placed into 10 mL of PBS buffer containing 500 μM of H2O2 or 10 mM of reduced glutathione (rGSH), and further incubated in water bath at 37 °C for 48 h. The cumulative release rate of DOX from the nanocapsules were calculated by determining the UV absorption of DOX at the wavelength of 485 nm.In vitro cellular uptake of the nanocapsules
Squamous carcinoma cell (SCC-15) cell line was obtained from American Type Culture Collection (ATCC). Cellular uptake of DOX-loaded PEG–PPS or PEG–PPS–cRGD nanocapsules was assessed via confocal laser scanning microscope (CLSM, LEICA, TCS-SP5, Germany) and flow cytometry (FCM, Beckman Coulter MoFloXDP). For CLSM imaging, SCC-15 cells were seeded in Lab-Tek chambered cover glass systems (8-wells) at a density of 7000 cells per well in 200 μL of complete DMEM medium, and cultured overnight at 37 °C in 5% CO2 atmosphere before use. Afterwards, the cells were incubated with 50 μL of DOX-loaded PEG–PPS or PEG–PPS–cRGD nanocapsules (final DOX concentration of 9 μg mL−1) for 4 h at 37 °C. After incubation, the cells were washed three times with PBS, and cell nuclei were further stained with 4′,6-diamidino-2-phenylindole (DAPI); the extent of internalization of the nanocapsules was visualized using a CLSM. For FCM analysis, 100000 cells in 2 mL of medium were plated per well in a 6-well plate and cultured overnight. After adding 500 μL of DOX-loaded PEG–PPS or PEG–PPS–cRGD nanocapsules for 4 h, cells were washed three times with PBS, trypsinized, centrifuged, re-suspended in PBS and analyzed using a FCM. A total of 10000 events was analyzed per sample.
To determine the effect of RGD receptor (αvβ3 integrin) on DOX-loaded PEG–PPS–cRGD nanocapsule uptake, cRGD peptide was added to the wells at a final concentration of 0.5 mM and pre-incubated with the cells for 30 min before adding the nanocapsules. Then, the cellular internalization was measured by CLSM and FCM, and the operation procedures were similar to those described in the section above.
In vitro MTT assay
The cytotoxicity of copolymer and DOX-loaded nanocapsules was assessed by using MTT assay. SCC-15 cells were plated into a 96-well plate at a cell density of 4000 cells per well in 100 μL of complete DMEM medium with 10% fetal bovine serum (FBS). The cells were cultured overnight at 37 °C in 5% CO2 before adding copolymer and DOX-loaded nanocapsules. Various concentrations of PEG–PPS, PEG–PPS–cRGD, free DOX, DOX-loaded PEG–PPS nanocapsules, and DOX-loaded PEG–PPS–cRGD nanocapsules were co-incubated with SCC-15 cells for 24 h at 37 °C. The polymer concentrations of PEG–PPS and PEG–PPS–cRGD copolymers ranged from 3.3 μg mL−1 to 330 μg mL−1. The free DOX, DOX-loaded PEG–PPS nanocapsules, and DOX-loaded PEG–PPS–cRGD nanocapsules were diluted in PBS with final DOX concentrations of 1.5 μg mL−1 to 18 μg mL−1. Further, 20 μL of MTT stock solution (5 mg mL−1) was added to the cell wells and incubated for another 4 h. Finally, the cell media were completely removed, and 100 μL of DMSO was added to the well to dissolve the formazan blue crystal. The absorbance of the solution was detected using a microplate reader (Thermo Fisher, MK3, America) at the wavelength of 570 nm. Cell viability was assessed via the formula:| Cell viability (%) = Asample/Acontrol × 100% |
In vitro live–dead assay
SCC-15 cells were seeded in Lab-Tek chambered cover glass systems (8-wells) at a density of 7000 cells per well in 200 μL of complete DMEM medium and cultured overnight at 37 °C in 5% CO2 atmosphere before use. Afterwards, the cells were incubated with 50 μL of free DOX and DOX-loaded nanocapsules (final DOX concentration of 18 μg mL−1) for 24 h at 37 °C. After incubation, the cells were washed three times with PBS and further stained with live/dead viability/cytotoxicity kit (Molecular Probes, Eugene, OR) using the manufacturer's protocol. Each gel was stained with 100 μL of the staining solution for 30 min at 37 °C in the dark and imaged with a CLSM.In vitro apoptosis assay
To determine whether the DOX-loaded nanocapsules induced apoptosis in tumor cells, an Annexin-V/propidium iodide (PI) double staining apoptosis detection assay (BD Biosciences, USA) was adopted. SCC-15 cells were collected after co-incubating with free DOX and DOX-loaded nanocapsules (final DOX concentration of 18 μg mL−1) for 48 h, and the cell concentration was adjusted to 1 × 106 cells per mL to make a single-cell suspension. Annexin-V/PI was used to stain the SCC-15 cells. The data were acquired on a FCM. All experiments were repeated three times (n = 3).44,45Furthermore, to further evaluate the in vitro apoptosis-inducing capabilities, western blot was also used to test the expression of cleaved caspase-3. SCC-15 cells were collected after co-incubating with free DOX and DOX-loaded nanocapsules (final DOX concentration of 18 μg mL−1) for 72 h. Protein extracts were prepared with RIPA lysis buffer and subjected to electrophoresis on 8% or 12% SDS-polyacrylamide gels and transferred to 0.45 μm nitrocellulose membranes. The membranes were stained with 0.2% Ponceau S Red to check equal protein loading and transfer. After blocking with 5% skimmed milk, the membranes were incubated with antibodies against caspase-3 overnight at 4 °C. The membranes were incubated with secondary antibody, GAPDH was used as the loading control, and the blots were visualized by UVITEC Cambridge western blotting detection reagents. All experiments were repeated three times (n = 3).45
In vitro hemolysis assay
To evaluate the hemolysis characteristics in vitro, distilled water (positive control group, abs100), saline (negative control group, abs0), PEG–PPS copolymers, and DOX-loaded nanocapsules were co-incubated with a 2% suspension of sheep red blood cells (Beijing Wobisen, China) in a water bath at 37 °C for 0.25, 0.5, 0.75, 1, 2, and 3 h, respectively. After incubation, the suspensions were centrifuged at 3000 rpm for 10 min, and the UV absorption of the supernatants was detected at the wavelength of 545 nm. The hemolysis rate was calculated using the following formula:| Hemolysis ratio = (abs − abs0)/(abs100 − abs0) × 100%. |
In vivo acute toxicity test
To evaluate the systemic toxicity in vitro, a limit test was adopted due to the physical characters and low toxicity of the nanocapsules. One hundred Kunming mice (Vital River, China) were randomly divided into 5 groups (each group contained 10 males and 10 females) for saline, free DOX, PEG–PPS copolymers, DOX-loaded PEG–PPS nanocapsules, and DOX-loaded PEG–PPS–cRGD nanocapsules, respectively. After intravenous administration of different samples (the same dose of DOX at 5 mg kg−1), mice were continuously observed for general behavioral changes, signs of toxicity and mortality for 1 h, then intermittently for 4 h, and thereafter over a period of 24 h. The mice were further observed and weighed for up to 14 days. The mice were sacrificed on the 14th day; 100 μL of whole blood was used for hematological determination (Siemens-Bayer ADVIA2120, German), which included white blood cell counts (WBC), lymphocyte counts (LYMPH), red blood cells (RBC), platelets (PLT), and 200 μL of plasma was used for biochemical examination (Toshiba Accute TBA-40FR, Japan), including glutamic-pyruvic transaminase (ALT), glutamic-oxalacetic transaminase (AST), creatinine (Cr), creatine kinase (CK) and creatine kinase MB (CK-MB), to evaluate the harm to the liver, kidney and heart. Heart, live, spleen, lung and kidney were embedded in paraffin and stained with hematoxylin–eosin (H&E) to observe histopathological changes.Animal model
All of the animal studies were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Beijing Vital River Laboratory Animal Technology Co., Ltd. and approved by the Institutional Animal Care and Use Committee of Beijing Vital River Laboratory Animal Technology Co., Ltd.In vivo pharmacokinetics
In vivo pharmacokinetics of free DOX and the DOX-loaded nanocapsules was investigated. Eighteen jugular vein catheter (JVC) SD rats were randomly divided into 3 groups (each group contained 3 males and 3 females) for free DOX, DOX-loaded PEG–PPS nanocapsules, and DOX-loaded PEG–PPS–cRGD nanocapsules. Then, 200 μL of blood sample was draw into heparinized polythene tubes at 0.25, 0.5, 1, 2, 4, 6, 12, and 24 h after intravenous administration (the same dose of DOX at 5 mg kg−1). After centrifugation at 10000 rpm for 8 min, 100 μL of plasma was collected and mixed with 300 μL of acetonitrile–methanol (9:1, v/v) and 10 μL of the IS working solution (2000 ng mL−1). The mixtures were vortexed for 1.0 min and spun in a centrifuge at 10000 rpm for 10 min. Then, 5 μL of supernatant was injected into the ultra-performance liquid chromatography (UPLC, DIONEX Ultimate 3000, USA) for analysis.46
Liquid chromatography was performed on an UPLC unit with Thermo Syncronis C18 column (2.1 × 100 mm 5 μm particle size). A gradient program was employed with the mobile phase combining solvent A (0.1% formic acid and 2 mmol L−1 ammonium formate in water) and solvent B (acetonitrile). A subsequent re-equilibration time (1 min) was performed before next injection. The flow rate was 0.30 mL min−1, and the injection volume was 5 μL. The column and sample temperature were maintained at 40 °C and 4 °C,47,48 respectively. An Thermo Q EXACTIVE mass spectrometer equipped with an electro-spray ionization (ESI) source was used for mass spectrometric detection. The detection was operated in Targeted-SIM mode. After optimization, the source parameters were set as follows: nebulizer gas, 35 arb; turbo gas, 10 arb; ion spray voltage, 2.8 kV; and temperature, 320 °C. Data acquiring and processing were performed using Analyst software (version 1.5, AB Sciex). The standard DOX working curve was set up as follows:
| Y = −1.5868 × 10−5 + 3.8283 × 10−6 × X … R2 = 0.9998 |
The plasma drug concentration–time data for each rat were analyzed by DAS 3.0 (BioGuider Co., Shanghai, China).48
In vivo imaging and biodistribution
To evaluate the targeted distribution of DOX-loaded nanocapsules in tumor-bearing mice, 24 BALB/c nude mice were injected with SCC-15 cells (5 × 106 cells per mouse) suspended in 100 μL of Matrigel (BD Biocoat Matrigel, USA) into the right back via a percutaneous approach. Tumor volume was calculated according to the formula:| (Tumor length) × (tumor width)2/2. |
When the tumor was approximately 200 mm3 in volume, the mice were randomly divided into 4 groups (each group contained 3 males and 3 females). The distribution images of DOX in vivo were taken at 1, 3, 6, 12, and 24 h after the intravenous administration of 5.0 mg kg−1 of DOX via tail vein using the ex/in vivo imaging system (CRi Maestro, USA) with a 503–535 nm excitation wavelength and 560–750 nm filter to collect the FL signals of DOX with auto-expose time.49,50 The mice were sacrificed after injection for 24 h, and the organs, including heart, liver, spleen, lung, kidneys and tumor, were collected for FL imaging. Then, DOX in various organs was extracted by chloroform–methanol (4/1, v/v), and the concentration of DOX was detected by UPLC-MS/MS system.
In vivo tumor growth inhibition
To evaluate the in vivo efficacy of DOX-loaded nanocapsules in tumor-bearing mice, the 24 tumor-bearing mice were randomly divided into 4 groups (each group contained 3 males and 3 females) for saline, free DOX, DOX-loaded PEG–PPS nanocapsules, and DOX-loaded PEG–PPS–cRGD nanocapsules. When the tumor reached approximately 200 mm3 in volume, the mice were treated with 5 mg kg−1 of DOX via tail vein 4 times (at day 0, 7, 14, 21). The mice were observed for general condition, and the tumor volumes were measured twice per week. After 28 days, the mice were sacrificed, and their organs and tumor were embedded in paraffin and stained with hematoxylin–eosin (H&E) to observe histopathological change.50,51 The inhibition rate of tumor (IRT) was calculated by the following formula:| IRT (%) = (1 − D/S) × 100% (D: tumor volume of treatment group; S: tumor volume of saline group). |
Statistical analysis
All statistical analyses were performed using Instat (GraphPad, San Diego, CA) and SPSS 20.0 (Chicago, USA). Experiments were statistically analyzed using the one-way ANOVA test to compare all pairs of data using a 95% confidence interval.Results and discussion
Characterization of the nanocapsules
The synthesized mechanism of the nanocapsules is illustrated in Fig. S6(a).† Four-arm PEG–PPS copolymer was self-crosslinked via disulfide exchange reaction of pyridine dithione groups with initiation of TEA. Fig. S6(b)† shows that the rise in UV absorption intensity of pyridine thione (absorbance at 342 nm) with reaction time was related to the released pyridine thione when the disulfide exchange reaction occurred among the end-capping group of pyridine dithione of PEG–PPS.42 Fig. S6(c)† shows dynamic light scattering (DLS) of the nanocapsules, indicating that the nanocapsules have an average particle size of approximately 100 nm, which is the preferable size for use in anticancer drug delivery. The TEM image also proved that the particle size of the nanocapsules was in the range of 50–100 nm (Fig. S6(d)†).DOX-loaded nanocapsules were prepared using a solvent–antisolvent process. Fig. 2(a) and (b) compare the sizes of DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules by DLS. In contrast to DOX-loaded PEG–PPS nanocapsules (particle size approximately 220 nm), the particle size of DOX-loaded PEG–PPS–cRGD nanocapsules increased to approximately 255 nm, indicating that the modification of hydrophilic chain of cRGD peptide on PEG–PPS resulted in the particle size becoming larger during the self-crosslinking process. Fig. 2(c) and (d) show TEM images of the DOX-loaded nanocapsules. The nanospheres with small dark drug particles dispersed among the gray polymer matrices are presented, demonstrating that DOX was successfully encapsulated inside polymer nanocapsules.
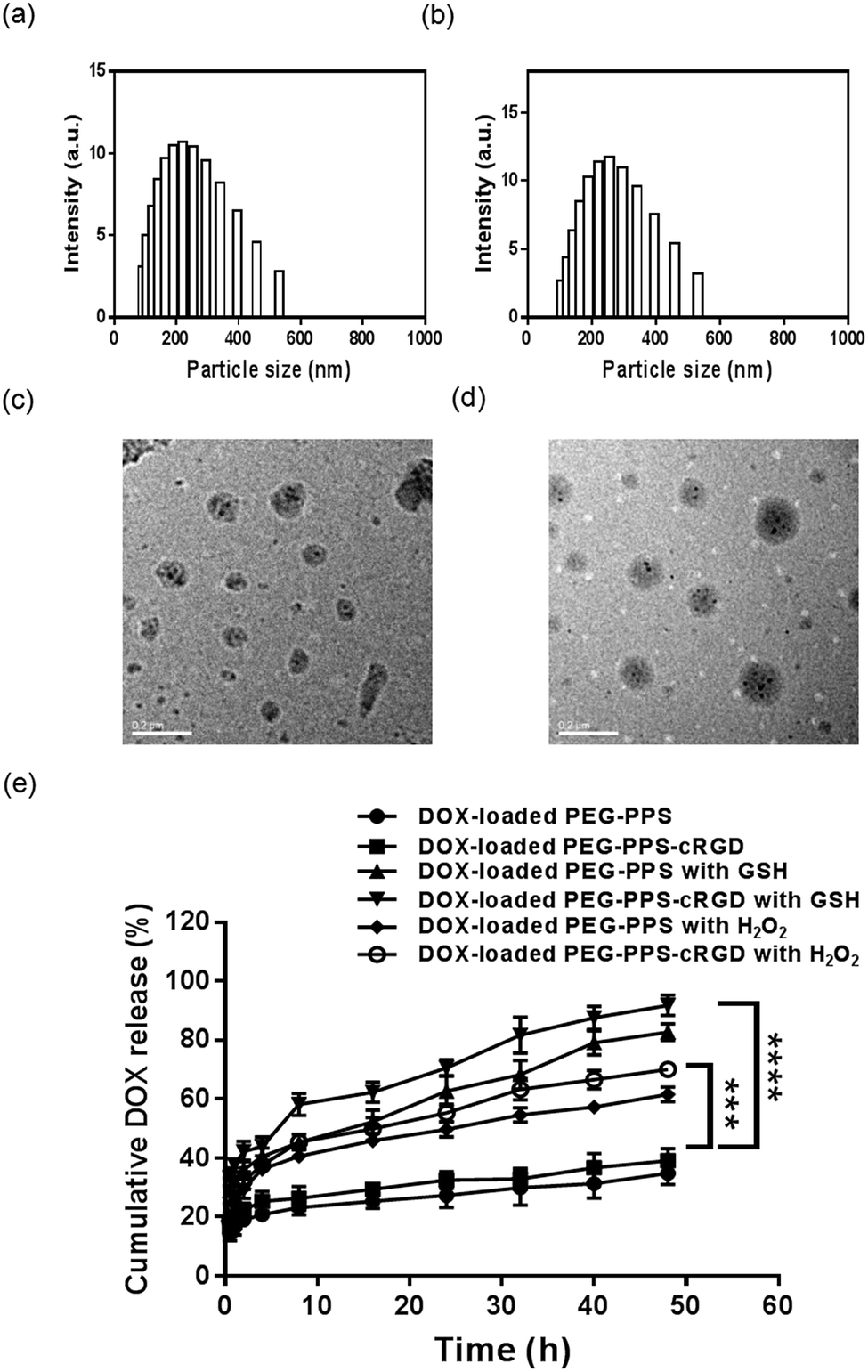 | ||
| Fig. 2 (a) and (b) Particle size distribution of DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules. (c) and (d) TEM images of DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules. (e) DOX-release profiles of DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules with or without 500 μM H2O2 or 10 mM of rGSH. Data represent means ± SD (n = 3). ***P < 0.001, ****P < 0.0001. | ||
Fig. S7† shows the stability test of DOX-loaded nanocapsules by DLS test. During the detection period of 48 h, the average particle size of the nanocapsules exhibited no obvious change in both PBS and complete DMEM medium, suggesting that these nanocapsules has well stability in biological mediums.
The drug-loading capacity (DLC) was calculated by redissolving the freeze-dried sample in DMSO for UV testing. The results showed that both DOX-loaded nanocapsules exhibited preferable drug-loading capacity with 4.4 wt% and 3.6 wt%, respectively. Moreover, to prove the nanocapsules have dual redox-responsive drug release property, we incubated DOX-loaded nanocapsules in PBS buffers with or without 500 μM H2O2 or 10 mM rGSH.52 Fig. 2(e) exhibits the drug release profiles. Without H2O2 and rGSH, only 34.9% and 39.1% of DOX was released from PEG–PPS and PEG–PPS–cRGD nanocapsules, respectively, during the testing period of 48 h. In contrast, the drug release rates of DOX-loaded nanocapsules co-incubated with H2O2 or rGSH were much faster, and cumulative release of 61.7% and 70.2% observed for PEG–PPS and PEG–PPS–cRGD nanocapsules co-incubated with H2O2, and 82.7% and 91.8% was observed for PEG–PPS and PEG–PPS–cRGD nanocapsules co-incubated with rGSH, suggesting that these nanocapsules are ultrasensitive to both oxidation and reduction stimuli and can be used as a promising carrier to prevent premature release of drugs as well as improve therapeutic efficiency of drugs in cancer chemotherapy.
In vitro cellular uptake of the nanocapsules
To prove that the cRGD targeting peptide can effectively direct the nanocapsules bonding to αvβ3 integrin-overexpressed squamous cells and further promote cellular uptake through the αvβ3 integrin receptor-mediated endocytosis pathway,41 we used CLSM and FCM to compare the cellular uptake of DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules in SCC-15 cells. As shown in Fig. 3(a), a bright red fluorescent signal of DOX was present in the cells incubated with DOX-loaded PEG–PPS–cRGD nanocapsules, and the fluorescence was localized in both the cytoplasm and nucleus. In contrast, weak fluorescence was observed in the cells incubated with cRGD-free nanocapsules. Fig. 3(b) and (c) assess the quantitative fluorescence intensity of the cells incubated with both nanocapsules by FCM. The average fluorescence intensity of the cells incubated with DOX-loaded PEG–PPS–cRGD nanocapsules was approximately 1.8-fold higher than the cells incubated with cRGD-free nanocapsules. Receptor blocking experiments were also performed. The free cRGD peptide was pre-incubated with the cells to block αvβ3 integrin before adding DOX-loaded PEG–PPS–cRGD nanocapsules. From Fig. 3(a), the red fluorescent signal of DOX was much weaker in the blocking cells compared to non-blocking cells. Otherwise, the results from FCM (Fig. 3(b) and (c)) were consistent with CLSM, and the average fluorescence intensity of the cells pre-incubated with free cRGD was dramatically decreased to 32% compared with non-blocking cells. These results confirmed that the modification of cRGD efficiently enhanced cellular uptake of the nanocapsules through αvβ3 integrin receptor-mediated endocytosis.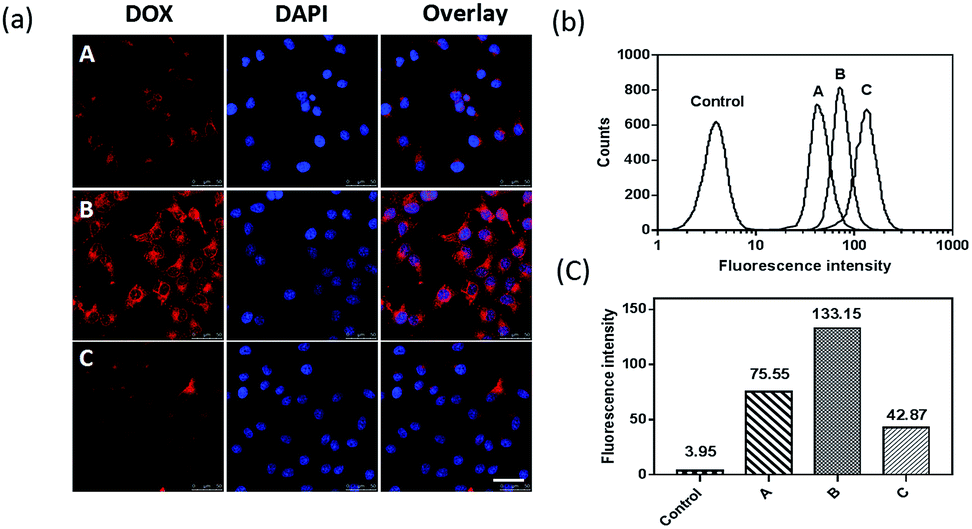 | ||
| Fig. 3 (a) CLSM and (b) and (c) FCM of SCC-15 cells after treatment with DOX-loaded PEG–PPS nanocapsules (A), DOX-loaded PEG–PPS–cRGD nanocapsules (B), and DOX-loaded PEG–PPS–cRGD nanocapsules with blocking αvβ3 integrin receptors by free cRGD peptide (C). | ||
In vitro cytotoxicity assay of the nanocapsules
To evaluate the in vitro cytotoxicity of copolymers and drug-loaded nanocapsules against SCC-15 cells, the MTT assay was adopted. As shown in Fig. 4(a), the MTT assay of both PEG–PPS and PEG–PPS–cRGD copolymers presented an excellent biocompatibility, with more than 80% cells remaining alive even after treatment with a highest concentration of 330 μg mL−1. The MTT assays of free DOX and DOX-loaded nanocapsules are displayed in Fig. 4(b). Both DOX-loaded nanocapsules present a concentration-related cytotoxicity. Compared to free DOX, DOX-loaded PEG–PPS and DOX-loaded PEG–PPS–cRGD nanocapsules show a significant cytotoxicity with 33.2% and 18.1% living cells at the highest DOX concentration of 18 μg mL−1.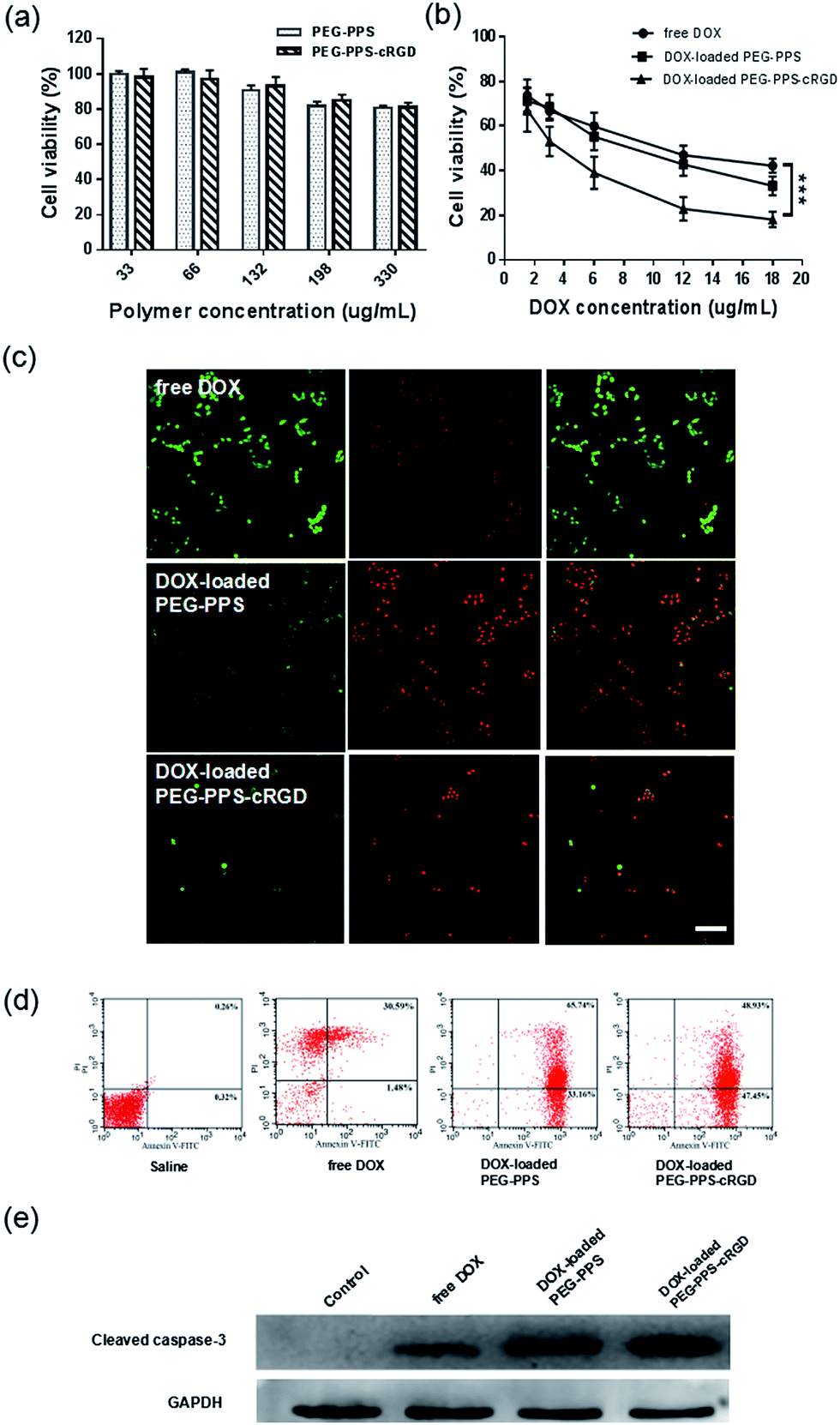 | ||
| Fig. 4 (a) MTT assay of SCC-15 cells after treatment with PEG–PPS and PEG–PPS–cRGD copolymers. (b) MTT assay of SCC-15 cells after treatment with free DOX, DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules. Data represent means ± SD (n = 3). ***P < 0.001. (c) Live–dead staining (green = live, red = dead) of SCC-15 cells after treatment with free DOX, DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules. Scale bar = 50 μm. (d) and (e) Apoptosis analysis of SCC-15 cells after treatment with free DOX, DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules by Annexin-V/PI staining and expression of cleaved caspase-3. | ||
To visually identify live and dead cells after treating with free DOX and DOX-loaded nanocapsules, the live–dead assay was utilized for staining live cells with green FL (ethidium homodimer-1) and dead cells with red FL (calcein-AM). Fig. 4(c) shows that there were more dead cells in the DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsule-treated sample than in the free DOX sample, proving that DOX-loaded nanocapsules had a better effect of inducing tumor cell death.
In vitro apoptosis assay of the nanocapsules
Annexin-V/PI double-staining labeled cells in each group showed the number of apoptotic or necrotic cells, which was quantified by FCM analysis in Fig. 4(d). The dot plot showed the viable cells that did not bind Annexin-V or PI in the D3 quadrant. Cells at early stages of apoptosis that bound Annexin-V but still had intact cell membranes and excluded PI are shown in the D4 quadrant. Cells with advanced stages of apoptosis or necrotic were both Annexin-V and PI positive and are shown in the D2 quadrant. Cells that lost their intact cell membranes that bound PI and excluded Annexin-V are shown in the D1 quadrant. The results showed that the SCC-15 cell apoptosis rate was significantly higher in the DOX-loaded nanocapsule groups than in the saline and free DOX groups.To further investigate the effect of DOX-loaded nanocapsules in apoptosis, the expression of cleaved caspase-3, which was used to evaluate the degree of apoptosis, was assessed by western blot assay. As shown in Fig. 4(e), after treating SCC-15 cells with both DOX-loaded nanocapsules, the expression of cleaved caspase-3 was significantly increased, especially for DOX-loaded PEG–PPS–cRGD nanocapsules, suggesting that more apoptosis might be induced in SCC-15 cells by DOX-loaded nanocapsules.53,54
Biosafety evaluation of the nanocapsules
Cancer chemotherapy with safety and low toxicity is a desired outcome. The hemolysis assay was used to evaluate the compatibility of DOX-loaded nanocapsules with erythrocytes. The hemolysis rate of DOX-loaded nanocapsules was less than 5% (in Table S1†), which met the standard requirements of intravenous administration.To assess the toxicity of DOX-loaded nanocapsules in vivo, healthy mice were intravenously injected with saline, free DOX, and both DOX-loaded nanocapsules for monitoring body weight, hematological determination, biochemical examination, and histological analysis. As presented in Table S2,† there was no significant difference in body weight among the saline group and both DOX-loaded nanocapsule groups, but the least weight gain was observed in the group treated with free DOX.55 The results of hematological determination are shown in Fig. 5(a). Compared with the saline negative group, WBC and LYMPH of DOX-loaded nanocapsule groups observed no significant decrease, suggesting that no myelosuppression occurred. The indexes of biochemical examination are illustrated in Fig. 5(b). The expression of CK and CK-MB in DOX-loaded nanocapsule groups exhibited no significant increase, suggesting that the treatment with DOX-loaded nanocapsule groups caused no obvious cardiac toxicity compared to the saline group. Furthermore, histological analysis of H&E staining demonstrated that the groups treated with both DOX-loaded nanocapsules had no noticeable histopathological change in any of the tested organs, but free DOX group showed disordered arrangement and swelling of muscle cells, fiber breakage and dissolving of sarcoplasm in cardiac tissue (Fig. 5(c)).
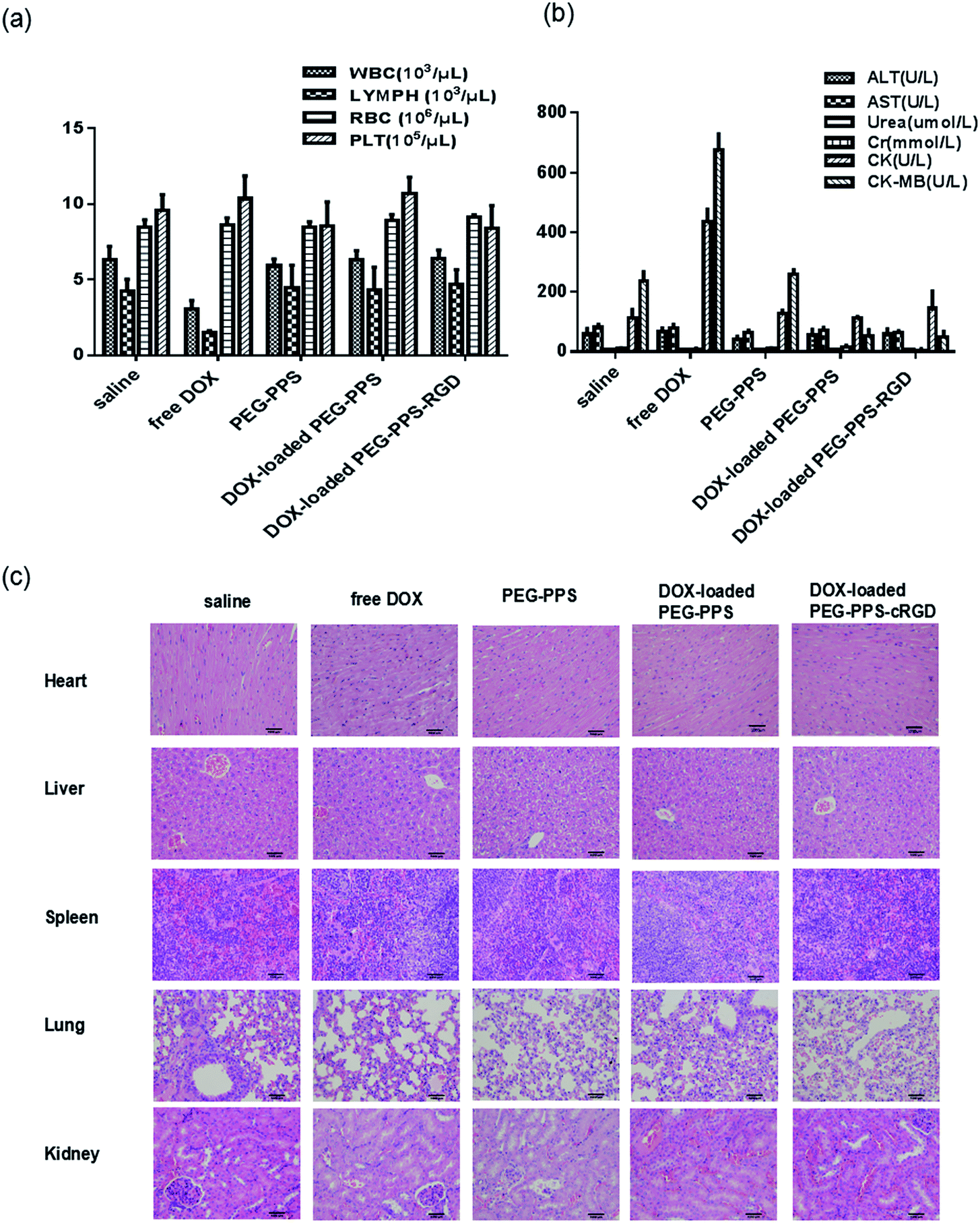 | ||
| Fig. 5 (a) Hematological determination, (b) biochemical examination, and (c) H&E staining of various organs at day 14 after treating the mice with saline, free DOX, DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules. | ||
Taken together, these biosafety evaluation results demonstrate that loading DOX into PEG–PPS or PEG–PPS–cRGD nanocapsules significantly alleviate the side effects of DOX due to preventing drug premature leakage and cardiac accumulation,56 making these nanocapsules valuable for cancer chemotherapy applications.
In vivo pharmacokinetics
After intravenously injecting the rats with free DOX and DOX-loaded nanocapsules at the same dose of 5 mg kg−1, 100 μL of blood plasma was collected at different times for pharmacokinetic testing. As shown in Fig. 6(a) and (b), the DOX concentration of both DOX-loaded nanocapsules in blood plasma was much higher than that of free DOX at the same time. Further, the main pharmacokinetic parameters from a three-compartment model analysis are summarized.57 As shown in Table 1, in vivo retention time of free DOX was short, and the half time (t1/2) was 4.02 h. When DOX was loaded in PEG–PPS and PEG–PPS–cRGD nanocapsules, t1/2 was prolonged by 2.35- and 2.68-fold, respectively, compared to free DOX. The area under the concentration–time curve (AUC) of DOX-loaded nanocapsules was 5.14- and 7.41-fold that of free DOX, respectively. The mean retention time (MRT) of DOX-loaded nanocapsules was 2.76- and 3.55-fold, respectively, compared with free DOX. The clearance (Cl) of DOX-loaded nanocapsules dramatically decreased. Therefore, the main pharmacokinetic parameters were superior to those of free DOX, indicating that DOX-loaded nanocapsules had better long-term blood circulation performance due to the structure of the nanocapsules containing PEG segment, which can play an important role for significantly prolonging the blood circulation time of the loading drugs.58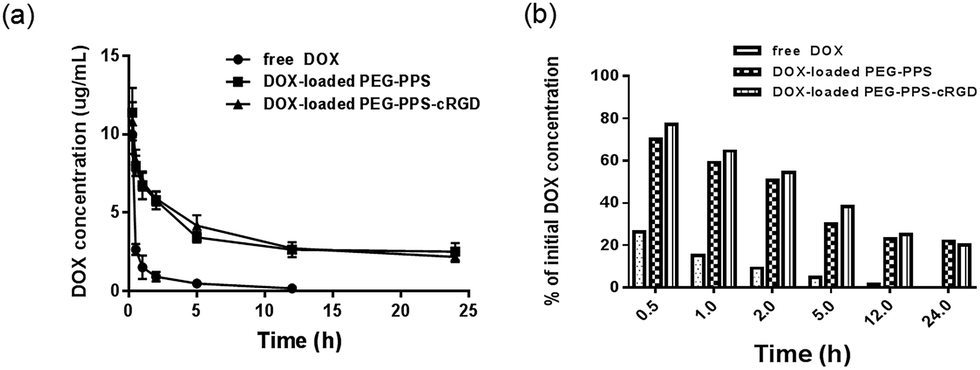 | ||
| Fig. 6 (a) Drug concentrations in the blood at different time points after treating the mice with free DOX, DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules. Data represent means ± SD (n = 6). (b) The percentage of rest drug comparing to initial drug concentration at different time points after injection. | ||
| AUC0→∞ (h μg L−1) | t1/2 (h) | Cl (L h−1 kg−1) | MRT0→∞ (h) | |
|---|---|---|---|---|
| Free DOX | 12.31 | 4.02 | 0.30 | 3.52 |
| DOX-loaded PEG–PPS | 63.29 | 9.44 | 0.06 | 9.72 |
| DOX-loaded PEG–PPS–cRGD | 91.19 | 10.78 | 0.05 | 12.51 |
In vivo imaging and biodistribution
To investigate the tumor-targeting abilities of DOX-loaded nanocapsules, the BALB/c nude squamous cell carcinoma- bearing mice were intravenously injected with saline, free DOX, DOX-loaded PEG–PPS nanocapsules, and DOX-loaded PEG–PPS–cRGD nanocapsules, and fluorescent images were subsequently recorded at different times by an ex/in vivo imaging system. As shown in Fig. 7(a), FL of DOX demonstrated a time-dependent biodistribution in the mice for all testing groups. In contrast to free DOX, both DOX-loaded nanocapsules showed higher FL signals in the tumor tissue at all of the detection time-points. After 24 h postinjection, the mice were sacrificed and various organs were isolated to image ex vivo for further monitoring the biodistribution of the nanocapsules. As shown in Fig. 7(b), although the higher FL signal appeared in the liver, spleen, lung, and kidney for both DOX-loaded nanocapsule groups, only the DOX-loaded PEG–PPS–cRGD nanocapsule group presented brighter FL signal in tumor tissue. Further, DOX concentrations in the organs were measured for quantitative drug distribution analysis (Fig. 7(c)). DOX concentrations in tumor tissue from DOX-loaded PEG–PPS–cRGD nanocapsule group were 2.9- and 1.7-fold higher than those from free DOX and DOX-loaded PEG–PPS nanocapsule groups, respectively. Overall, these results clarified that the nanocapsules with the tumor-targeting cRGD peptide effectively accumulate in the tumor site, not only through passive targeting by the EPR effect but also through active targeting by the cRGD receptor-mediated effect.41 | ||
| Fig. 7 (a) In vivo fluorescence imaging of tumor-bearing mice after intravenous injection with saline, free DOX, DOX-loaded PEG–PPS and PEG–PPS–cRGD nanocapsules at different times. (b) Ex vivo fluorescence imaging of various organs and tumor tissue. (c) DOX concentrations in various organs and tumor tissue. (d) and (e) The volume growth curves of tumors, and tumor inhibitory rate. Data represent means ± SD (n = 6). **P < 0.01, ***P < 0.001, ****P < 0.0001. (f) H&E staining for pathological changes in tumor sections. | ||
In vivo tumor growth inhibition
The in vivo tumor inhibition efficacy of the DOX-loaded nanocapsules in the treatment of xenograft squamous cell carcinoma was investigated by using the BALB/c nude tumor-bearing mice. As shown in Fig. 7(d), compared to saline group, tumor growth was significantly inhibited after injecting free DOX and DOX-loaded nanocapsules. In particular, the mice in the DOX-loaded PEG–PPS–cRGD nanocapsule group exhibited the greatest effect of tumor growth inhibition and the inhibition rate was 51.9% on 28th day (Fig. 7(e)). To further assess the in vivo antitumor effect, the histological analysis of tumor is shown in Fig. 7(f). Compared with the three other groups, the DOX-loaded PEG–PPS–cRGD nanocapsule group exhibited more obvious nuclear condensation and fragmentation in the H&E images, indicating that DOX-loaded PEG–PPS–cRGD nanocapsules had a better antitumor efficiency. Together, loading DOX into PEG–PPS–cRGD nanocapsules could significantly inhibit tumor growth as well as improve the chemotherapy effect of DOX for squamous cell carcinoma.Conclusions
In summary, a self-crosslinked nanocapsule with smart dual redox-responsiveness and tumor targeting was successfully prepared using multifunctional PEG–PPS–cRGD branched copolymers. DOX-loaded nanocapsules exhibited spherical structures with small drug particles dispersed among the polymer nanocapsules. The in vitro drug release studies showed that DOX-loaded nanocapsules had a robust drug release profile under both oxidation and reduction environment. Cellular uptake experiments presented that DOX-loaded nanocapsules are efficiently taken up into the cells by receptor-mediated endocytosis. Furthermore, the in vitro cytotoxicity investigations demonstrated that DOX-loaded nanocapsules exhibit serious cytotoxicity against the SCC-15 cell line. Finally, the in vivo studies also proved that DOX-loaded nanocapsules effectively target the tumor sites in xenograft models of squamous cell carcinoma in nude mice, and extraordinarily inhibit tumor growth with high biological safety. Moreover, we believe that our work developed promising polymer nanocarriers, which are not limited to use with DOX, and can be adapted to a variety of chemotherapy drugs for different cancer treatments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation of China (Grant 51303009, Grant 21476018 and Grant 21622601), and the Fundamental Research Funds for the Central Universities (Grant PYBZ1714).References
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- F. M. Kievit and M. Zhang, Adv. Mater., 2011, 23, H217–H247 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS.
- Y. Bae and K. Kataoka, Adv. Drug Delivery Rev., 2009, 61, 768–784 CrossRef CAS PubMed.
- C. Cui, Y. N. Xue, M. Wu, Y. Zhang, P. Yu, L. Liu, R. X. Zhuo and S. W. Huang, Biomaterials, 2013, 34, 3858–3869 CrossRef CAS PubMed.
- C. K. Huang, C. L. Lo, H. H. Chen and G. H. Hsiue, Adv. Funct. Mater., 2007, 17, 2291–2297 CrossRef CAS.
- S. Sant, V. Nadeau and P. Hildgen, J. Controlled Release, 2005, 107, 203–214 CrossRef CAS PubMed.
- L. Jing, X. Liang, X. Li, Y. Yang and Z. Dai, Acta Biomater., 2013, 9, 9434–9441 CrossRef CAS PubMed.
- P. Xu, E. Gullotti, L. Tong, C. B. Highley, D. R. Errabelli, T. Hasan, J. X. Cheng, D. S. Kohane and Y. Yeo, Mol. Pharm., 2009, 6, 190–201 CrossRef CAS PubMed.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- J. H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- M. Oishi, H. Hayashi, M. Iijima and Y. Nagasaki, J. Mater. Chem., 2007, 17, 3720–3725 RSC.
- Q. Sun, M. Radosz and Y. Shen, J. Controlled Release, 2012, 164, 156–169 CrossRef CAS PubMed.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Nano Today, 2012, 7, 467–480 CrossRef CAS.
- F. Meng, R. Cheng, C. Deng and Z. Zhong, Mater. Today, 2012, 15, 436–442 CrossRef CAS.
- B. Städler, A. D. Price and A. N. Zelikin, Adv. Funct. Mater., 2011, 21, 14–28 CrossRef.
- K. Liang, G. K. Such, A. P. R. Johnston, Z. Zhu, H. Ejima, J. J. Richardson, J. Cui and F. Caruso, Adv. Mater., 2014, 26, 1901–1905 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. Zhao, A. Biswas, B. Hu, K.-I. Joo, P. Wang, Z. Gu and Y. Tang, Biomaterials, 2011, 32, 5223–5230 CrossRef CAS PubMed.
- J. Li, M. Huo, J. Wang, J. Zhou, J. M. Mohammad, Y. Zhang, Q. Zhu, A. Y. Waddad and Q. Zhang, Biomaterials, 2012, 33, 2310–2320 CrossRef CAS PubMed.
- Y. Wang, G. Wei, X. Zhang, F. Xu, X. Xiong and S. Zhou, Adv. Mater., 2017, 29, 1605357 CrossRef PubMed.
- K. M. Poole, C. E. Nelson, R. V. Joshi, J. R. Martin, M. K. Gupta, S. C. Haws, T. E. Kavanaugh, M. C. Skala and C. L. Duvall, Biomaterials, 2015, 41, 166–175 CrossRef CAS PubMed.
- M. K. Gupta, J. R. Martin, T. A. Werfel, T. Shen, J. M. Page and C. L. Duvall, J. Am. Chem. Soc., 2014, 136, 14896–14902 CrossRef CAS PubMed.
- Q. Li, Y. Wen, J. Wen, Y.-P. Zhang, X.-D. Xu, A. Victorious, R. Zavitz and X. Xu, RSC Adv., 2016, 6, 38984–38989 RSC.
- M. K. Gupta, T. A. Meyer, C. E. Nelson and C. L. Duvall, J. Controlled Release, 2012, 162, 591–598 CrossRef CAS PubMed.
- W. Wu, W. Wang and J. Li, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS.
- J. Wang, F. Zhang, W. P. Tsang, C. Wan and C. Wu, Biomaterials, 2017, 120, 11–21 CrossRef CAS PubMed.
- X. Wu, C. He, Y. Wu, X. Chen and J. Cheng, Adv. Funct. Mater., 2015, 25, 6744–6755 CrossRef CAS.
- D. Hong, Y. Ken-Tye, R. Indrajit, H. Rui, W. Fang, Z. Lingling, L. Wing-Cheung, Z. Weiwei, J. Wei, L. Liwei, J. B. Earl and N. P. Paras, Nanotechnology, 2011, 22, 165101 CrossRef PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295–4312 RSC.
- N. P. Truong, J. F. Quinn, A. Anastasaki, D. M. Haddleton, M. R. Whittaker and T. P. Davis, Chem. Commun., 2016, 52, 4497–4500 RSC.
- Z. Li, S. Yin, L. Cheng, K. Yang, Y. Li and Z. Liu, Adv. Funct. Mater., 2014, 24, 2312–2321 CrossRef CAS.
- Y. Wang and D. S. Kohane, Nat. Rev. Mater., 2017, 2, 17020 CrossRef CAS.
- H. Y. Yoon, H. Koo, K. Y. Choi, S. J. Lee, K. Kim, I. C. Kwon, J. F. Leary, K. Park, S. H. Yuk, J. H. Park and K. Choi, Biomaterials, 2012, 33, 3980–3989 CrossRef CAS PubMed.
- M. M. J. Kamphuis, A. P. R. Johnston, G. K. Such, H. H. Dam, R. A. Evans, A. M. Scott, E. C. Nice, J. K. Heath and F. Caruso, J. Am. Chem. Soc., 2010, 132, 15881–15883 CrossRef CAS PubMed.
- E. Lieb, M. Hacker, J. Tessmar, L. A. Kunz-Schughart, J. Fiedler, C. Dahmen, U. Hersel, H. Kessler, M. B. Schulz and A. Göpferich, Biomaterials, 2005, 26, 2333–2341 CrossRef CAS PubMed.
- S. Raha, T. Paunesku and G. Woloschak, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 269–281 CrossRef CAS PubMed.
- M. Li, Y. Gao, Y. Yuan, Y. Wu, Z. Song, B. Z. Tang, B. Liu and Q. C. Zheng, ACS Nano, 2017, 11, 3922–3932 CrossRef CAS PubMed.
- J. Zhang, T. Tokatlian, J. Zhong, Q. K. T. Ng, M. Patterson, W. E. Lowry, S. T. Carmichael and T. Segura, Adv. Mater., 2011, 23, 5098–5103 CrossRef CAS PubMed.
- Y.-T. Chiang, Y.-W. Yen and C.-L. Lo, Biomaterials, 2015, 61, 150–161 CrossRef CAS PubMed.
- D. Li, N. Wang, L. Zhang, Z. Hanyu, B. Xueyuan, B. Fu, C. Shaoyuan, W. Zhang, S. Xuefeng, R. Li and X. Chen, Stem Cell Res. Ther., 2013, 4, 103 CrossRef PubMed.
- H. Zhang, J. Y. Yang, F. Zhou, L. H. Wang, W. Zhang, S. Sha and C. F. Wu, J. Evidence-Based Complementary Altern. Med., 2011, 2011, 965016 Search PubMed.
- J. Y. Lee, J. H. Park, J. J. Lee, S. Y. Lee, S. J. Chung, H. J. Cho and D. D. Kim, Carbohydr. Polym., 2016, 151, 68–77 CrossRef CAS PubMed.
- H. Niu, M. Xu, S. Li, J. Chen, J. Luo, X. Zhao, C. Gao and X. Li, Med. Sci. Monit. Basic Res., 2017, 23, 150–158 CrossRef PubMed.
- W. Zhang, R. Li, M. Sun, D. Hu, J. Qiu and Y. Yan, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 965, 107–111 CrossRef CAS PubMed.
- L. Zhang, S. Gao, F. Zhang, K. Yang, Q. Ma and L. Zhu, ACS Nano, 2014, 8, 12250–12258 CrossRef CAS PubMed.
- M. Zheng, C. Yue, Y. Ma, P. Gong, P. Zhao, C. Zheng, Z. Sheng, P. Zhang, Z. Wang and L. Cai, ACS Nano, 2013, 7, 2056–2067 CrossRef CAS PubMed.
- C. Qian, J. Yu, Y. Chen, Q. Hu, X. Xiao, W. Sun, C. Wang, P. Feng, Q. D. Shen and Z. Gu, Adv. Mater., 2016, 28, 3313–3320 CrossRef CAS PubMed.
- Y. E. Kurtoglu, R. S. Navath, B. Wang, S. Kannan, R. Romero and R. M. Kannan, Biomaterials, 2009, 30, 2112–2121 CrossRef CAS PubMed.
- D. R. Schultz and W. J. Harrington Jr, Semin. Arthritis Rheum., 2003, 32, 345–369 CrossRef CAS PubMed.
- F. Wang, L. Wang, Y. Zhao, Y. Li, G. Ping, S. Xiao, K. Chen, W. Zhu, P. Gong, J. Yang and C. Wu, Mol. Oncol., 2014, 8, 1640–1652 CrossRef CAS PubMed.
- Y. Chen, W. Yang, B. Chang, H. Hu, X. Fang and X. Sha, Eur. J. Pharm. Biopharm., 2013, 85, 406–412 CrossRef CAS PubMed.
- D. Outomuro, D. R. Grana, F. Azzato and J. Milei, Int. J. Cardiol., 2007, 117, 6–15 CrossRef PubMed.
- J. Wang, D. Zheng, Y. Wang, C. Zhang and X. Sun, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1059, 35–42 CrossRef CAS PubMed.
- S. Huang, S. Duan, J. Wang, S. Bao, X. Qiu, C. Li, Y. Liu, L. Yan, Z. Zhang and Y. Hu, Adv. Funct. Mater., 2016, 26, 2532–2544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10499e |
| This journal is © The Royal Society of Chemistry 2017 |