
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalytic asymmetric Michael addition reaction of L-arginine to fumarate for the green synthesis of N-(([(4S)-4-amino-4-carboxy-butyl]amino)iminomethyl)-L-aspartic acid lithium salt (L-argininosuccinic acid lithium salt)†
B. Schoenenbergera,
A. Wszolekb,
R. Meiera,
H. Brundiekb,
M. Obkirchera and
R. Wohlgemuth *a
*a
aSigma-Aldrich, Member of Merck Group, Industriestrasse 25, CH-9470 Buchs, Switzerland. E-mail: roland.wohlgemuth@sial.com
bEnzymicals, Walther-Rathenau-Strasse 49a, 17489 Greifswald, Germany
First published on 16th October 2017
Abstract
The basic natural amino acid L-argininosuccinate containing two chiral centers occurs in L-alanine, L-arginine, L-aspartate, L-glutamate and L-proline metabolic pathways and plays a role in the biosynthesis of secondary metabolites and other amino acids. It is a precursor for arginine in the urea cycle or the citrulline–NO cycle as well as a precursor to fumarate in the citric acid cycle via argininosuccinate lyase. We aimed to run part of the urea cycle in reverse by catalyzing not the elimination but the addition reaction of L-arginine to fumarate in order to synthesize L-argininosuccinate. Argininosuccinate lyase (ASL) from Saccharomyces cerevisiae has been chosen as the catalyst for this addition reaction. The selected ARG4 gene was synthesized and homogeneously expressed in E. coli leading to a highly active argininosuccinate lyase. The ASL-catalyzed addition reaction of L-arginine to fumarate has been successfully developed at gram scale. After a standard workup procedure the pure final product L-argininosuccinate has been isolated in good yield and high purity.
Introduction
The construction of new bonds by the addition reaction named after Arthur Michael, who described it in 1887,1 has become a key methodology of synthetic organic chemistry. Its further development and its catalytic asymmetric versions continue to attract much interest.2 The discovery of selective organocatalytic and metal-catalyzed asymmetric Michael additions has inspired interest in utilizing this versatile reaction for a broad range of synthetic applications. High enantio- and diastereo-selectivity has been achieved in the catalytic synthesis of intermediates and more complex natural products,3–11 the catalytic generation of quaternary all-carbon stereocenters12,13 and in the formation of new bonds from carbon to nitrogen, oxygen or sulfur atoms in catalytic aza-, oxa- or sulfa-Michael addition reactions.14 A highly practical and efficient catalytic asymmetric Michael addition procedure has been scaled up for the large-scale synthesis of enantiomerically pure (R)-3-[bis-(methoxycarbonyl)-methyl]-cyclohexanone.15 A number of different Michael addition reactions have been integrated into cascades like multiple Michael additions,16 the Mukaiyama–Michael addition17 or the triple cascade Michael/Michael/aldol condensation with control of four stereocenters in the synthesis of tetra-substituted cyclohexenecarbaldehydes.18Green chemistry approaches have focussed on developing hydrolases for Michael-type additions in organic solvents19 and on replacing organic solvents, e.g. by green solvents20 and ionic liquids,21 solvent-free reactions in ball mills22 and reactions on water.23 As catalytic asymmetric Michael addition reactions in water represent an ideal green synthetic methodology for creating molecular complexity from simple building blocks by the formation of new carbon–carbon bonds, the corresponding highly selective and suitable catalysts need to be developed. The discovery of a polyketide synthase-mediated Michael addition in the polyketide-chain branching of the rhizoxin biosynthetic pathway has therefore been a milestone.24 A computationally designed retro-aldolase has been shown to catalyse asymmetric Michael addition of ethyl 2-cyanoacetate to (E)-4-(4-methoxyphenyl)but-3-en-2-one with a S/R enantiomeric ratio of the product of 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.25 Two enantiocomplementary Michaelases, which have been redesigned from 4-oxalocrotonate tautomerase, catalyse the asymmetric addition of acetaldehyde to various nitroolefins for the synthesis of both enantiomers of γ-nitro-aldehydes.26 When we aimed at developing a highly selective, green and straightforward synthesis of the metabolite N-(([(4S)-4-amino-4-carboxybutyl]amino)iminomethyl)-L-aspartic acid 1, we turned to the natural enzyme argininosuccinate lyase (ASL) involved in the metabolic pathway of L-arginine. The non-proteinogenic natural amino acid N-(([(4S)-4-amino-4-carboxybutyl]amino)iminomethyl)-L-aspartate 1 is a key metabolite of L-arginine metabolism and is commonly referred to in the literature as L-argininosuccinate without a complete description of the metabolite's two chiral centers (Fig. 1). Not only is it interconvertible to amino acids like L-alanine, L-aspartate, L-glutamate and L-proline, but it also serves as a precursor for L-arginine, creatine, agmatine and urea in the urea cycle or for nitric oxide in the citrulline–nitric oxide cycle.27–30 As a precursor, L-argininosuccinate 1 connects the urea cycle with the citric acid cycle via the aspartate–argininosuccinate shunt, and is also connected to the malate–aspartate cycle via its product fumarate. The arginine metabolism has been recognized as being of increasing importance for the study of tumor development and progression, since many tumor cells have lost the ability to produce L-arginine de novo.31 As expression of the urea cycle enzyme argininosuccinate synthetase is decreased in melanoma, mesothelioma and cancers of the liver, kidney, pancreas, prostate and ovary,32 L-argininosuccinate formation is reduced and aspartate is accumulated, enhancing the proliferation of cancer cells.33 Nitric oxide (NO), generated through the L-arginine metabolism, has been linked to cardiovascular diseases.34 L-Arginine and its metabolic pathways affect also the immune system by its action on human T cells, T cell receptors and its key role in the inflammatory function of macrophages, where the nitric oxide synthesis pathway is associated with the inflammatory M1 phenotype, while the arginase pathway is associated with the tolerant M2 cell phenotype.35 Argininosuccinic aciduria (MIM 207900) is an inborn error of metabolism caused by a deficiency of argininosuccinate lyase36 and the second most common urea cycle disorder in humans.37 As this leads to elevated levels of L-argininosuccinate 1 in plasma and urine, its analysis in newborn screenings is highly valuable.27,38
:2.25 Two enantiocomplementary Michaelases, which have been redesigned from 4-oxalocrotonate tautomerase, catalyse the asymmetric addition of acetaldehyde to various nitroolefins for the synthesis of both enantiomers of γ-nitro-aldehydes.26 When we aimed at developing a highly selective, green and straightforward synthesis of the metabolite N-(([(4S)-4-amino-4-carboxybutyl]amino)iminomethyl)-L-aspartic acid 1, we turned to the natural enzyme argininosuccinate lyase (ASL) involved in the metabolic pathway of L-arginine. The non-proteinogenic natural amino acid N-(([(4S)-4-amino-4-carboxybutyl]amino)iminomethyl)-L-aspartate 1 is a key metabolite of L-arginine metabolism and is commonly referred to in the literature as L-argininosuccinate without a complete description of the metabolite's two chiral centers (Fig. 1). Not only is it interconvertible to amino acids like L-alanine, L-aspartate, L-glutamate and L-proline, but it also serves as a precursor for L-arginine, creatine, agmatine and urea in the urea cycle or for nitric oxide in the citrulline–nitric oxide cycle.27–30 As a precursor, L-argininosuccinate 1 connects the urea cycle with the citric acid cycle via the aspartate–argininosuccinate shunt, and is also connected to the malate–aspartate cycle via its product fumarate. The arginine metabolism has been recognized as being of increasing importance for the study of tumor development and progression, since many tumor cells have lost the ability to produce L-arginine de novo.31 As expression of the urea cycle enzyme argininosuccinate synthetase is decreased in melanoma, mesothelioma and cancers of the liver, kidney, pancreas, prostate and ovary,32 L-argininosuccinate formation is reduced and aspartate is accumulated, enhancing the proliferation of cancer cells.33 Nitric oxide (NO), generated through the L-arginine metabolism, has been linked to cardiovascular diseases.34 L-Arginine and its metabolic pathways affect also the immune system by its action on human T cells, T cell receptors and its key role in the inflammatory function of macrophages, where the nitric oxide synthesis pathway is associated with the inflammatory M1 phenotype, while the arginase pathway is associated with the tolerant M2 cell phenotype.35 Argininosuccinic aciduria (MIM 207900) is an inborn error of metabolism caused by a deficiency of argininosuccinate lyase36 and the second most common urea cycle disorder in humans.37 As this leads to elevated levels of L-argininosuccinate 1 in plasma and urine, its analysis in newborn screenings is highly valuable.27,38
 | ||
| Fig. 1 Argininosuccinate lyase-catalyzed reaction between L-arginine 2 and fumarate 3 to N-(([(4S)-4-amino-4-carboxybutyl]amino)imino-methyl)-L-aspartate 1 (in short: L-argininosuccinate) as part of the urea cycle running in reverse. | ||
In the urea cycle L-argininosuccinate is the product of the ATP dependent and rate-limiting reaction of citrulline and aspartate, catalysed by the enzyme argininosuccinate synthase (ASS).
Subsequently, L-argininosuccinate 1 is cleaved into fumarate 3 and L-arginine 2 by the enzyme argininosuccinate lyase (ASL).39–43 This ATP independent enzyme is highly conserved in bacteria and eukaryotes and has been studied in a wide range of organisms.44,45 Interestingly, ASL can also catalyse the reverse reaction to form L-argininosuccinate 1 from L-arginine 2 and fumarate 3 as shown by Ratner et al.,46 when fumarate and arginine had been used in high concentrations to obtain a 75% conversion when equilibrium is reached.
We aimed at developing a synthesis of pure L-argininosuccinate 1 by simply running part of the urea cycle in reverse using the ASL from Saccharomyces cerevisiae as catalyst (Fig. 1). For this reason, we studied the recombinant expression of the ARG4 gene as biocatalyst and its application for the condensation reaction of L-arginine 2 and fumarate 3 to L-argininosuccinate 1 at preparative scale for the first time in order to obtain the pure L-argininosuccinate lithium salt.
Results and discussion
The purpose of this study was to develop a simple biocatalytic procedure for the production of L-argininosuccinate 1 in gram-scale. We chose a recombinant overexpression strategy for argininosuccinate lyase (ASL) employing the ARG4 gene from Saccharomyces cerevisiae (Uniprot P04076, ARLY_YEAST). Thus, ARG4 was cloned and expressed via a synthetic gene cloning strategy, employing E. coli BL21 (DE3) as expression system. The recombinant ARG4 protein was overexpressed in E. coli as a soluble protein in high yield, i.e. around 50% of the protein was localized in the soluble protein fraction of E. coli. As active enzyme, ARG4 is a tetramer, composed of four active sites. The size of one monomer is approximately 52 kDa, as determined by SDS-PAGE analysis (Fig. 2). Since the recombinant protein carried an N-terminal his-tag, ARG4 was successfully purified by IMAC purification (Fig. 2).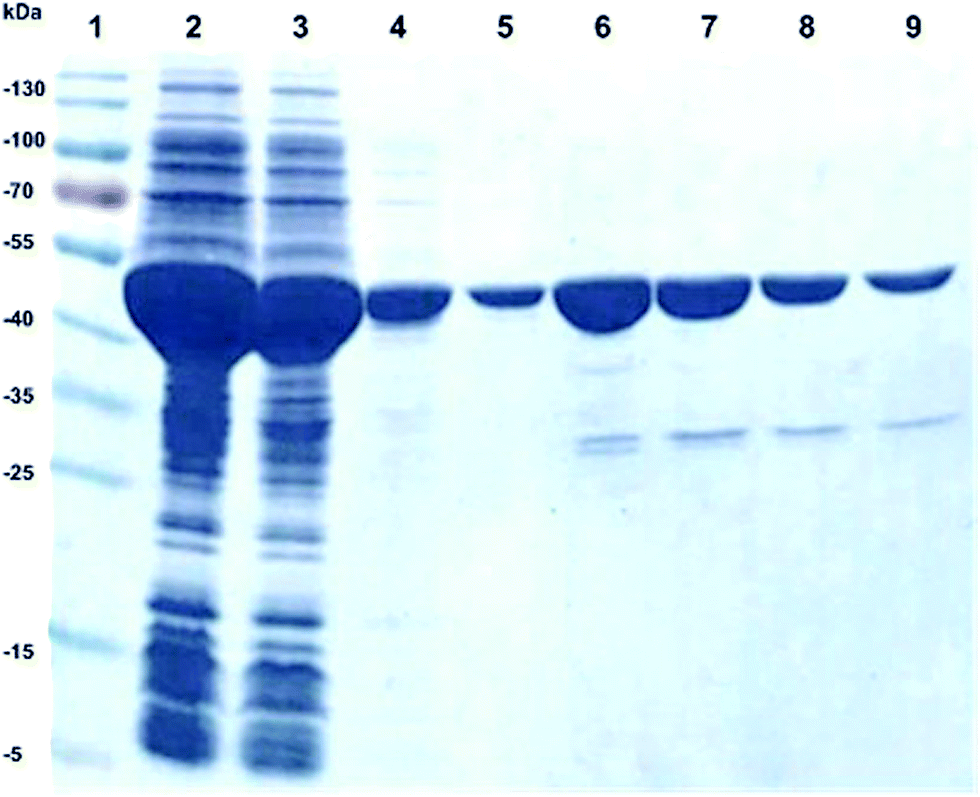 | ||
| Fig. 2 SDS-PAGE of His-tag purification of recombinant ARG4, lane 1: marker, lane 2: soluble fraction of cell crude extract from E. coli BL21(DE3), lane 3: flow through, lane 4: washing fraction, lane 5–9: elution of purified ARG4, MW of ARG4: ∼52 kDa. | ||
The obtained yield of the purified ARG4 was 20 mg L−1 of E. coli culture. The specific activity was about 20 U mg−1 of purified protein and 206 U mL−1 in the crude extract (Table 1). In contrast to this, the specific activity of argininosuccinate lyase purified directly from Saccharomyces cerevisiae crude extract was only 0.081 U mg−1.47
| Protein content | 0.92 mg mL−1 |
| Activity of cell crude extract (soluble fraction) | 206 U mL−1 |
| Activity of purified ARG4 | 20 U mg−1 |
Since storage stability can be an issue for insufficient activity, when employing a biocatalyst in preparative scale, we studied the activity of argininosuccinate lyase over one month in different formulations and storage temperatures (4 °C and −20 °C). The storage temperature and the addition of 30% glycerol had apparently no influence on the enzyme activity. Hence, the argininosuccinate lyase, ARG4, can be stored as powder as well as crude extract (Fig. 3). In contrast, in a previous study48 argininosuccinate lyase was isolated from bovine brain in a long and complicated procedure and only 6 mg of homogeneous enzyme were obtained from about 80 kg tissue. A drastic loss of activity of brain and liver isolated enzymes has been observed after 6 months of storage.48
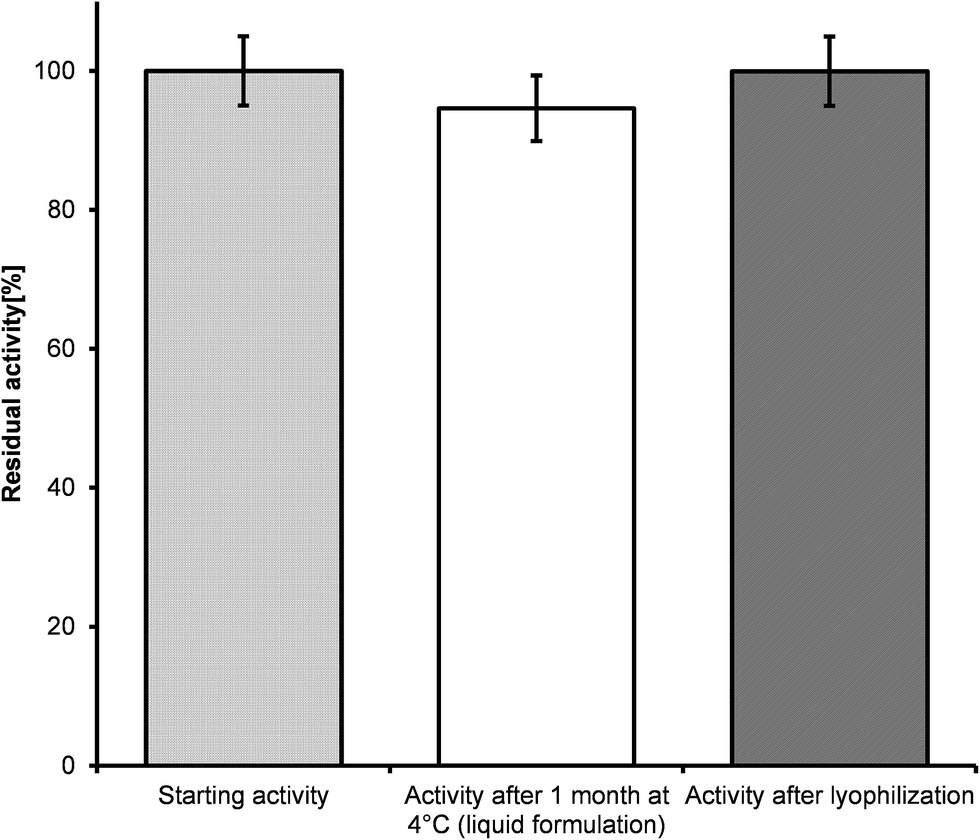 | ||
| Fig. 3 Activities of different formulations of argininosuccinate lyase. | ||
To evaluate the enzyme activity under reaction conditions the biocatalytic Michael addition reaction was analyzed as a function of time directly in the NMR tube. 1H-NMR signals at 4.17 ppm (proton at the newly formed asymmetric carbon atom on the succinate portion) and 6.44 ppm (fumarate protons) were diagnostic for the progress of the reaction (Fig. 4). An enzyme activity of roughly 4 U mL−1 was calculated for the cell crude extract (soluble fraction) from the fumarate turnover rate during the first 5 hours of the reaction. The time course of the biocatalytic Michael addition reaction can be seen from the increase of the 1H-NMR-signal for the proton at the asymmetric succinate carbon and the decrease in the 1H-NMR-signal for the olefinic protons of fumarate (Fig. 5).
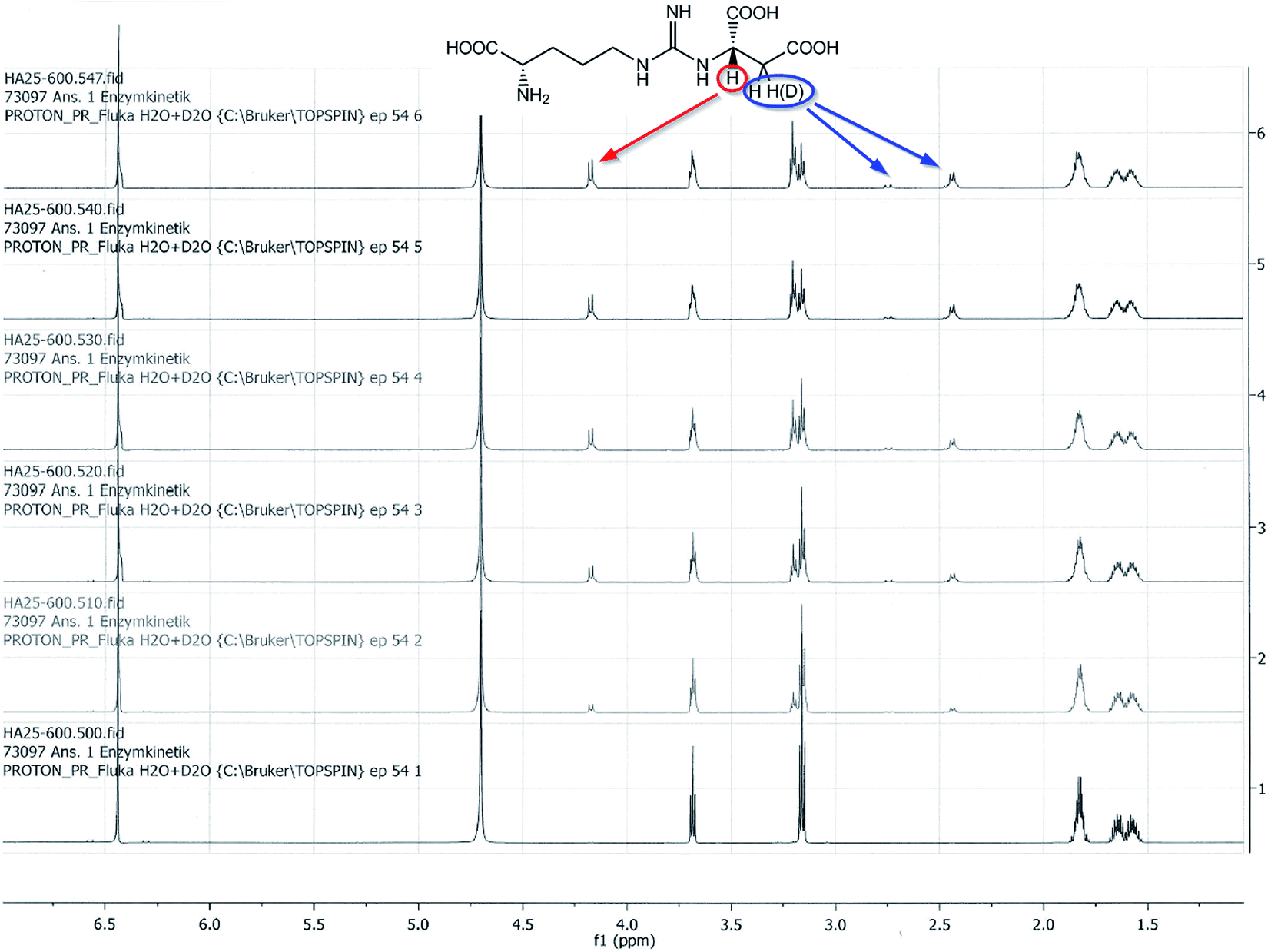 | ||
| Fig. 4 1H-NMR of the biocatalytic Michael addition reaction of L-arginine 2 to fumarate 3 in D2O. 1H-NMR signals at 2.75 and 2.43 ppm show a too small integral due to insertion of deuterium from the solvent (D2O). | ||
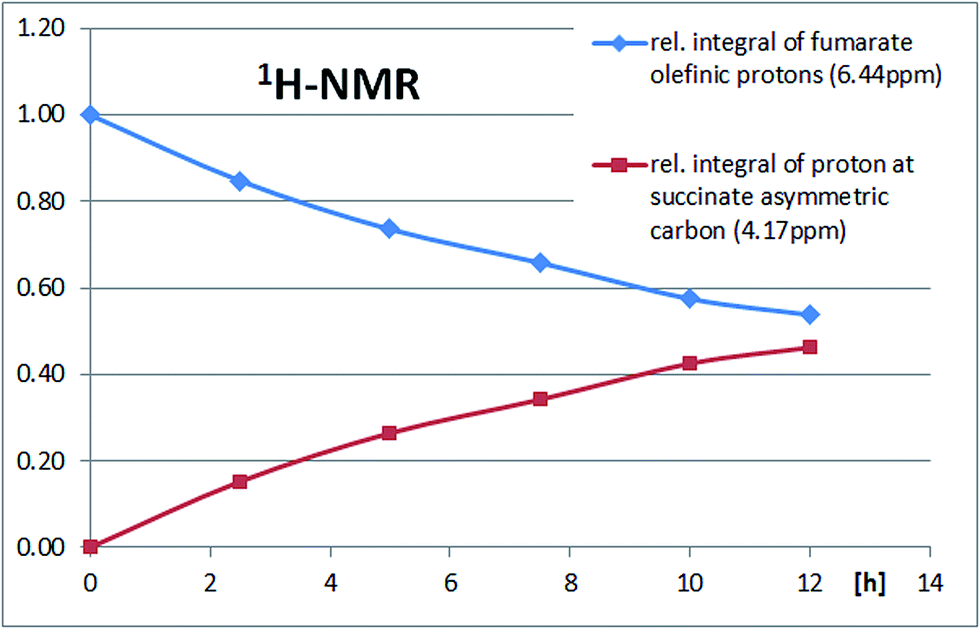 | ||
| Fig. 5 Timecourse of a biocatalytic Michael addition reaction using a solution of 50 μmol fumarate, 100 μmol L-arginine in 1 mL D2O, adjusted to pH 7.5 with NaOH and 20 μL argininosuccinate lyase (ASL) solution (soluble fraction). | ||
Based on the NMR investigations, a synthetic procedure for the biocatalytic asymmetric Michael addition of L-arginine to fumarate using the new recombinant argininosuccinate lyase (ASL) has been successfully developed. This has enabled a simple and scalable synthesis of the pure metabolite N-(([(4S)-4-amino-4-carboxybutyl] amino)iminomethyl)-L-aspartic acid 1 as its lithium salt (L-argininosuccinic acid lithium salt) for the first time, with 70% yield (based on the isolated pure product) and excellent purity at gram scale, as described in the experimental section and the ESI.† This not only provides a straightforward one-step route to L-argininosuccinic acid lithium salt without side-products, but also opens great future opportunities for utilizing biocatalytic asymmetric Michael addition reactions for simple synthetic routes with high molecular economy to complex metabolites using simple building blocks.
Experimental
Gene synthesis and subcloning of ARG4
The argininosuccinate lyase gene ARG4 was derived from Saccharomyces cerevisiae (strain ATCC 204508/S288c) (UniProtKB: P04076) as described by Beacham and coworkers.49 The synthetic gene was codon optimized for E. coli and equipped with an N-terminal 6x-his-tag and a TEV protease cleavage site for optional removal of the affinity tag. The gene was synthesized and subcloned into pET24a(+) via NdeI and NotI by Genscript (USA), giving vector pET24-His(TV)-ARG4-E. coli(op).Recombinant expression of ARG4 in E. coli
E. coli strain BL21 (DE3) was transformed with the derived plasmid using standard procedures.50 Selected E. coli transformants were used for expression tests, in which different media, induction times and temperatures were optimized. The selected conditions were later used for the overexpression of ARG4 in E. coli BL21 (DE3). Firstly, an E. coli overnight culture was prepared in 5 mL LB-medium51 supplemented with 50 μg mL−1 kanamycin and incubated at 30 °C and 180 rpm. Next a 50 mL pre-culture was prepared at 37 °C and 150 rpm in a 250 mL Erlenmeyer flask with baffles. Therefore, 50 mL of Terrific Broth (TB) medium supplemented with 50 μg mL−1 kanamycin51 was inoculated with 500 μL of the according overnight culture. When the pre-culture reached mid-log phase, the main culture was started. Hence, 400 mL of fresh TB medium in a 2 L Erlenmeyer flask was inoculated with 20 mL of the E. coli pre-culture. After the culture reached an OD600 of 1.3 (optical density), protein expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). The main culture was shaken at 25 °C at 110 rpm for 20 h. Cells were harvested by centrifugation at 5000 rpm for 30 min at 4 °C and washed once with an ice-cold 50 mM sodium phosphate buffer at pH 7.5. The E. coli cell pellets containing the overexpressed enzyme (recombinant ARG4) were frozen at −20 °C.Cell disruption
For cell disruption, frozen cells were adjusted to an OD600 of 40 in 50 mM potassium phosphate buffer pH 7.5 and mixed with glass beads (Ø 0.25–0.5 μm) at an approximate ratio of 1:1. Then, the cells were disintegrated mechanically by a cell disruptor (Retsch Mixer Mill MM 200: two cycles for 5 min at 600 rpm). The samples were cooled on ice and insoluble cell debris was removed by centrifugation (5000 rpm, 30 min, at 4 °C). The supernatant, which contained the soluble protein fraction, was immediately used for activity determination.
IMAC-purification and protein content determination of rec. ASL
To purify the recombinant ARG4 by IMAC (immobilized metal ion chromatography), the soluble protein fraction was incubated with Talon® resin on an orbital shaker (30 min, 4 °C). Then, a standard gravity flow protocol was applied to purify the target protein. Therefore, the resin was washed at 4 °C with 10 column volumes (CV) of 50 mM potassium phosphate buffer pH 7, 5 containing 10 mM imidazole to get rid of unspecifically bound protein. The target protein ARG4 was eluted with 1.5 CV of elution buffer (50 mM potassium phosphate buffer pH 7.5, 150 mM imidazole). Samples were desalted on desalting columns against 50 mM potassium phosphate buffer pH 7.5 in order to remove imidazole from the purified protein samples (Zeba Spin columns, Pierce). The purified protein was immediately used for activity determination. The protein content of the purified protein was determined with the BCA method according to the manufacturer's protocol (Pierce Protein Research Products, Thermo Scientific) using bovine serum albumin (BSA) as a standard. The analysis of ARG4 overexpression and the purity was performed using 12.5% SDS-polyacrylamide gels. The gels were stained with Coomassie Blue.Determination of argininosuccinate lyase activity
The analysis of the argininosuccinate lyase activity of ARG4 was based on the production of fumarate according to Kuchel et al.52 and Havir et al.53 and measured spectrophotometrically by the absorbance at a wavelength of 240 nm. ARG4 was assayed at 25 °C by recording at 240 nm the formation of fumarate during the reaction (ε = 2.44 × 103 M−1 cm−1). The assay mixture contained 50 mM potassium phosphate buffer pH 7.5 and 1 mM of potassium L-argininosuccinate, in a total volume of 1 mL. The reaction was initiated by the addition of the recombinant protein. One unit of enzyme activity is defined as the amount of enzyme catalyzing the conversion of 1 μmol of L-argininosuccinate substrate per minute at pH 7.5 and 25 °C. All assays were performed in triplicate.Enzyme stability investigation
Following the determination of the catalytic activity of the argininosuccinate lyase aliquots of the ARG4 in 50 mM potassium phosphate buffer pH 7.5 were transferred into two test tubes. One aliquot of the enzyme was stored as a liquid at 4 °C for one month, the other was freeze-dried (CHRIST beta 1–8 freeze dryer, Martin Christ Freeze Dryers, Osterode am Harz, Germany), where the liquid was frozen at −80 °C. The dehydration step lasted for 20 h at a temperature of −53 °C until dried powder formed. The resulting lyophilized powders were rehydrated to its original volume at room temperature with 50 mM potassium phosphate buffer pH 7.5. Then the samples were subjected to the subsequent argininosuccinate lyase activity tests as described above and the residual enzyme activities were measured on the same day.Reaction kinetics by NMR
The enzymatic reactions were monitored by NMR as shown in Fig. 4 and 5.Gram scale synthesis of N-(([(4S)-4-amino-4-carboxy-butyl]amino) iminomethyl)-L-aspartic acid lithium salt (L-Argininosuccinic acid lithium salt) 1
A mixture of 5 mmol each of L-arginine and fumaric acid in 100 mL of H2O was adjusted to pH 7.5 with 1 M LiOH and to this solution were added 500 μL of the ASL enzyme solution (soluble fraction). The solution was stirred at room temperature and the progress of the reaction was monitored periodically by 1H-NMR and TLC (silica plates, n-propanol:H2O = 2:1, UV 254 and ninhydrin). After 6 days the conversion ceased and the mixture was concentrated on a rotary evaporator at high vacuum and room temperature, which gave a white solid. This was dissolved in 5 mL H2O and then 10 mL n-propanol were added. The resulting emulsion was flash-chromatographed on silica with n-propanol:H2O = 2:1. The early fractions contained unreacted fumarate, then the L-argininosuccinate 1 was eluted. These fractions were pooled, filtered, frozen and lyophilised to give 1.07 g (∼70%) of the pure product N-(([(4S)-4-amino-4-carboxy-butyl]amino)iminomethyl)-L-aspartic acid 1 lithium salt (L-argininosuccinic acid 1 lithium salt) in the form of a colorless solid.
Analytical data for N-(([(4S)-4-amino-4-carboxy-butyl]amino) imino-methyl)-L-aspartic acid 1 lithium salt (L-Argininosuccinic acid 1 lithium salt)
NMR: δH (400 MHz; D2O) 4.14 (1H, dd, J1 3.4, J2 9.9, succinyl–CH–NH), 3.65 (1H, dd, J1 = J2 6.2, arginyl-CαH), 3.17 (2H, t, J 6.8, arginyl-CδH2), 2.71 (1H, dd, J1 3.4, J2 16.1, succinyl-CH), 2.41 (1H, dd, J1 9.9, J2 16.1, succinyl-CH′), 1.80 (2H, m, arginyl-CβH2), 1.58 (2H, m, arginyl-CγH2); δC (100.6 MHz; D2O) 178.9, 177.1, 174.4 (3 COOH), 155.8 (guanidino-C), 55.1, 54.3 (2 NH2–C–COOH), 40.6 (succinyl-CH2 and arginyl-Cδ), 27.6 (arginyl-Cβ), 23.9 (arginyl-Cγ). H2O content (Karl Fischer titration): 14.1% elemental analysis, found: C, 33.3; H, 6.2; N, 15.5%. Calculated for C10H16N4O6Li2. 14.1% H2O: C, 35.2; H, 6.2; N 16.4% TLC, purity: 95.1% (n-propanol:NH4OH(25%):H2O = 6:3:1; ninhydrin; Rf 0.4) qNMR, content: 94.4% (calculated as di-lithium salt including 14.1% H2O).
Conclusions
A simple and straightforward biocatalytic asymmetric Michael addition reaction has been established for the synthesis of the key metabolite N-(([(4S)-4-amino-4-carboxybutyl]amino)imino methyl)-L-aspartic acid, commonly referred to as L-argininosuccinate. This one-step addition reaction has been developed by running part of the urea cycle in reverse. Argininosuccinate lyase from Saccharomyces cerevisiae has been selected as biocatalyst. The corresponding gene ARG4 was expressed in E. coli for the production of the highly active and stable argininosuccinate lyase (ASL) biocatalyst. The use of this argininosuccinate lyase and reaction monitoring by NMR enabled the development of a biocatalytic asymmetric Michael addition reaction as a novel green chemistry route with high molecular economy for the synthesis of this important metabolite at gram scale.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Namtso Reichlin for NMR-measurements.Notes and references
- A. Michael, J. Prakt. Chem., 1887, 35, 349–356 CrossRef; A. Michael, J. Am. Chem. Soc., 1887, 9, 112–115 Search PubMed.
- E. Reyes, U. Uria, J. L. Vicario and L. Carrillo, Org. React., 2016, 90, 1–898 Search PubMed.
- M. Sawamura, H. Hamashima and Y. Ito, J. Am. Chem. Soc., 1992, 114, 8295–8296 CrossRef CAS.
- H. Sasai, T. Arai and M. Shibasaki, J. Am. Chem. Soc., 1994, 116, 1571–1572 CrossRef CAS.
- K. Funabashi, Y. Saida, M. Kanai, T. Arai, H. Sasai and M. Shibasaki, Tetrahedron Lett., 1998, 39, 7557–7558 CrossRef CAS.
- J. M. Betancort and C. F. Barbas, Org. Lett., 2001, 3, 3737–3740 CrossRef CAS PubMed.
- D. Enders and A. Seki, Synlett, 2002, 26–28 CrossRef CAS.
- B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423–2425 CrossRef CAS PubMed.
- H. Huang and E. N. Jacobsen, J. Am. Chem. Soc., 2006, 128, 7170–7171 CrossRef CAS PubMed.
- C. Hui, F. Pu and J. Xu, Chem.–Eur. J., 2016, 23, 4023–4036 CrossRef PubMed.
- A. Ueda, T. Umeno, M. Doi, K. Akagawa, K. Kudo and M. Tanaka, J. Org. Chem., 2016, 81, 6343–6356 CrossRef CAS PubMed.
- Y. Hamashima, D. Hotta, N. Umebayashi, Y. Tsuchiya, T. Suzuki and M. Sodeoka, Adv. Synth. Catal., 2005, 347, 1576–1586 CrossRef CAS.
- X. Gu, Y. Dai, T. Guo, A. Franchino, D. J. Dixon and J. Ye, Org. Lett., 2015, 17, 1505–1508 CrossRef CAS PubMed.
- Aza-Michael addition: M. Sánchez-Roselló, J. L. Aceña, A. Simón-Fuentes and C. del Pozo, Chem. Soc. Rev., 2014, 43, 7430–7453 RSC Oxa-Michael addition: C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988–999 RSC Sulfa-Michael addition: P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807–8864 CrossRef CAS PubMed.
- Y. Xu, K. Ohori, T. Ohshima and M. Shibasaki, Tetrahedron, 2002, 58, 2585–2588 CrossRef CAS.
- M. Ozeki, N. Hayama, S. Fukutome, H. Egawa, K. Arimitsu, T. Kajimoto, S. Hosoi, H. Iwasaki, N. Kojima, M. Node and M. Yamashita, ChemistrySelect, 2016, 1, 2565–2569 CrossRef CAS.
- J.-S. Yu, F.-M. Liao, W.-M. Gao, K. Liao, R.-L. Zuo and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 7381–7385 CrossRef CAS PubMed.
- D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861–863 CrossRef CAS PubMed.
- Z. Guan, L.-Y. Lia and Y.-H. He, RSC Adv., 2015, 5, 16801–16814 RSC.
- J. Du, B. Shuai, M. Tao, G. Wang and W. Zhang, Green Chem., 2016, 18, 2625–2631 RSC.
- S. Luo, X. Mi, L. Zhang, S. Liu, H. Xu and J.-P. Cheng, Angew. Chem., Int. Ed., 2006, 45, 3093–3097 CrossRef CAS PubMed.
- Y. F. Wang, R. X. Chen, K. Wang, B. B. Zhang, Z. B. Li and D. Q. Xu, Green Chem., 2012, 14, 893–895 RSC.
- H. Y. Bae and C. E. Song, ACS Catal., 2015, 5, 3613–3619 CrossRef CAS.
- B. Kusebauch, B. Busch, K. Scherlach, M. Roth and C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 5001–5004 CrossRef CAS PubMed.
- X. Garrabou, T. Beck and D. Hilvert, Angew. Chem., Int. Ed., 2015, 54, 5609–5612 CrossRef CAS PubMed.
- J.-Y. van der Meer, H. Poddar, B.-J. Baas, Y. Miao, M. Rahimi, A. Kunzendorf, R. van Merkerk, P. G. Tepper, E. M. Geertsema, A.-M. W. H. Thunnissen, W. J. Quax and G. J. Poelarends, Nat. Commun., 2016, 7(1), 1–16 Search PubMed.
- B. C. Chen, L. H. Ngu and M. Y. Zabedah, Malays. J. Pathol., 2010, 32, 87–95 CrossRef PubMed.
- C.-Y. Wu, S.-T. Chen, S.-H. Chiou and K.-T. Wang, Biotechnol. Lett., 1991, 13, 405–410 CrossRef CAS.
- P. L. Hartlage, M. E. Coryell, W. K. Hall and D. A. Hahn, J. Pediatr., 1974, 85, 86–88 CrossRef CAS PubMed.
- B. Levin, H. M. M. Mackay and V. G. Oberholzer, Arch. Dis. Child., 1961, 36, 622–632 CrossRef CAS PubMed.
- F. Qiu, Y.-R. Chen, X. Liu, C.-Y. Chu, L.-J. Shen, J. Xu, S. Gaur, H. J. Forman, H. Zhang, S. Zheng, Y. Yen, J. Huang, H.-J. Kung and D. K. Ann, Sci. Signaling, 2014, 7, ra31 CrossRef PubMed.
- B. Delage, D. A. Fennell, L. Nicholson, I. McNeish, N. R. Lemoine, T. Crook and P. W. Szlosarek, Int. J. Cancer, 2010, 126, 2762–2772 CAS.
- S. Rabinovich, L. Adler, K. Yizhak, A. Sarver, A. Silberman, S. Agron, N. Stettner, Q. Sun, A. Brandis, D. Helbling, S. Korman, S. Itzkovitz, D. Dimmock, I. Ulitsky, S. C. S. Nagamani, E. Ruppin and A. Erez, Nature, 2015, 527, 379–383 CrossRef CAS PubMed.
- S. Moncada and A. Higgs, N. Engl. J. Med., 1993, 329, 2002–2012 CrossRef CAS PubMed.
- L. A. J. O'Neill, R. J. Kishton and J. Rathmell, Nat. Rev. Immunol., 2016, 16, 553–565 CrossRef PubMed.
- A. Erez, Genet. Med., 2013, 15, 251–257 CrossRef CAS PubMed.
- L. Hu, A. V. Pandey, S. Eggimann, V. Rüfenacht, D. Möslinger, J. M. Nuoffer and J. Häberle, J. Biol. Chem., 2013, 288, 34599–34611 CrossRef CAS PubMed.
- L. Hu, A. V. Pandey, C. Balmer, S. Eggimann, V. Rüfenacht, J. M. Nuoffer and J. Häberle, J. Inherited Metab. Dis., 2015, 38, 815–827 CrossRef CAS PubMed.
- W. E. O'Brien and R. H. Barr, Biochemistry, 1981, 20, 2056–2060 CrossRef.
- K. Farrell and S. Overton, Biochem. J., 1987, 242, 261–266 CrossRef CAS PubMed.
- H. Nakamura, T. Yada, T. Saheki, T. Noda and S. Nakagawa, Brain Res., 1991, 539, 312–315 CrossRef CAS PubMed.
- J. F. Feng, T. M. Chen, Y. A. Wen, J. Wang and Z. G. Tu, J. Clin. Lab. Anal., 2008, 22, 220–227 CrossRef CAS PubMed.
- S. C. S. Nagamani, A. Erez and B. Lee, Genet. Med., 2012, 14, 501–507 CrossRef CAS PubMed.
- Y.-L. Cao, G.-L. Li, K.-T. Wang, H.-Y. Zhang and L.-F. Li, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 682–684 CrossRef CAS PubMed.
- R. Tischner, M. Galli, Y. M. Heimer, S. Bielefeld, M. Okamoto, A. Mack and N. M. Crawford, FEBS J., 2007, 274, 4238–4245 CrossRef CAS PubMed.
- S. Ratner, Methods Enzymol., 1957, 3, 643–647 Search PubMed.
- J.-C. Jauniaux, L. A. Urrestarazu and J.-M. Wiame, J. Bacteriol., 1978, 133, 1096–1107 CAS.
- K. Murakami-Murofushi and S. Ratner, Anal. Biochem., 1979, 95, 139–155 CrossRef CAS PubMed.
- I. R. Beacham, B. W. Schweitzer, H. M. Warrick and J. Carbon, Gene, 1984, 29, 271–279 CrossRef CAS PubMed.
- D. Hanahan, J. Mol. Biol., 1983, 166, 557–580 CrossRef CAS PubMed.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular Cloning, Cold Spring Harbor Laboratory Press, New York, 1989, vol. 2 Search PubMed.
- P. W. Kuchel, L. W. Nichol and P. D. Jeffrey, Biochim. Biophys. Acta, Enzymol., 1975, 397, 478–488 CrossRef CAS.
- E. A. Havir, H. Tamir, S. Ratner and R. C. Warner, J. Biol. Chem., 1965, 240, 3079–3088 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical Data. See DOI: 10.1039/c7ra10236d |
| This journal is © The Royal Society of Chemistry 2017 |