DOI:
10.1039/C7RA10148A
(Paper)
RSC Adv., 2017,
7, 47767-47774
Restorable, high-strength poly(N-isopropylacrylamide) hydrogels constructed through chitosan-based dual macro-cross-linkers with rapid response to temperature jumps†
Received
12th September 2017
, Accepted 4th October 2017
First published on 11th October 2017
Abstract
To address the poor mechanical performance of conventional thermo-responsive poly(N-isopropylacrylamide) (pNIPAM) hydrogels, methacryloyl covalently-modified chitosan (methacryloylchitosan, MACS) was conceived as the chemical macro-cross-linker and employed cooperatively with chitosan, as the physical macro-cross-linker, to construct dually macro-cross-linked pNIPAM hydrogels for the first time. The employed MACS was synthesized through acylation of chitosan using methacrylic acid activated by N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxy-succinimide (NHS). The influence of the preparation conditions, including the solids content, the dosage and component of the cross-linkers as well as the amount of the initiator, on the mechanical performance and swelling/deswelling properties of the obtained hydrogels was investigated systematically. The prepared dual-cross-linked pNIPAM hydrogels exhibit high mechanical strength, manifested in the achieved compressive stress of 8.75 MPa at 98% strain and tensile strength of 0.105 MPa. The high recovery efficiency (beyond 95%) from cyclic tensile tests reveals the good recoverability of these hydrogels. Moreover, they exhibit excellent thermo-responsiveness, to be more specific, rapid deswelling rates, i.e., most can release 50% of the absorbed water in ∼10 minutes on the temperature jump across the phase transition temperature. In particular, the deswelling rate increases with the increase of either the cross-linkers or MACS. The prominent mechanical performance and rapid responsiveness of the novel pNIPAM hydrogels is highly conductive for their future applications.
Introduction
Hydrogels are insoluble, three-dimensional networks constructed from hydrophilic polymers by virtue of chemical and/or physical cross-linking. They have a porous microstructure that holds a huge quantity of water, resulting in a wet and soft nature resembling biological tissues. They show appealing potential in environmental science, catalysis, food industry, hygienic products and especially biomedical products. Hereinto, smart hydrogels which can undergo distinct physical changes towards a small environmental change are much concerned due to the huge prospect for the applications in targeted controlled release, biosensors and actuators, soft robotics, intelligent switches and so on. Significant attention has been paid to smart hydrogels from both scientific and technological researchers since the first report on thermo-responsive poly(N-isopropylacrylamide) (pNIPAM) hydrogel in 1978.1 Thermo-responsive hydrogels can drastically alter their volume at a certain temperature termed as the volume-phase transition temperature (VPTT) through shrinking/swelling in response to the temperature change. The VPTT of pNIPAM hydrogel is around 32 °C, which is close to the normal body temperature of human being and can be modulated feasibly via manipulating hydrophilic/hydrophobic balance through incorporation of various co-monomers.2,3 The feature has attracted the attention of researchers from materials science and biomedical field all the time since Tanaka's pioneering work.1 However, the fragility and weakness of conventional pNIPAM hydrogels seriously impeded their actual utilization, owing to the inhomogeneous network structure usually caused by small molecular cross-linkers.
Researchers have long committed to developing hydrogels with high strength, and huge progress has been made from the beginning of this century. Several types of highly mechanical hydrogels have been developed, including nano-composite (NC) hydrogels,4–8 double-network (DN) hydrogels,9,10 slide-ring (SR) hydrogels,11 tetra-armed polyethylene glycol-based hydrogel,12 and macromolecular micro-sphere composite (MMC) hydrogels.13 The strategies for some of the above hydrogels have been attempted in fabricating high-strength pNIPAM hydrogels. Nano-composite technology has become a common tactics for boosting the performance of polymer materials including soft-wet hydrogels. In effect, the first report on NC hydrogels involves pNIPAM-based NC hydrogels with high tensile strength and stretchability by exploiting clay as the nano-fillers.4 Thenceforth, silica particles,14 graphene oxide,15 starch,16 and pNIPAM nano-particles17 were employed in fabricating high-strength pNIPAM NC hydrogels in succession. The strengthening effect of nano-particles in NC hydrogels originates primarily from the multifunctional cross-linking joints on the nano-particle's surface; endow them the name multifunctional cross-linkers. DN technique, as a feasible and widely adopted strategy in constructing hydrogels with high strength, has been explored in pNIPAM-based hydrogels. Fei et al. firstly reported pNIPAM–pNIPAM DN hydrogel with a maximum compressive strength of 0.452 MPa,18 which comprises two pNIPAM networks of different cross-linking density using N,N′-methylenebisacrylamide as the cross-linking agents. Later they improved the compressive strength significantly through copolymerizing NIPAM with 2-acrylamido-2-methylpropanesulfonic acid during the formation of the first network, but the obtained DN hydrogels retained higher water after deswelling.19 Li et al. attained a thermo- and pH-responsive DN hydrogel with compressive strength up to 3.6 MPa using pNIPAM as the 1st network, polyacrylic acid (PAA) as the 2nd network and graphene oxide (GO) as the reinforcing nano-filler.20 Other strategies, such as SR and MMC technique were also reported in fabricating high-strength pNIPAM hydrogels. Take for examples, Imran et al. reported the SR pNIPAM hydrogels with high tensile strength of 40.9 kPa and fracture elongation of 912% using α-cyclodextrin threaded in polyethylene glycol (α-CD–PEG) complex as the crosslinker.21 Wang et al. prepared thermo-responsive pNIPAM hydrogels employing peroxidized macromolecular microspheres as polyfunctional initiating species and the macro-cross-linker, the compressive strength of which exceed 10 MPa (at 95% strain) with 60% water content.22
Although several strategies have been attempted to promote the mechanical performance of pNIPAM hydrogels, there remains a long distance for pNIPAM hydrogels to meet the need of practical applications.23 It is still challenging to develop pNIPAM hydrogels with comprehensive excellent properties, including rapid response rate and excellent mechanical performance, such as compressive and tensile properties, the recoverability during repeated loading–unloading and etc. Recently, we developed a series of hydrogels with high tensile and compressive strength, high fracture elongation and toughness employing polycationic chitosan as a physical macro-cross-linker,24 including poly(methacrylic acid) (PMAA) hydrogel, poly(acrylic acid) (PAA) hydrogel, poly(acrylamide) (PAM) hydrogel and so on. However, pNIPAM hydrogel physically macro-cross-linked by chitosan cannot achieve satisfactory mechanical properties comparative to PMAA, PAA and PAM hydrogels, being too soft and somewhat sticky. Just as already indicated, ionic interactions serve as the primary cross-links to maintain the shape of the hydrogels physically cross-linked by chitosan, some secondary interactions such as H-bonding also have significant effects in promoting the hydrogel's mechanical performance.24 The softness of pNIPAM hydrogel can be attributed to the weak secondary H-bonding between pNIPAM molecules ascribed to the steric hindrance of large isopropyl groups. Thus, additional interactions must be introduced to enhance the mechanical performance of pNIPAM hydrogel except for ionic interactions. As the strongest one among all interactions, covalent cross-linking should be the optimum strategy. Within this context, to avert the fragility brought about usually by small molecular cross-linkers, a multifunctional macro-cross-linker, namely methacryloylchitosan, was conceived to construct dually macro-cross-linked pNIPAM hydrogels in conjunction with chitosan.
The internal microstructure, mechanical performance, temperature-sensitivity and swelling/deswelling kinetics of the prepared hydrogels were investigated and presented herein.
Experimental
Materials
Chitosan (degree of deacetylation >90.0%, molecular weight of 700–800 kDa, Shanghai Lanji Technology Development Co., Ltd), N-(3-dimethylaminopropyl)-N′-ethyl-carbodiimide hydrochloride (EDC, 98.0%, Aladdin Industrial Corporation, Shanghai, China), N-hydroxysuccinimide (NHS, 98.0%, Aladdin Industrial Corporation, Shanghai, China), N-iso-propylacrylamide (NIPAM, >99%, Acros Organics), potassium persulfate (KPS, A.R., Shanghai Chemical Reagent Corporation, Shanghai), methacrylic acid (MAA, >99%, Aladdin Industrial Corporation, Shanghai, China), were all used as received.
Fabrication of dually macro-cross-linked pNIPAM hydrogels
Methacryloylchitosan (MACS) was synthesized according to literature (see the ESI†).25 To fabricate dually macro-cross-linked pNIPAM hydrogels, the synthesized MACS, chitosan, MAA (with equivalent –COOH groups to –NH2 groups in chitosan) and NIPAM were dissolved in distilled water under magnetic stirring until a transparent solution was obtained. Then KPS dissolved in a small amount of water was added in and stirred for a few seconds. The solution was fed separately into cylindrical glass tubes of 16 mm and 10 mm in diameter. Afterwards, the tubes were sealed and immersed in a thermostat water bath after being degassed through bubbling N2 for 30 min. Finally, the temperature of the water bath was controlled at 27.5 °C for 48 h to allow the formation of hydrogels through free-radical polymerization.
Internal microstructure of the pNIPAM hydrogels
The as-prepared hydrogels were cut into small pieces, soaked in distilled water for 48 h (with the water being refreshed every 8 h). After being freeze-dried, the hydrogel slices were fixed onto the object stage and deposited an ultrathin conductive gold coating by low vacuum sputter coating. The micro-structure of the prepared hydrogels was visualized on a TM3000 scanning electron microscope (SEM) at the acceleration voltage of 15 kV.
Mechanical performance of the as-prepared pNIPAM hydrogels
Compressive tests. The unidirectional compressive tests were performed on an electronic universal testing machine (WDW-5T, Jinan HengRui Jin Testing Machine Co., Ltd, China) furnished with a 5000 N load cell. The as-prepared hydrogels were cut into cylindrical samples of 16 mm in diameter and 10 mm in height. To avoid water evaporation during testing, each sample was covered with a thin layer of silicon oil in advance. The translational speed of the pressure plate was kept at 5 mm min−1 in all measurements. The compressive stress (σc) and strain (εc) were calculated according to eqn (1) and (2) respectively.| | |
εc = (H0 − Hi)/H0 × 100%
| (2) |
where Fc, S denoted the applied compressive force (N) and the cross-sectional area of the initial sample respectively. H0 and Hi represented the sample height before and after compression. To ensure accuracy, the final results were the average of at least three measurements.
Tensile tests. The unidirectional tensile tests were performed on the same machine furnished with a 100 N load cell. The specimens for tensile tests were cylindrical with a diameter of 10 mm and a length of 80 mm. Each sample was covered with a thin layer of silicon oil in advance to avoid water evaporation during testing. In each testing, the gauge distance was ∼10 mm and the crosshead speed was 100 mm min−1. The tensile stress (σt) and strain (εt) was calculated according to eqn (3) and (4) respectively.| | |
εt = [(l − l0)/l0] × 100%
| (4) |
where S was the cross-sectional area of the initial hydrogel sample, Ft was the value of the applied force, l0 was the initial gauge length, l was the gauge length during testing. The tensile strength (σb) and fracture elongation (εb) corresponds to the stress and strain at break.
Recoverability of the as-prepared pNIPAM hydrogels
The recoverability of the hydrogels was determined by the tensile loading–unloading cycles, carried through performing subsequent loading on the same specimen immediately after unloading. The crosshead speeds of both loadings and unloadings were all kept at 100 mm min−1. The recovery efficiency was evaluated based on the stress ratio of the nth loading to the first one at the same strain.
Swelling and deswelling kinetics of the pNIPAM hydrogels
Both swelling and deswelling kinetics was determined by means of gravimetry. The swelling kinetics was investigated at 15 °C. In brief, slices of dry samples were weighed and soaked in distilled water of 15 °C. During swelling, each sample was regularly taken out, blotted carefully the residual water on the surface with filter paper and weighed, till the weight no longer changed. The swelling ratio (WS) of the hydrogel sample at different time was calculated according to eqn (5),where Wd and Wt denoted the weight of the dry sample and the sample at time t respectively. To ensure accuracy, the final results were the average of five measurements.
The hydrogel sample upon swelling equilibrium at 15 °C was transferred to water at 37 °C for deswelling. During deswelling, each sample was regularly taken out, blotted carefully the residual water on the surface and weighed, till the weight no longer changed. The water retention ratio (WR) was calculated according to eqn (6),
| | |
WR = (Wt − Wd)/(We − Wd)
| (6) |
where
We represented the sample weight at swelling equilibrium at 15 °C,
Wd and
Wt denoted the weight of the dry sample and the sample at time
t. To ensure accuracy, the final results were the average of five repetitions.
Temperature-sensitivity of the pNIPAM hydrogels
The temperature-sensitivity was investigated by means of gravimetry at elevated temperature. The hydrogel sample reaching swelling equilibrium at 15 °C was incubated in distilled water of different temperature in succession. The temperature of the distilled water ranges from 15 to 55 °C with a 5 °C interval. Under each temperature, the hydrogel was allowed to deswell to equilibrium, taken out and weighed after being blotted the excess surface water carefully. The water retention (WR) at different temperatures was obtained from eqn (7),| | |
WR = (WT − Wd)/(W15 − Wd)
| (7) |
where WT was the equilibrium weight of the hydrogel at temperature T, W15 was the equilibrium weight at 15 °C, Wd was the dry hydrogel weight. To ensure accuracy, the final results were the average of five repetitions.
Results and discussion
Fabrication of dually macro-cross-linked pNIPAM hydrogels
The main reaction occurred in the synthesis of MACS is depicted in Fig. SI1.† MACS was optimized by varying the molar ratio of MAA to chitosan. The structure of the obtained MACS was verified by IR technique (Fig. SI2†). Solubility experiments indicate that the obtained product is not only soluble in water, but also in dilute aqueous solution of NaOH, suggesting that covalent bonding is formed between chitosan and MAA instead of ionic interactions between –NH2 and –COOH groups. The influence of different MACS obtained on the mechanical performance of the corresponding pNIPAM hydrogels was investigated (Fig. SI3†), and the optimal one synthesized with a molar ratio of MAA to chitosan of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was utilized in later fabrication of pNIPAM hydrogels.
:1 was utilized in later fabrication of pNIPAM hydrogels.
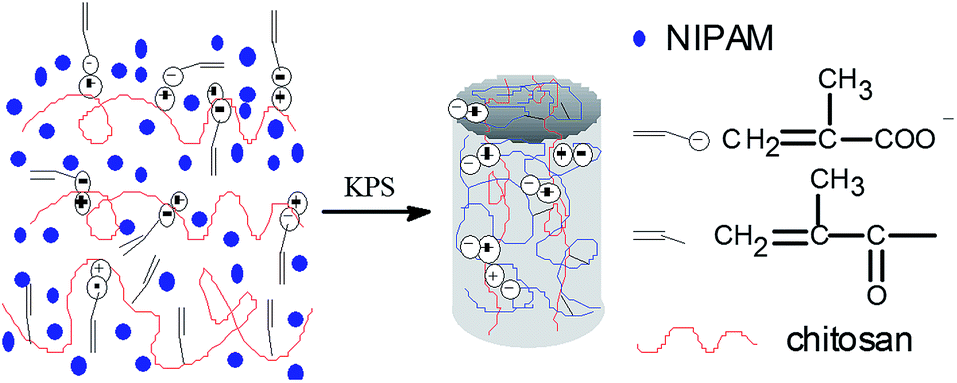
In the fabrication of dually macro-cross-linked pNIPAM hydrogels, chitosan plays the role of a physical macro-cross-linker via ionic complex with MAA which become part of the hydrogel network through copolymerization with NIPAM, as described previously.24 Methacryloyl covalently-modified chitosan functions as a chemical macro-cross-linker. The formation of the pNIPAM hydrogels is outlined in Scheme 1. A series of pNIPAM hydrogels were fabricated by varying the solids content (x wt%, denoted as SC x), the weight percentage of the cross-linkers to NIPAM (y wt%, denoted as Cr y), the weight percentage of MACS in the cross-linkers (z wt%, denoted as MACS z), and the molar ratio of KPS to NIPAM (w mol%, denoted as KPS w). Their internal microstructure, mechanical properties, swelling/deswelling kinetics and thermo-responsiveness were investigated systematically.
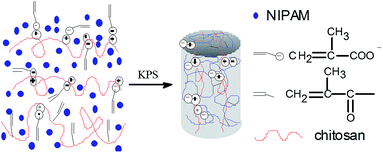 |
| | Scheme 1 Formation mechanism of the dually macro-cross-linked hydrogels. | |
Internal morphology of the dually macro-cross-linked pNIPAM hydrogels
Fig. 1 presents SEM micrographs of pNIPAM hydrogels with different amount of cross-linkers and MACS. It is observed that channel-like pores are formed inside the hydrogel network, forming a tubular microstructure. The pore sizes decrease and more pores are formed with the increase of the total cross-linkers. This is readily comprehensible since the cross-linking density increases with increasing the cross-linkers. At a fixed amount of the cross-linkers, the internal microstructure alters in the same manner with the increase of MACS as with the cross-linkers. This might be ascribed to the increasing, stable covalent cross-linking with increasing MACS, restraining the dissociation of ionic interactions upon swelling. The channel-like porous microstructure will be advantageous for water's penetrating in and out hydrogel network.
 |
| | Fig. 1 SEM images with varying cross-linkers and MACS (fixed SC at 20 wt% and KPS at 0.22 mol% for all; Cr at 6.4 wt% (up), MACS at 20 wt% (down) respectively). | |
Mechanical performance of the as-prepared pNIPAM hydrogels
The mechanical performance is crucial for smart hydrogels' applications in many fields, and is dependent on many factors such as the network structure, the nature of the polymeric matrix, the water content and so on. To optimize the fabrication conditions for achieving hydrogels with optimum properties, the influence of major parameters (including the dosage and formulation of the cross-linkers, the solids content and the amount of KPS) on the properties of the hydrogels was investigated and is presented here.
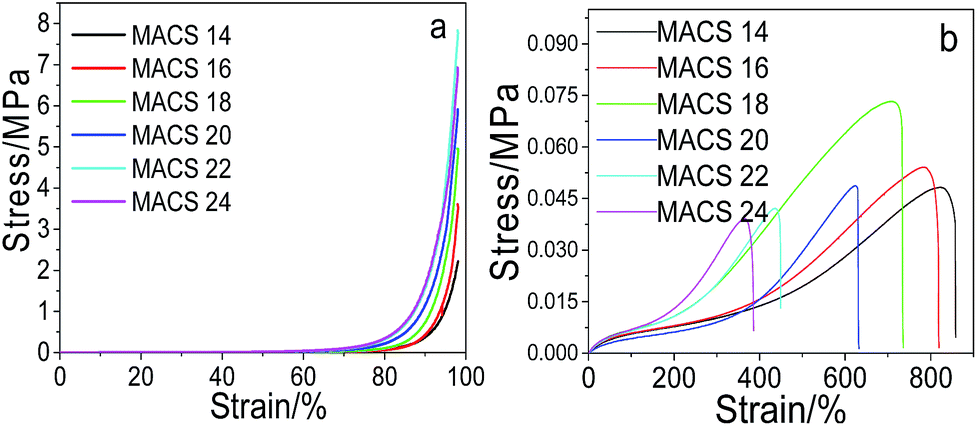
Effect of MACS on the mechanical performance. Fig. 2 shows the compressive (a) and tensile (b) stress–strain curve of pNIPAM hydrogels with different amount of MACS. It can be seen from Fig. 2a that the compressive stress (at 98% strain) increases with the increase of MACS in the first place, and then decreases with further increase of MACS. When fixed the SC at 20 wt%, the amount of the cross-linkers at 5.7 wt% and the KPS ratio at 0.22 mol%, a maximum compressive stress (at 98% strain) of 7.9 MPa is attained at 22 wt% MACS. The changing trend of the tensile strength resembles to the compressive stress, i.e., increases initially and then decreases with increasing MACS, reaching a maximum of ∼0.074 MPa at 18 wt% MACS, according to Fig. 2b. Yet the elongation at break decreases monotonously with the increase of MACS.
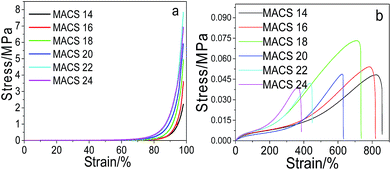 |
| | Fig. 2 Compressive (a) and tensile (b) stress–strain curves of the prepared hydrogels with varying MACS (at fixed SC of 20 wt%, Cr of 5.7 wt%, KPS of 0.22 mol%). | |
The changing trends of the compressive and tensile properties with MACS are plausible. At a constant dosage of the cross-linkers, increase of MACS (as the chemical macro-cross-linker) implies decrease of chitosan (as the physical macro-cross-linker), which brings about an increase in covalent cross-linking and a decrease of ionic interactions in the hydrogel network. The increase in covalent cross-linking enhances the rigidity of the hydrogels owing to the high strength of covalent bonds, leading to decrease of the fracture elongation on one side and increase of compressive stress and tensile strength initially on the other side. However, too much covalent bonding will result in fragility of the hydrogels and hence decrease of the compressive stress and tensile strength.
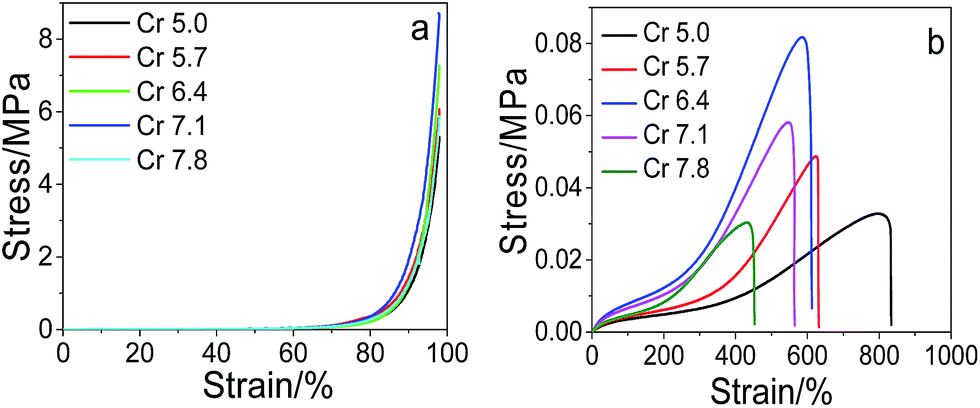
Effect of the cross-linkers on the mechanical performance. The changing trends of the compressive stress, the tensile strength and elongation at break with the total cross-linkers are similar to that with MACS (see Fig. 3). When the SC is fixed at 20 wt%, the amount of MACS is fixed at 20 wt% and the dosage of KPS is 0.22 mol%, the maximum compressive stress attained is 8.75 MPa at 7.1 wt% cross-linkers and a maximum tensile strength of 0.082 MPa is attained at 6.4 wt% cross-linkers. Since increase in the cross-linkers raises the cross-linking density of the network and decreases the flexibility of the polymer chains, thus leading to the decrease of the fracture elongation on one hand and the increase of the compressive stress and tensile strength initially on the other hand. However, too many cross-linkers will result in fragility of the hydrogels and hence decrease of the compressive stress and tensile strength. The results indicate much better mechanical performance of the prepared dually macro-cross-linked pNIPAM hydrogels than conventional one cross-linked by N,N′-methylenebisacrylamide (Fig. SI4†).
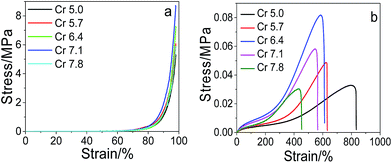 |
| | Fig. 3 Compressive (a) and tensile (b) stress–strain curves of the prepared hydrogels with varying crosslinkers (at fixed SC of 20 wt%, MACS of 20 wt%, KPS of 0.22 mol%). | |
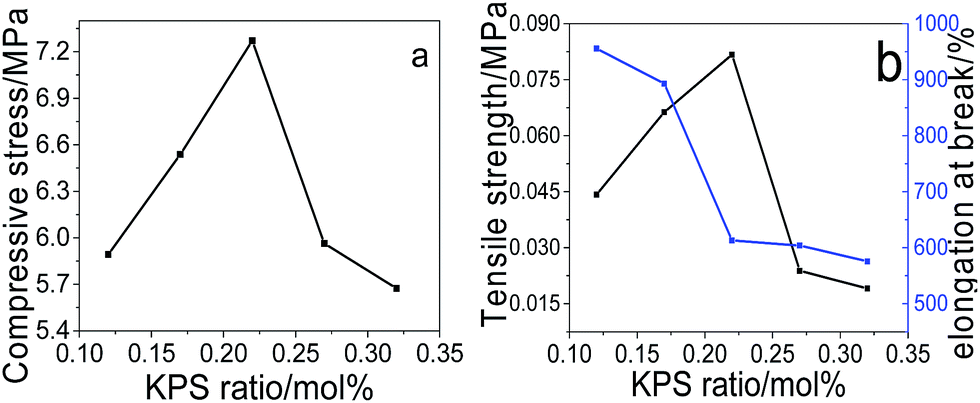
Effect of KPS ratio on the mechanical performance. Fig. 4 demonstrates the compressive (a) and tensile (b) performance of pNIPAM hydrogels fabricated under the initiating of different amount of KPS, obtained from the measured compressive and tensile stress–strain curves (Fig. SI5†). It is found from Fig. 4 that both too much and too little amount of KPS are not favourable for the mechanical strength of the prepared hydrogels, both the maximum compressive stress (at 98% strain) and the tensile strength are achieved at 0.22 mol% of KPS to NIPAM. Since the polymerization cannot proceed completely under the initiation of too little amount of KPS, leading to low density of polymers in hydrogel network, hence resulting in low strength of the hydrogels. However, usage of too much KPS leads to large amounts of primary radicals during initiation, bringing about low-molecular-weight polymers, hence contributing to the decrease of the mechanical strength.
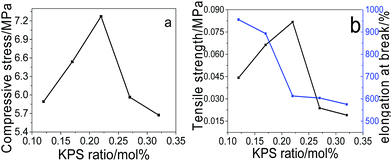 |
| | Fig. 4 Effects of KPS on the compressive (a) and tensile (b) performance of the prepared hydrogels (at fixed SC of 20 wt%, MACS of 20 wt%, Cr of 6.4 wt%). | |
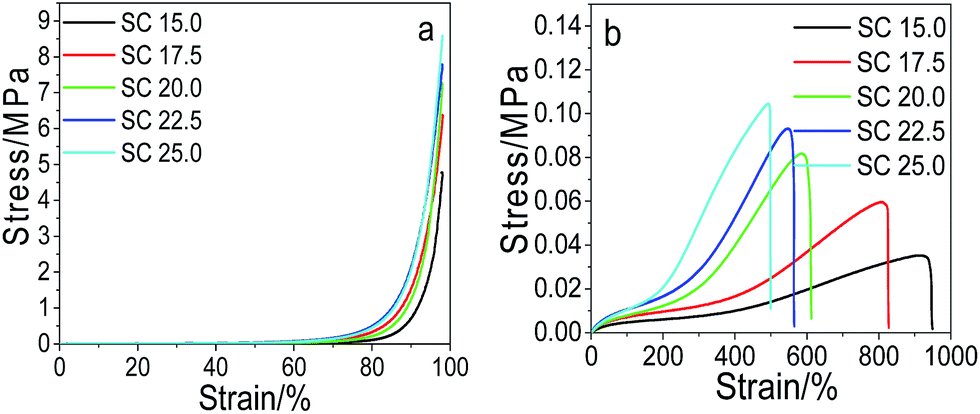
Effect of the solids content on the mechanical performance. The effect of the solids content on the mechanical performance of pNIPAM hydrogels is presented in Fig. 5. It can be seen that both the compressive stress at 98% strain and the tensile strength increases with the increase of the solids content. At a fixed dosage of the cross-linkers of 6.4 wt%, MACS of 20 wt% and KPS of 0.22 mol%, the maximum compressive stress and tensile strength are both attained at 25 wt% SC, being 8.65 MPa and 0.105 MPa respectively. However, the fracture elongation decreases with the increase of the SC. As increase in the SC leads to rise in the polymer density in the hydrogels, thus contributes to more entanglement between polymers, consequently enhancing the mechanical strength. Nevertheless, the entanglement between polymers reduces the flexibility of the polymers, consequently reducing the elongation at break.
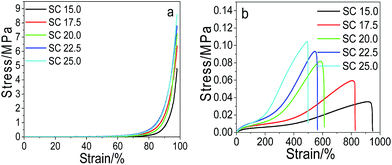 |
| | Fig. 5 Compressive (a) and tensile (b) stress–strain curves of the prepared hydrogels with varying SC (at fixed MACS of 20 wt%, Cr of 6.4 wt%, KPS of 0.22 mol%). | |
Recoverability of the as-prepared pNIPAM hydrogels
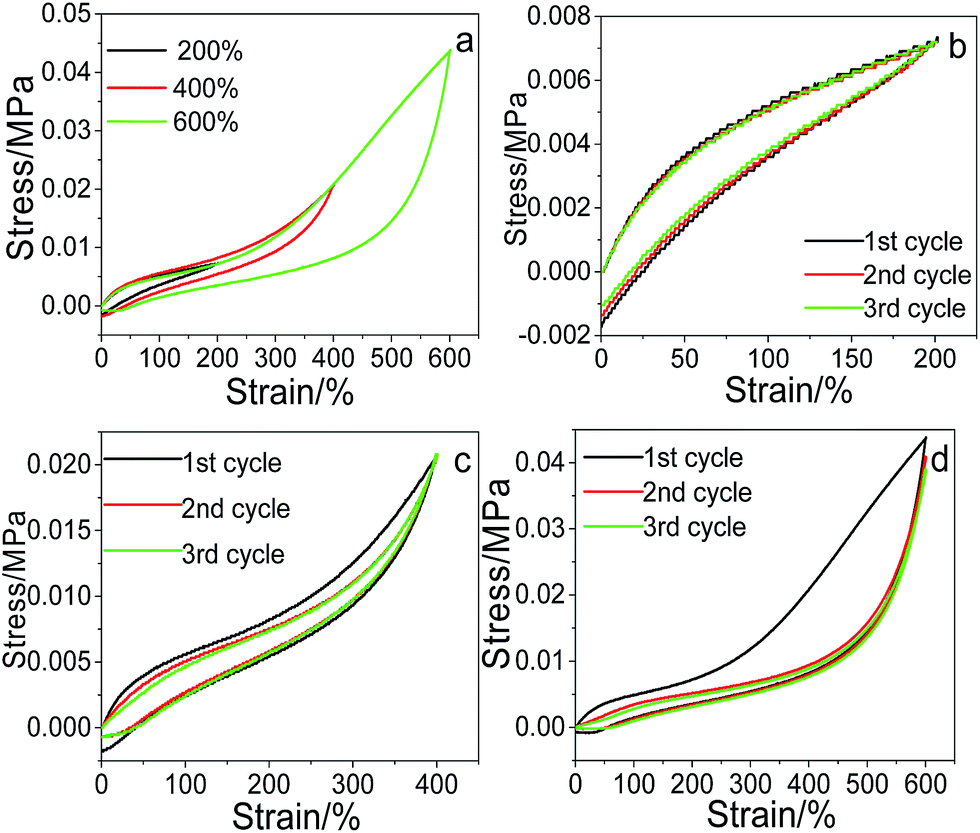
The recoverability of the as-prepared pNIPAM hydrogels was evaluated through cyclic tensile tests, taking pNIPAM hydrogel (with 20 wt%, 5.7 wt%, 20 wt% and 0.22 mol% of SC, Cr, MACS and KPS respectively) as an example. The comparative tensile loading–unloading behaviour under various strains of 200%, 400% and 600%, together with the cyclic loading–unloading behaviour under each strain was investigated respectively, and the results are displayed in Fig. 6. The loading–unloading stress–strain curve of the hydrogel at different strains (see Fig. 6a) shows that the hysteresis loops of the loading–unloading become larger with increasing strain, indicating more energy dissipated during the stretching process.
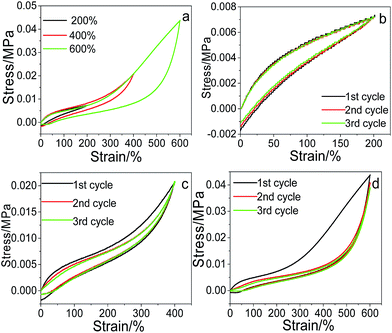 |
| | Fig. 6 Cyclic tensile stress–strain curves, one loading–unloading cycle at different strain (a), three cycles at 200% strain (b), 400% strain (c) and 600% strain (d). | |
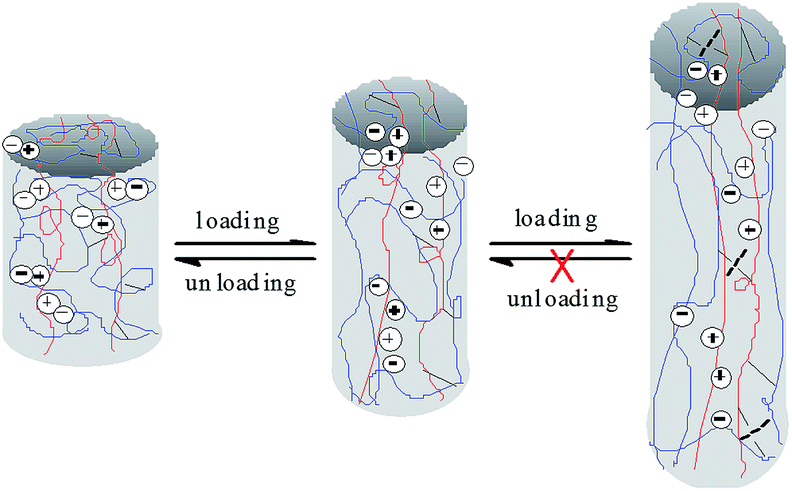
The three loading–unloading cycles overlap well at 200% strain, but with the increase of the tensile strain, the 2nd and 3rd loading–unloading cycles begins to deviate from the 1st cycle. The larger the tensile strain, the more serious the deviation. The recovery efficiency can be assessed according to the tensile stress ratio of the nth cycle to the first one. The calculated recovery efficiency of the 2nd and 3rd cycles are both 95.64 at 200% tensile strain, and are 99.37% and 99.27% respectively at 400% tensile strain, suggesting good recoverability of the prepared hydrogels at low strain. However, when the tensile strain is increased to 600%, the recovery efficiency of the 2nd and 3rd cycles is calculated to be 93.15% and 88.13% respectively, lower than those at 200% and 400% strain, and tends to decline with the cycle index. This is reasonable since at low strain, mainly the physical links (e.g., ionic interactions) in the network are destroyed upon loading. The physical links can recover rapidly upon unloading due to the reversibility of physical interactions, thus displaying high recovery efficiency. With the increase of the tensile strain, the covalent links in the network begin to be disrupted. The broken covalent bonds cannot be recovered due to the irreversibility (Scheme 2), resulting in the decrease of the recovery efficiency.
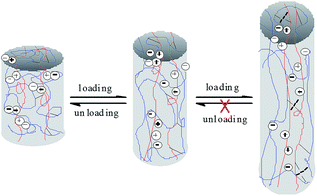 |
| | Scheme 2 Schematic recovery mechanism of the prepared hydrogels. | |
Swelling/deswelling behavior of the dually macro-cross-linked pNIPAM hydrogels
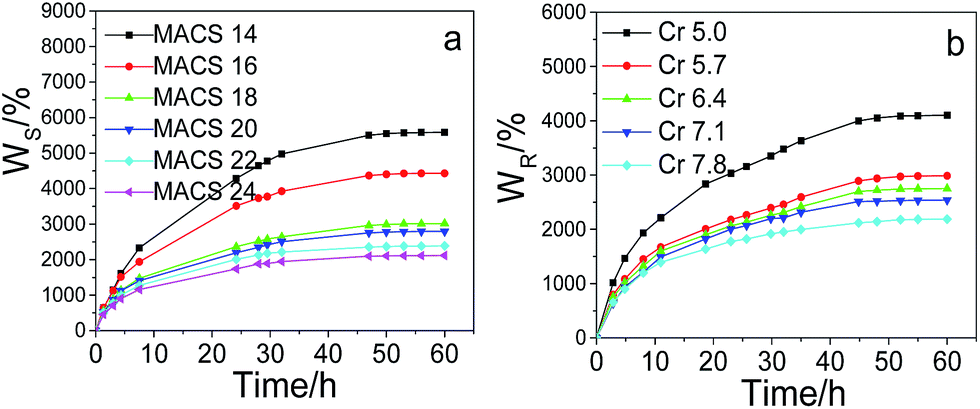
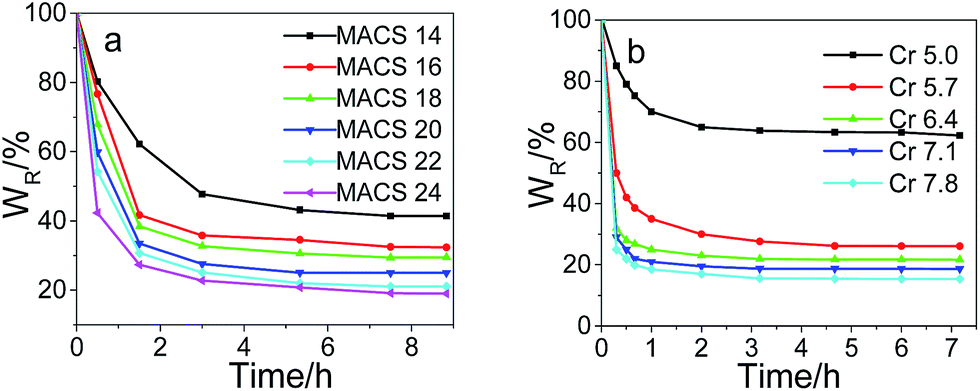
The swelling kinetics of the dual-cross-linked pNIPAM hydrogels with different amount of MACS and cross-linkers was investigated; the results are depicted in Fig. 7. It is observed all the dually macro-cross-linked pNIPAM hydrogels swell rapidly at the early stage, and the swelling rate decreases with time. The time required to equilibrium shortens with the increase of MACS or Cr, which might be caused by the increasing channels formed inside the gel network. The hydrogels approach swelling equilibrium in 24 h and almost reach swelling equilibrium in 32 h for MACS 24 and Cr 7.8. The equilibrium swelling ratios decrease with the increase in either MACS or the cross-linkers, due to the increasingly denser networks. The deswelling kinetics changing with the cross-linkers and MACS is plotted in Fig. 8. The results in Fig. 8a indicate that all the dually macro-cross-linked pNIPAM hydrogels exhibit rapid deswelling rate. When the amount of the cross-linkers is fixed at 6.4 wt%, the deswelling rates at 37 °C increase with the increase of MACS, so does the dehydration ratio. In specific, most hydrogels approach deswelling equilibrium in 1 h and reach equilibrium in 3 h, and the one with 24 wt% MACS released 50% of the absorbed water in ∼15 min. The effects of the cross-linkers on the deswelling rate and water retention are analogous but more significant (Fig. 8b). At a fixed MACS loading of 20 wt%, the hydrogels with the cross-linkers ranging from 6.4 wt% to 7.8 wt% liberate more than 50 wt% of the absorbed water in no more than 15 min. In particular, the hydrogel with 7.8 wt% cross-linkers lose more than 80% of the absorbed water in 10 min. All the investigated hydrogels deswell to equilibrium in 2 h, and from that with 6.4 wt% cross-linkers on, the hydrogels reach deswelling equilibrium in only 1 h. The rapid deswelling rates of the dually macro-cross-linked pNIPAM hydrogels are closely related to the channel-like porous microstructure formed inside the hydrogels, which contributes to the feasibility of water releasing. The more cross-linkers or MACS used, the more water-releasing channels formed inside the hydrogels (concluded from the SEM results), hence water molecules are squeezed out more easily from the hydrogels. The rapid deswelling rates will be very beneficial for future use, e.g., in soft robotics and on–off switches which require rapid response toward temperature change.
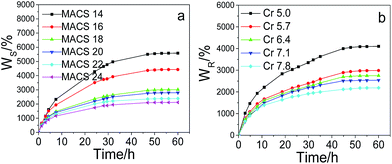 |
| | Fig. 7 Swelling kinetics of the hydrogels with different MACS (a), and cross-linkers (b) (fixed SC at 20 wt% and KPS at 0.22 mol%; Cr at 5.7 wt%, MACS at 20 wt% respectively). | |
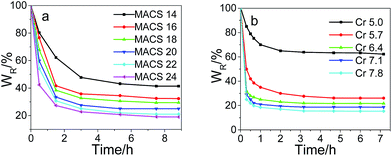 |
| | Fig. 8 Deswelling kinetics of the hydrogels with different MACS (a), and cross-linkers (b) (fixed SC at 20 wt% and KPS at 0.22 mol%; Cr at 6.4 wt%, MACS at 20 wt% respectively). | |
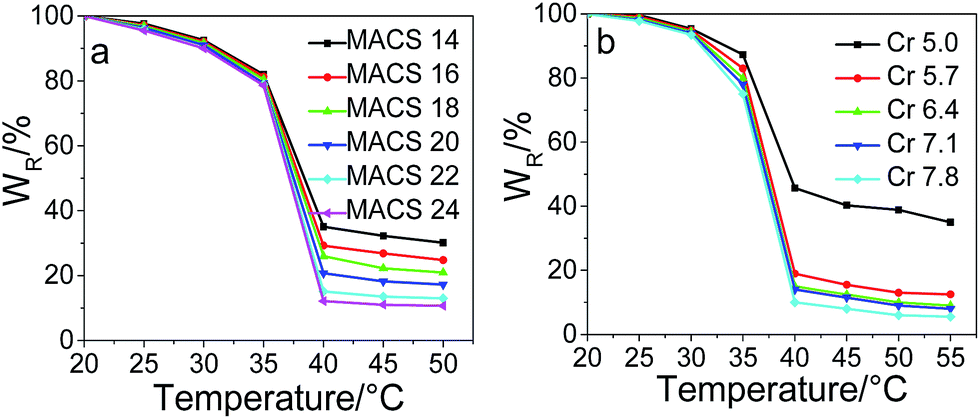
Temperature-sensitivity of the dually macro-cross-linked pNIPAM hydrogels
The water retention changing with temperature is plotted in Fig. 9 for pNIPAM hydrogels with different dosage of the cross-linkers and MACS. In the experimental temperature range of 20 to 55 °C, the water retention of each hydrogel decreases slowly with elevating temperature first, and an abrupt decrease is observed from 35 to 40 °C, revealing the good temperature-sensitivity of the prepared hydrogels. The VPTTs of the hydrogels are 37.5, 37.4, 37.4, 37.3, 37.1 and 37.0 °C with elevating MACS and 38.0, 37.4, 37.3, 37.3, 37.2 °C with elevating Cr, evaluated by the inflexion in the plot of the first derivative of the water retention versus temperature; the obtained results suggest a tiny decrease with the increase of either MACS or the cross-linkers. Furthermore, the more MACS or cross-linkers used, the more water liberated during the volume phase transition. In specific, at the fixed cross-linkers of 6.4 wt%, the water released at 40 °C increases from 65% to 88% as the amount of MACS increases from 14 to 24 wt%; while at the fixed MACS of 20 wt%, the water released at 40 °C increases from 57% to 90% as the cross-linkers increase from 5.0 to 7.8 wt%. The results are in line with the internal microstructure observed by SEM, as elucidated in the above section.
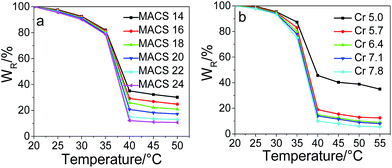 |
| | Fig. 9 Water retention versus temperature of hydrogels with varying MACS (a) and cross-linkers (b) (SC being 20 wt% and KPS 0.22 mol%; Cr being 6.4 wt%, MACS 20 wt% respectively). | |
Both Fig. 8 and 9 show that the hydrogel with 5.0 wt% cross-linkers displays significantly low deswelling ratio, which might attributes on one side to the much less channels formed inside the gel network, and on the other side to a more feasibility of collapsing at the initial deswelling stage which restricts water being squeezed out the hydrogel.
Conclusions
In conclusion, we fabricated a series of thermo-sensitive pNIPAM hydrogels of a single network dually cross-linked via a physical and a chemical macro-cross-linker based on chitosan. The strong, covalent cross-linkings arising from the chemical macro-cross-linker endows the hydrogels high strength, while the reversible ionic interactions between chitosan and gel molecules endow them excellent recoverability towards repeated loading–unloadings. Additionally, the obtained hydrogels demonstrate excellent thermo-sensitivity, especially rapid deswelling rate originating from the channel-like porous microstructure inside the networks. The comprehensive excellent properties of these thermo-responsive hydrogels are very instrumental for potential applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support from the National Science Foundation of China (No. 51503110, No. 51403113); the Collaborative Innovation Centre for Marine Biomass Fibres, Materials and Textiles of Shandong Province.
Notes and references
- M. Shibayama and T. Tanaka, Adv. Polym. Sci., 1993, 109, 3 CrossRef.
- X. M. Ma, J. Y. Xi, X. B. Huang, X. Zhao and X. Z. Tang, Mater. Lett., 2004, 58, 3400 CrossRef CAS.
- X. M. Ma, J. Y. Xi, X. Zhao and X. Z. Tang, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3575 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS.
- R. Liu, S. Liang, X. Z. Tang, D. Yan, X. Li and Z. Z. Yu, J. Mater. Chem., 2012, 22, 14160 RSC.
- A. K. Gaharwar, S. A. Dammu, J. M. Canter, C. J. Wu and G. Schmidt, Biomacromolecules, 2011, 12, 1641 CrossRef CAS PubMed.
- L. Carlsson, S. Rose, D. Hourdet and A. Marcellan, Soft Matter, 2010, 6, 3619 RSC.
- J. Yang, C. R. Han, X. M. Zhang, F. Xu and R. C. Sun, Macromolecules, 2014, 47, 4077 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurakawa and Y. Osada, Adv. Mater., 2003, 15, 1155 CrossRef CAS.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171 CrossRef CAS PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485 CrossRef CAS.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379 CrossRef CAS.
- T. Huang, H. G. Xu, K. X. Jiao, L. P. Zhu, H. R. Brown and H. L. Wang, Adv. Mater., 2007, 19, 1622 CrossRef CAS.
- N. Miyamoto, K. Shimasaki, K. Yamamoto, M. Shintate, Y. Kamachi, B. P. Bastakoti, N. Suzuki, R. Motokawa and Y. Yamauchi, Chem.–Eur. J., 2014, 20, 14955 CrossRef CAS PubMed.
- Z. C. Zhu, Y. Li, H. Xu, X. Peng, Y. N. Chen, C. Shang, Q. Zhang, J. Q. Liu and H. L. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 15637 CAS.
- Y. Tan, K. Xu, P. X. Wang, W. B. Li, S. M. Sun and L. S. Dong, Soft Matter, 2010, 6, 1467 RSC.
- L. W. Xia, R. Xie, X. J. Ju, W. Wang, Q. Chen and L. Y. Chu, Nat. Commun., 2013, 4, 2226 Search PubMed.
- R. Fei, J. T. George, J. Park and M. A. Grunlan, Soft Matter, 2011, 8, 481 RSC.
- R. Fei, J. T. George, J. Park, A. K. Means and M. A. Grunlan, Soft Matter, 2013, 9, 2912 RSC.
- Z. Li, J. Shen, H. Ma, X. Lu, M. Shi, N. Li and M. Ye, Mater. Sci. Eng., C, 2013, 33, 1951 CrossRef CAS PubMed.
- A. B. Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef PubMed.
- J. Zhao, K. X. Jiao, J. Yang, C. C. He and H. L. Wang, Polymer, 2013, 54, 1596 CrossRef CAS.
- M. A. Haq, Y. L. Su and D. J. Wang, Mater. Sci. Eng., C, 2017, 70, 842 CrossRef PubMed.
- X. M. Ma, L. Guo, Q. Ji, Y. Q. Tan, Y. C. Xing and Y. Z. Xia, Polym. Chem., 2016, 7, 26 RSC.
- P. Guo, J. D. Anderson, J. J. Bozell and S. Zivanovic, Carbohydr. Polym., 2016, 140, 171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. SI1–SI4. See DOI: 10.1039/c7ra10148a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
R. Lia,
J. Rena,
X. C. Lva,
X. H. Zhaoab,
Q. Jibc and
Y. Z. Xia*b
*ab,
R. Lia,
J. Rena,
X. C. Lva,
X. H. Zhaoab,
Q. Jibc and
Y. Z. Xia*b