DOI:
10.1039/C7RA09944D
(Paper)
RSC Adv., 2017,
7, 48745-48753
Synthesis of bis-(benzocyclohexan-ketoimino) Ni(II) with different electron groups and their catalytic copolymerization of norbornene and polar norbornene†
Received
6th September 2017
, Accepted 4th October 2017
First published on 17th October 2017
Abstract
Four N,O-chelating type bidentate ligand nickel(II) complexes, Ni{C10H8(O)C[ArN]CH3}2, two of them containing electron-donating groups (Ni1: Ar = p-PhCH3, Ni2: Ar = p-PhOCH3) and another two containing electron-withdrawing groups (Ni3: Ar = p-PhF, Ni4: Ar = p-PhCF3), were synthesized and the molecular structures of Ni2, Ni3 and Ni4 were determined by X-ray crystallography. Copolymerization of norbornene (NB) and 5-norbornene-2-carboxylic acid methyl ester (NB-COOCH3) were carried out in toluene with the above complexes as catalyst precursors and B(C6F5)3 as the co-catalyst. All complexes showed high catalytic activity toward the copolymerization (up to 2.80 × 105 gpolymer molNi−1 h−1) and the complexes with electron-withdrawing groups (Ni3 and Ni4) showed higher catalytic activity, and the activity was enhanced with the increased electron-withdrawing ability of the substituent (Ni4 > Ni3). The obtained poly(NB-co-NB-COOCH3)s were confirmed to be vinyl-addition copolymers and non-crystalline. All copolymers exhibited better solubility in common organic solvents, and showed high molecular weights (up to the grade of 105 g mol−1) and good thermal stability (Td > 370 °C). Meanwhile, variation of the substituted electron group could slightly affect the above-mentioned copolymers' properties.
1. Introduction
Cyclic olefin copolymers (COCs) as a good candidate for engineering plastics and optical apparatus have aroused great attention from both science and industry over the past decade.1,2 Cyclic olefin polymers are usually produced by addition polymerization and ring-opening metathesis polymerization, and the addition polymerization will not destroy the ring structure of the monomer itself. Norbornene (NB) and its derivatives are polymerizable owing to the existence of ring strain, thus norbornene has become one of the most active participants in cyclo-olefin copolymerization.3,4 Vinyl-type polynorbornene (PNB) does not contain any double bonds and with a rotationally strongly constrained configuration,5 which consists of rigid alicyclic polymer backbone, it features many advantageous qualities, such as high thermal stability, high glass transition temperatures (Tg), high optical transparency, large refractive index, low birefringence, low dielectric constant, low moisture absorption as well as low cost. These advantages have been widely applied in many microelectronic and optical applications.6 However, the widespread application of PNB has been hampered by its low solubility, mechanical brittleness as well as poor processability. It has been reported that PNB obtained by nickel catalysts could be dissolved in chlorobenzene at room temperature in the 2000s, and the solubility and processability of PNB have been improved.5,7 Introducing polar groups8–11 or flexible chains12,13 into the PNB backbone has become an effective means of solving the problem. The design and synthesis of particularly efficient and stable catalysts is very important for the polymerization. It is generally known that the vinyl-type polynorbornene can be efficiently obtained by using traditional Ziegler–Natta catalysts (TiCl4, TiCl3),14,15 metallocene catalysts (Ti, Zr, Hf),16,17 and late transition metal catalysts (Ni, Pd, Fe, Co, Cu).18–29 Among these catalysts, the late transition metal catalysts have attracted more attention owing to their lower oxophilicity, higher catalytic activity, and better stability in polar solvents. The most common late transition metal catalysts are Ni(II) and Pd(II) complexes. Ni(II) complexes endow the catalysts with the ability to produce copolymers with a variety of polar functionalized olefins, allowing the preparation of complex functional polymer architectures.30 Ni(II) complexes are the most frequently used catalyst species because of the better catalytic performances and stability than that of Pd(II) complexes and illustrated reaction mechanisms. In recent years, β-ketoimino metal complexes have drawn wide attention from researchers owing to the remarkable simplicity of ligand syntheses and rich opportunities for fine-tuning of their steric and electronic properties. These complexes exhibit excellent activity and good tolerance to polar groups toward the functionalized vinyl-type polynorbornene in the presence of MAO, modified MAO (MMAO), or B(C6F5)3.31–33 Our group have also synthesized and characterized a series of β-ketoimino nickel and palladium complexes that could effectively catalyze copolymerization of norbornene (NB) and NB derivatives.34–39 However, the important ligand-designing parameters, steric and electronic factors, were not completely involved in our previous reports. In this work, we aimed to further examine the impact of electronic effects on the catalytic activity. To this end, we prepared four β-ketoimino nickel complexes (Ni1–Ni4), of which two contain electron-donating groups (Ni1, Ni2) and another two contain electron-withdrawing groups (Ni3, Ni4), and compared their activities via the copolymerization of NB and NB-COOCH3 upon activation with B(C6F5)3. All complexes appeared to be highly active catalysts, and the catalytic activities of Ni3 and Ni4 are higher than those of Ni1 and Ni2. The properties of the obtained polymers, such as molecular weight, solubility and thermal stability, were slightly affected by variation of the substituted electron group. X-ray structures of Ni2, Ni3 and Ni4 were acquired.
2. Experimental
2.1 Materials
All operations with water and oxygen-sensitive compounds were performed under a dry nitrogen atmosphere using Schlenk lines or a glove box (M Braun), and all solvents were dehydrated before use. Toluene and tetrahydrofuran (THF) were dried over sodium/benzophenone and distilled under dry nitrogen. Norbornene (NB) was purchased from Alfa Aesar and purified by drying over sodium and distilling at 110 °C under nitrogen atmosphere, used as a solution (0.4 g mL−1) in toluene. 5-Norbornene-2-carboxylic acid methyl ester (NB-COOCH3) was purchased from energy chemical and purified by distillation over CaH2 at a reduced pressure under dry nitrogen atmosphere.
2.2 Measurements
The structures of the ligands and polymers were characterized by 1H and 13C NMR spectra, which were recorded on a Bruker ARX 400 NMR spectrometer at room temperature in CDCl3 using tetramethylsilane (TMS) as the internal standard. The FTIR spectra were recorded using a Shimadzu IR Prestige-21 FTIR spectrophotometer and average data were obtained from the standard wave number range from 500 to 4000 cm−1. The molecular weight and molecular weight distribution of the polymers were determined by gel permeation chromatography (PL-GPC220), with a refractive index detector using tetrahydrofuran as the eluent with a flow rate of 0.5 mL min−1 and the detector at 40 °C.
The wide-angle X-ray diffraction (WXRD) curves were obtained on a Bruker D8 Focus X-ray diffractometer, operating at 40 kV and 40 mA with a copper target (λ = 1.54 Å) at a scanning rate of 2° min−1 from 3° to 50°. Thermal gravimetric analysis (TGA) was performed on a PerkinElmer TA-600 under nitrogen atmosphere from room temperature to 650 °C at a heating rate of 10 °C min−1, and 5% loss of the polymer weight was used to evaluate its thermal stability. The differential scanning calorimetry (DSC) measurements were obtained on a Shimadzu DSC-60 with a heating–cooling rate of 10 °C min−1 under a nitrogen atmosphere.
2.3 Crystal structure determination
The X-ray diffraction data of single crystals of the complexes were obtained using the ω–2θ scan mode on a Bruker Smart APEX II CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073). The structures were solved by direct methods, and further refinements with full-matrix least-squares on F2 were obtained with the SHELXTL program package. All non-hydrogen atoms were refined anisotropically.
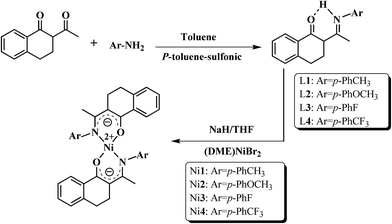
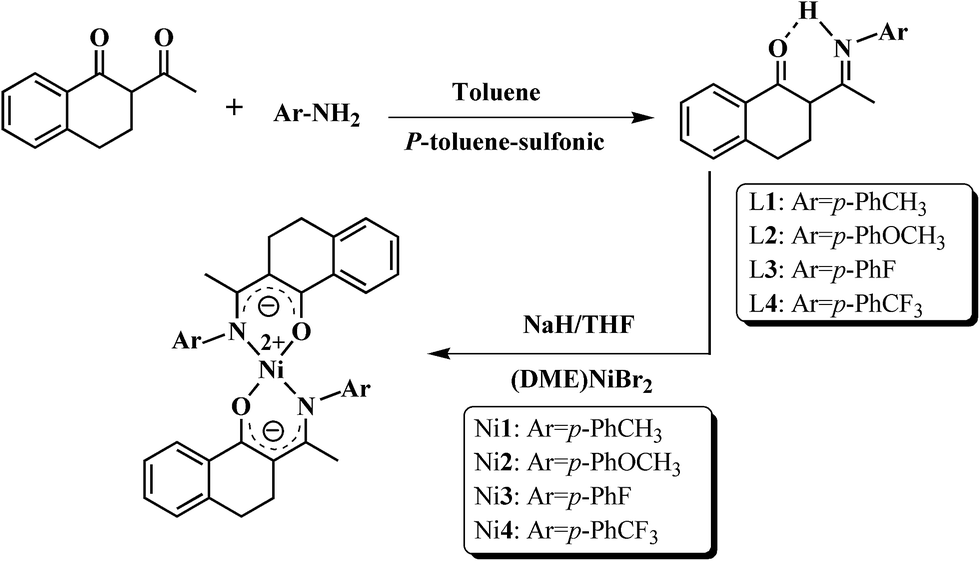
2.4 Syntheses of ligands (L1–L4) and nickel complexes (Ni1–Ni4)
N,O-Chelating type ligands and their bidentate nickel complexes were synthesized referring to the synthetic procedure reported by our group40 and shown in Scheme 1.
 |
| | Scheme 1 The synthetic routes for the benzocyclohexan-ketoimine ligands (L1–L4) and bis-(benzocyclohexan-ketoimino) Ni(II) complexes (Ni1–Ni4). | |
Synthesis of C10H8(O)C[HN(p-PhCH3)]CH3 (L1). To a 250 mL, round-bottomed flask equipped with a magnetic stir bar was added 2-acetyl-1-tetralone (1.88 g, 0.01 mol), p-methylaniline (1.07 g, 0.01 mol), a catalytic amount of p-toluene-sulfonic acid and toluene (150 mL). The mixture was heated to reflux for 5 h, while water was removed as a toluene azeotrope at 125 °C using a water separator. The resulting solution was evaporated under vacuum to remove the residual toluene (concentrated to 4–5 mL), then the raw mixture was purified by column chromatography with n-hexane and ethyl acetate (20/1 in v/v). After concentration, yellow crystals were finally obtained.Yield: 1.68 g (57.4%). 1H NMR (CDCl3, δ, ppm): 14.06 (s, 1H, O⋯H–N); 7.27–8.07 (m, 4H, C10H8); 6.97–7.22 (m, 4H, C6H4); 2.56–2.96 (m, 4H, C10H8); 2.35 (m, 3H, R-CH3); 2.10 (s, 3H, CH3).
Synthesis of C10H8(O)C[HN(p-PhOCH3)]CH3 (L2). Analogous to the synthetic route of L1, p-methylaniline was substituted by p-methoxyaniline and yellow crystals were obtained.Yield: 1.49 g (48.4%). 1H NMR (CDCl3, δ, ppm): 13.99 (s, 1H, O⋯H–N); 7.12–8.06 (m, 4H, C10H8); 6.81–7.10 (m, 4H, C6H4); 2.59–2.94 (m, 4H, C10H8); 3.80 (s, 3H, OCH3); 2.06 (s, 3H, CH3).
Synthesis of C10H8(O)C[HN(p-PhF)]CH3 (L3). Analogous to the synthetic route of L1, p-methylaniline was substituted by p-fluoroaniline and yellow crystals were obtained.Yield: 1.92 g (68.4%). 1H NMR (CDCl3, δ, ppm): 14.01 (s, 1H, O⋯H–N); 7.26–8.01 (m, 4H, C10H8); 6.98–7.22 (m, 4H, C6H4); 2.58–2.93 (m, 4H, C10H8); 2.08 (s, 3H, CH3).
Synthesis of C10H8(O)C[HN(p-PhCF3)]CH3 (L4). Analogous to the synthetic route of L1, p-methylaniline was substituted by p-trifluoromethylaniline and yellow crystals were obtained.Yield: 2.06 g (60.3%). 1H NMR (CDCl3, δ, ppm): 14.13 (s, 1H, O⋯H–N); 7.27–8.08 (m, 4H, C10H8); 6.67–7.23 (m, 4H, C6H4); 2.50–2.95 (m, 4H, C10H8); 2.23 (s, 3H, CH3).
Synthesis of Ni{C10H8(O)C[N(p-PhCH3)]CH3}2(Ni1). To a 100 mL, round-bottomed flask equipped with a magnetic stir bar was added L1 (0.554 g, 0.002 mol) and THF (20 mL) under nitrogen protection. When the ligand dissolved completely, 0.08 g of NaH (60%, 0.002 mol) was added under a dry nitrogen atmosphere. After being stirred vigorously for 8 h at room temperature, the solution changed to orange color and the excess THF was removed in vacuo. After that, (DME)NiBr2 was introduced under an atmosphere of dry and oxygen-free nitrogen, then dichloromethane (20 mL) was injected and the reacting mixture immediately turned to green color. After being stirred vigorously for 24 h at room temperature, the precipitate was separated by filtration and concentrated, and the dark-green nickel complex (Ni1) was obtained in 70% yield.
Synthesis of Ni{C10H8(O)C[N(p-PhOCH3)]CH3}2 (Ni2). Ni2 was prepared according to a similar procedure to that for Ni1 and was obtained as a dark-green crystal in 58% yield.
Synthesis of Ni{C10H8(O)C[N(p-PhF)]CH3}2 (Ni3). Ni3 was prepared according to a similar procedure to that for Ni1 and obtained as a green crystal in 64% yield.
Synthesis of Ni{C10H8(O)C[N(p-PhCF3)]CH3}2 (Ni4). Ni4 was prepared according to a similar procedure to that for Ni1 and was obtained as a dark-green crystal in 44% yield.
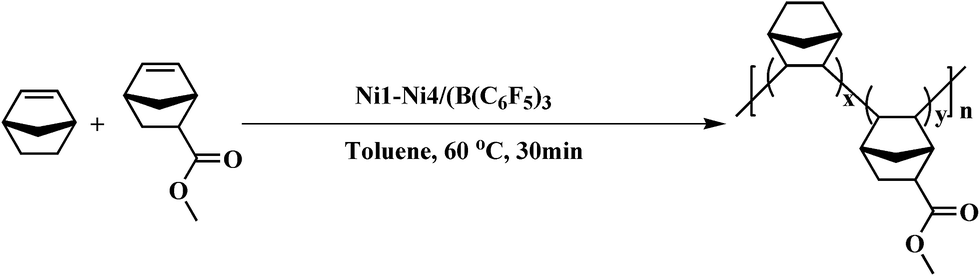
2.5 Polymerization
All experimental manipulations were performed under a dry nitrogen atmosphere. NB and NB-COOCH3 (the total amount was kept at 0.01 mol) were added to a 100 mL two-necked round-bottom flask containing a magnetic stir bar. The nickel complex Ni1–Ni4 (5 × 10−6 mol) solutions were added by syringe followed by B(C6H5)3 (1.0 × 10−4 mol). The solution was continuously stirred for 30 min at 60 °C. The reaction was terminated by addition of methanol/hydrochloric acid (Vm/VH = 9/1) and was stayed over 12 h. After filtering and washing with methanol several times, the products were collected and dried in a vacuum oven at 40 °C to a constant weight. The routes of copolymerization are shown in Scheme 2.
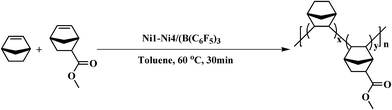 |
| | Scheme 2 Copolymerization of NB and NB-COOCH3 catalyzed by Ni1–Ni4/B(C6F5)3. | |
3. Results and discussion
3.1 Structures of catalysts
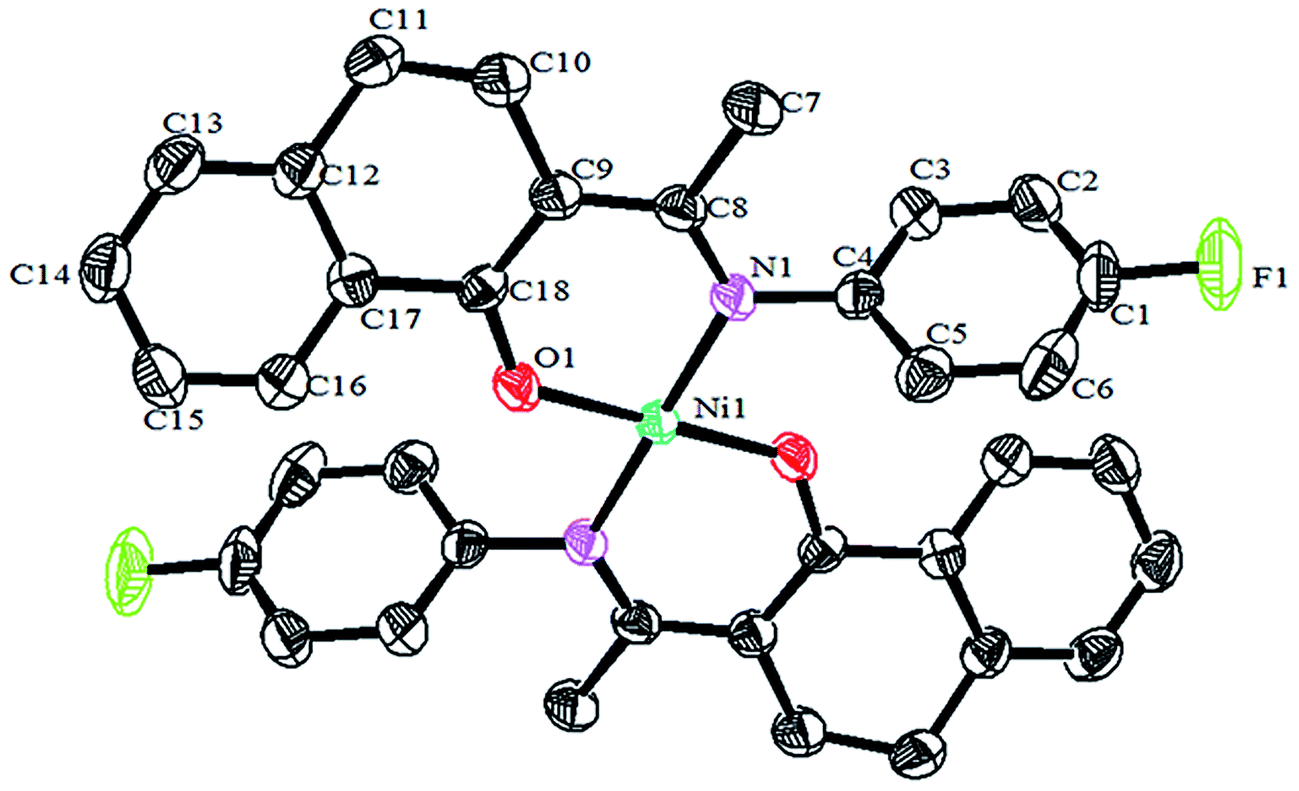
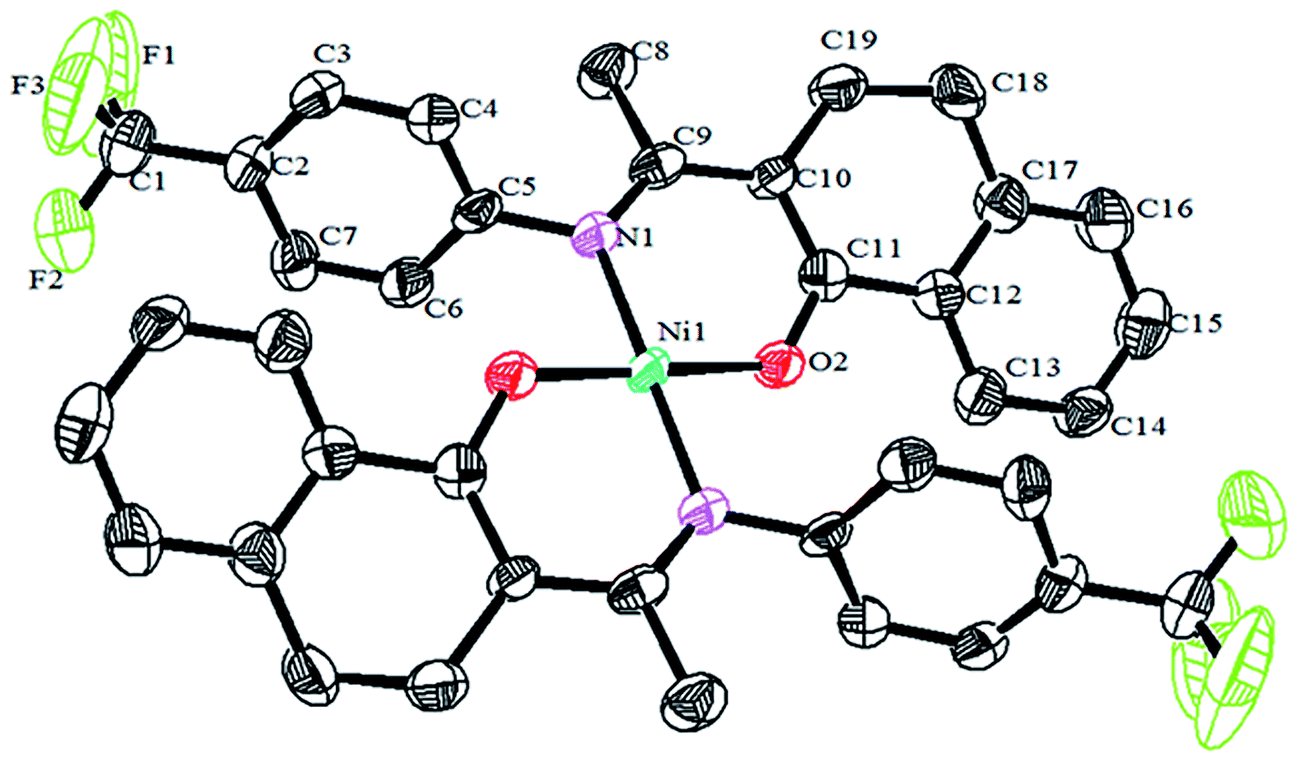
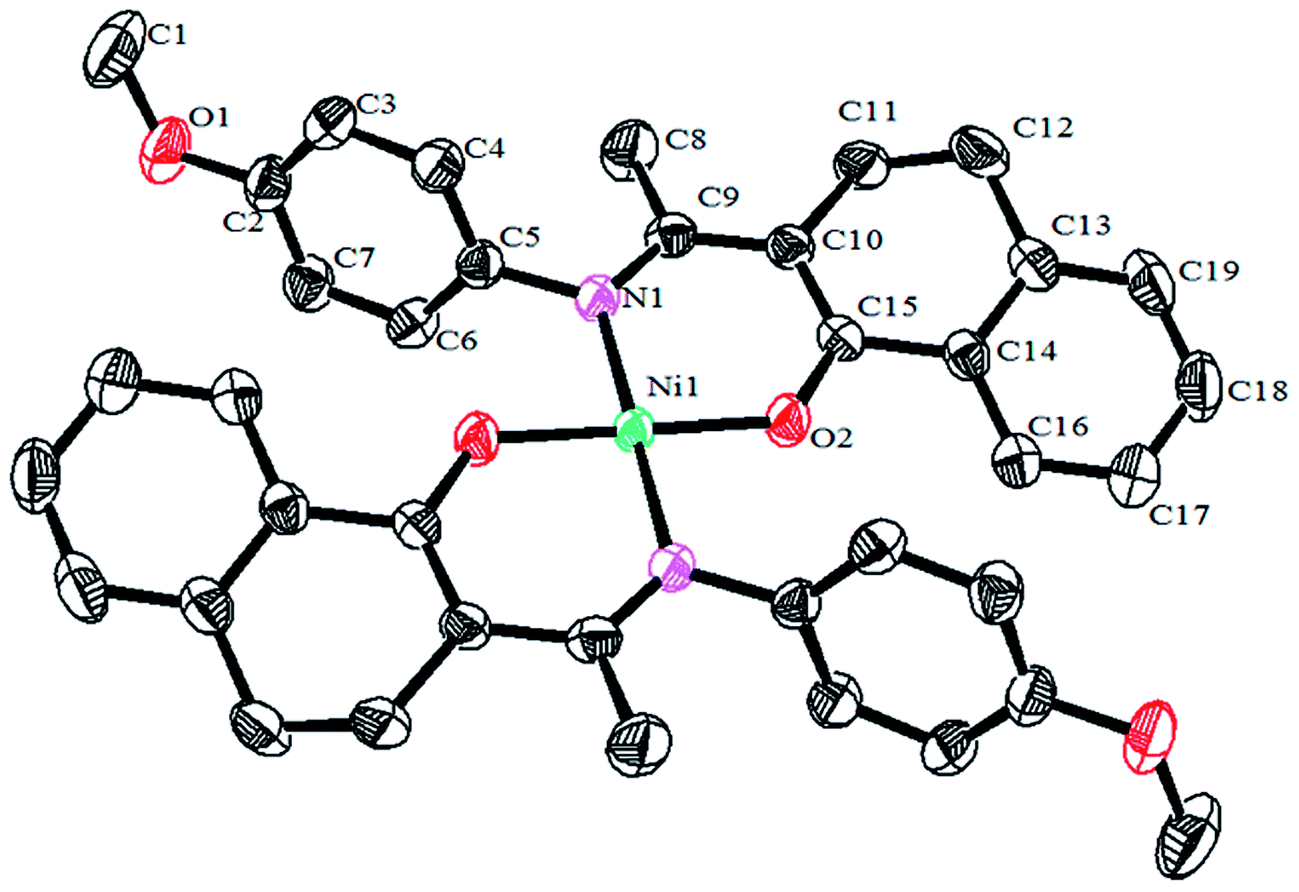
Crystals of the catalysts suitable for single crystal X-ray diffraction were obtained by slow evaporation from dichloromethane solution. The ORTEP plots of Ni2–Ni4 are shown in Fig. 1–3, respectively. The crystallographic data are summarized in Table 1. The data showed that the crystal systems of Ni2, Ni3 and Ni4 were monoclinic. Selected bond lengths and angles are listed in Table 2. The structure of Ni2 is mononuclear and a four-coordinate square planar configuration. The center metal ion (Ni2+) and the N,O-chelator form two stable six-member metallacyclic chelate rings (NiOCCCN). In the solid state, the coordination geometries around nickel atoms are typically square plane where the N or O atoms lie in the trans-position and are similar to some known nickel(II) complexes. The benzocyclohexan-ketoimino ligand is stable rigid structure and the catalyst structure is stable and symmetrical, which will be conducive to stable catalytic activity. Crystal structure of Ni2 was shown in Fig. 1. The structures of Ni3 and Ni4 are similar to that of Ni2, and crystal structures are shown in Fig. 2 and 3, respectively.
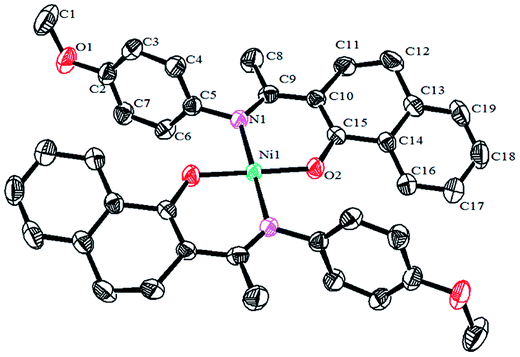 |
| | Fig. 1 ORTEP plots of Ni2 showing the crystal structure. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. | |
Table 1 Crystal data and structure refinement details for Ni2, Ni3 and Ni4
| |
Ni2 |
Ni3 |
Ni4 |
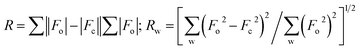 . . |
| Empirical formula |
C38H36N2NiO4 |
C36H30F2N2NiO2 |
C38H30F6N2NiO2 |
| Formula weight |
643.40 |
619.31 |
719.35 |
| Crystal color |
Green |
Green |
Green |
| Temperature (K) |
293(2) |
296(2) |
100 |
| Wavelength (Å) |
0.71076 |
0.71073 |
0.71076 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/n |
P21/n |
| a (Å) |
14.6652(13) |
8.916(3) |
8.719(3) |
| b (Å) |
14.8520(13) |
7.111(3) |
7.351(3) |
| c (Å) |
7.0027(6) |
21.905(8) |
24.392(10) |
| α (deg) |
90 |
90 |
90 |
| β (deg) |
94.988(2) |
93.538(9) |
94.484(10) |
| γ (deg) |
90 |
90 |
90 |
| Volume (Å3) |
1519.5(2) |
1386.2(9) |
1558.6(10) |
| Z |
2 |
2 |
2 |
| Dcala (g m−3) |
1.406 |
1.484 |
1.533 |
| Abs coeff. (mm−1) |
0.684 |
0.752 |
0.698 |
| F(000) |
676.0 |
644.0 |
740.0 |
| Crystal size (mm) |
0.22 × 0.16 × 0.15 |
0.24 × 0.16 × 0.14 |
0.20 × 0.16 × 0.12 |
| θ Range (deg) |
3.108 to 25.500 |
3.42 to 26.00 |
3.239 to 24.795 |
| Limiting indices |
−17 ≤ h ≤ 17, −17 ≤ k ≤ 13, −8 ≤ l ≤ 8 |
−10 ≤ h ≤ 10, −7 ≤ k ≤ 8, −27 ≤ l ≤ 22 |
−10 ≤ h ≤ 10, −8 ≤ k ≤ 8, −24 ≤ l ≤ 28 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
2810/0/207 |
2632/0/197 |
2618/6/225 |
| Goodness-of-fit on S(F2)a |
1.061 |
0.950 |
1.116 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0304, WR2 = 0.0729 |
R1 = 0.0478, WR2 = 0.092 |
R1 = 0.0958, WR2 = 0.2224 |
| R Indices (all data) |
R1 = 0.0404, WR2 = 0.0759 |
R1 = 0.0585, WR2 = 0.1003 |
R1 = 0.1224, WR2 = 0.2299 |
| Largest diff peak and hole (eÅ−3) |
0.182 and −0.872 |
0.571 and −0.424 |
1.162 to −0.543 |
Table 2 Selected bond lengths (Å) and angles (deg) for Ni2, Ni3 and Ni4
| Ni2 |
Ni3 |
Ni4 |
| Bond |
Length |
Bond |
Length |
Bond |
Length |
| Ni(1)–O(1) |
1.8279(11) |
Ni(1)–O(1) |
1.830(2) |
Ni(1)–O(1) |
1.822(5) |
| Ni(1)–O(2) |
1.8279(11) |
Ni(1)–O(2) |
1.830(2) |
Ni(1)–O(2) |
1.822(5) |
| Ni(1)–N(1) |
1.9158(14) |
Ni(1)–N(1) |
1.914(2) |
Ni(1)–N(1) |
1.916(7) |
| Ni(1)–N(1) |
1.9158(14) |
Ni(1)–N(2) |
1.914(2) |
Ni(1)–N(2) |
1.916(7) |
| N(1)–C(5) |
1.447(2) |
C(4)–N(1) |
1.443(4) |
N(1)–C(5) |
1.430(10) |
| Bond |
Angle |
Bond |
Angle |
Bond |
Angle |
| O(2)–Ni(1)–O(1) |
180.0 |
O(2)–Ni(1)–O(1) |
180.0(11) |
O(1)–Ni(1)–O(2) |
180.0(3) |
| O(2)–Ni(1)–N(2) |
87.69(6) |
O(2)–Ni(1)–N(2) |
88.03(10) |
O(2)–Ni(1)–N(2) |
88.20(3) |
| O(1)–Ni(1)–N(2) |
92.31(6) |
O(1)–Ni(1)–N(2) |
91.97(10) |
O(1)–Ni(1)–N(2) |
91.8(3) |
| O(2)–Ni(1)–N(1) |
92.31(6) |
O(2)–Ni(1)–N(1) |
88.03(10) |
O(2)–Ni(1)–N(1) |
91.8(3) |
| O(1)–Ni(1)–N(1) |
87.69(6) |
O(1)–Ni(1)–N(1) |
91.97(10) |
O(1)–Ni(1)–N(1) |
88.2(3) |
| N(2)–Ni(1)–N(1) |
180.0 |
N(1)–Ni(1)–N(2) |
180.0(15) |
N(1)–Ni(1)–N(2) |
180.0 |
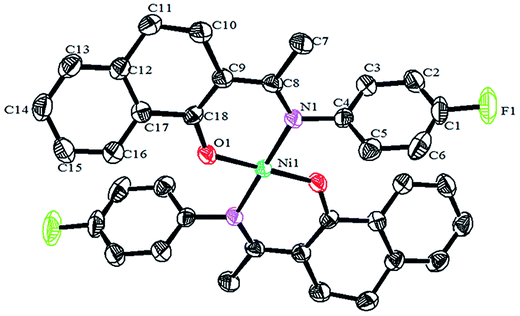 |
| | Fig. 2 ORTEP plots of Ni3 showing the crystal structure. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. | |
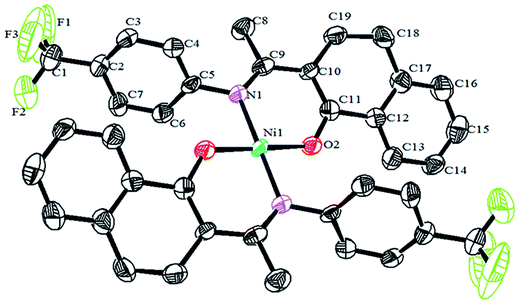 |
| | Fig. 3 ORTEP plots of Ni4 showing the crystal structure. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. | |
The ligand electronic effect led to different charge distribution on the nickel metal atom, and the catalytic activity predominantly increased with an increase in the electrophilicity of the nickel metal center. The steric effect of the aryl ring effectively shielded the axial plane of the central metal and suppressed the side reaction of chain growth. The results indicated that the electronic effect of the para-position substituent on the arylimino groups and the steric effect of the aryl ring had a great influence on the catalytic activity.
3.2 Copolymerization of NB and NB-COOCH3
To further examine the impact of electronic effects on the catalytic activity, Ni1–Ni4 were applied in homo-polymerization of NB as well as copolymerization of NB and NB-COOCH3 upon activation with B(C6F5)3. The detailed data are summarized in Table 3 and were collected under similar conditions. All of the complexes are quite similar and showed high catalytic activity for the copolymerization of NB and NB-COOCH3 (up to 2.80 × 105 gpolymer molNi−1 h−1). Comparing the activities of these catalysts, we found that the activities of Ni3 and Ni4 containing electron-withdrawing groups were higher than those of Ni1 and Ni2 containing electron-donating groups, and the activity of Ni4 containing the –CF3 substituent was higher than that for Ni3 containing the –F substituent, which illustrated that the electron-withdrawing effect was beneficial to the improvement of the catalytic performance. Therefore, the reactivity and polymer yield increased with the increasing electron-withdrawing ability of the substituents on the arylimino groups. Beyond that, the activities and polymer yields decreased with the content of NB-COOCH3 increasing in co-monomers. The phenomenon could be explained by oxygen atoms competing with the double-bond for coordination, and a high concentration of oxygen atoms impedes the coordination of the double-bond.
Table 3 Copolymerization of NB and NB-COOCH3 catalyzed by Ni1–Ni4 combined with B(C6F5)3a
| Catalysts |
NB/NB-COOCH3 (mol mol−1) |
Yield (%) |
Activityb |
Mwc (g mol−1) |
Mw/Mnc |
NB-COOCH3 incorp.d (mol%) |
| Reaction conditions: c[Ni] = 5 mmol L−1; n[NB] + n[NB-COOCH3] = 0.01 mol; solvent: toluene; reaction time: 30 min; temperature: 60 °C; n[B]/n[Ni] = 20/1. In units of gpolymer mol−1Ni h−1. Determined by GPC vs. polystyrene standards in THF. Determined by 1HNMR spectroscopy in CDCl3. |
| Ni1 |
100/0 |
79.2 |
2.98 × 105 |
Insoluble |
— |
0 |
| Ni1 |
90/10 |
52.9 |
2.11 × 105 |
1.34 × 105 |
1.92 |
7.30 |
| Ni1 |
70/30 |
27.3 |
1.21 × 105 |
1.19 × 105 |
1.69 |
24.9 |
| Ni1 |
50/50 |
7.90 |
3.90 × 104 |
1.07 × 105 |
1.83 |
35.2 |
| Ni2 |
100/0 |
84.6 |
3.08 × 105 |
Insoluble |
— |
0 |
| Ni2 |
90/10 |
58.8 |
2.35 × 105 |
1.59 × 105 |
1.79 |
8.70 |
| Ni2 |
70/30 |
38.1 |
1.69 × 105 |
1.24 × 105 |
1.66 |
22.4 |
| Ni2 |
50/50 |
18.3 |
9 × 104 |
1.18 × 105 |
1.74 |
38.7 |
| Ni3 |
100/0 |
89.5 |
3.33 × 105 |
Insoluble |
— |
0 |
| Ni3 |
90/10 |
62.8 |
2.51 × 105 |
1.63 × 105 |
1.83 |
8.20 |
| Ni3 |
70/30 |
45.1 |
2.01 × 105 |
1.45 × 105 |
2.41 |
19.3 |
| Ni3 |
50/50 |
22.8 |
1.12 × 105 |
1.21 × 105 |
1.94 |
42.1 |
| Ni4 |
100/0 |
91.2 |
3.52 × 105 |
Insoluble |
— |
0 |
| Ni4 |
90/10 |
70.0 |
2.80 × 105 |
1.96 × 105 |
2.83 |
7.90 |
| Ni4 |
70/30 |
51.3 |
2.29 × 105 |
1.84 × 105 |
3.47 |
11.3 |
| Ni4 |
50/50 |
26.3 |
1.30 × 105 |
1.67 × 105 |
2.58 |
34.1 |
The poly(NB-co-NB-COOCH3)s were soluble in common organic solvents (such as CHCl3, CH2Cl2, cyclohexane, and THF) as well as o-dichlorobenzene at room temperature, which exhibited better solubility compared with the PNB.
3.3 GPC curves of copolymers
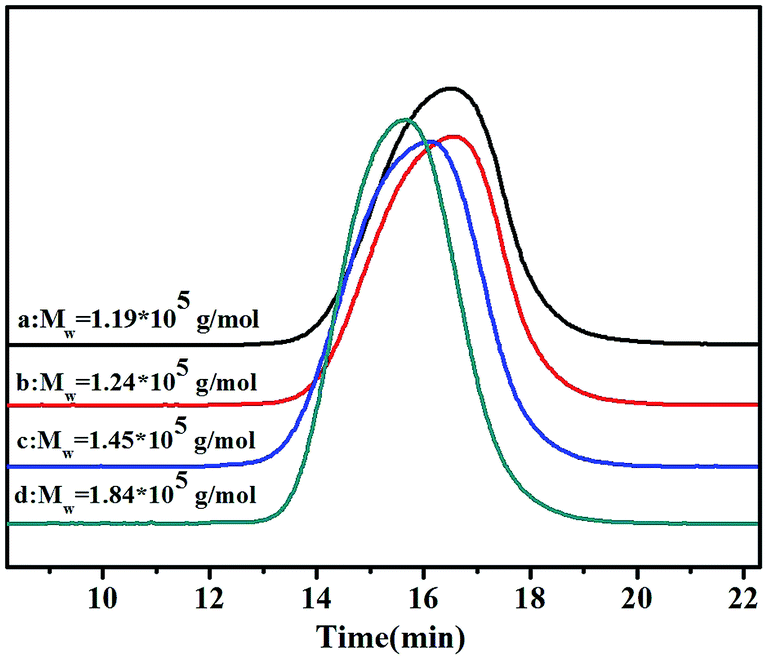
GPC curves of the poly(NB-co-NB-COOCH3)s with different NB-COOCH3 incorporation ratios achieved using the Ni1–Ni4/B(C6F5)3 systems are shown in Fig. 4. The molecular weights (Mw) of the copolymers were all up to the grade of 105 and decreased with increasing NB-COOCH3 content. The poly-dispersity of these copolymers was in the range of 1.66–3.47. The copolymers obtained by Ni4 with wider molecular weight distribution indicated that the higher activity results in different chain growth and termination of polymerization.7 Moreover, all copolymers were single modal in the GPC chromatograms, which indicated that copolymerization occurred at single active sites and the products were true copolymers rather than blends of homopolymers.
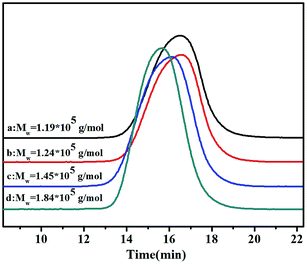 |
| | Fig. 4 The GPC curves of poly(NB-co-NB-COOCH3)s with (a) 24.9, (b) 22.4, (c) 19.3, and (d) 11.3 mol% NB-COOCH3 incorporation ratios. (a), (b), (c) and (d) obtained with Ni1–Ni4/B(C6F5)3 systems, respectively. | |
3.4 1H and 13C NMR spectra of copolymers
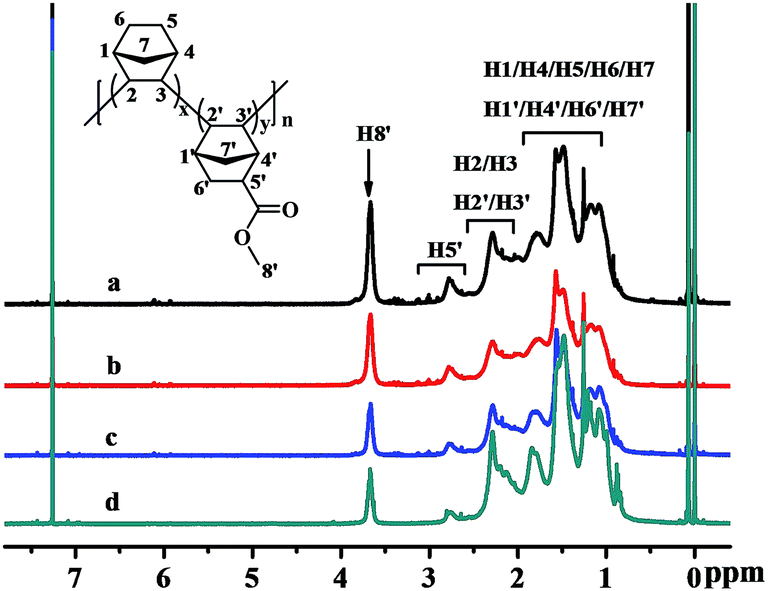
The structures of the poly(NB-co-NB-COOCH3)s were characterized by 1H NMR spectra and are shown in Fig. 5, which demonstrated that the polymerization was carried out via vinyl-addition type mechanism by the absence of the resonance of the proton hydrogen connecting to the double bond at about 5.3–6.0 ppm.41 The peaks at 3.5–3.8 ppm could be assigned to the hydrogens of –COOCH3 (H8′) and it could be seen that the intensity of the peaks increased with increasing polar comonomer feed ratios. The characteristic peaks at 2.5–2.8 ppm could be attributed to the methine hydrogen corresponding to H5′, which indicated that the NB-COOCH3 had accessed to the backbone of norbornene successfully. The peaks at 2.0–2.5 ppm were assigned to the methine hydrogen corresponding to H2/H3/H2′/H3′ and those at 1.6–2.0 ppm were assigned to the methine hydrogen corresponding to H1/H4/H1′/H4′. The peaks at 0.7–1.6 ppm could be attributed to the methene hydrogen corresponding to H5/H6/H7/H6′/H7′. The molar fractions of NB-COOCH3 were calculated by 1H NMR analyses and to be 24.9, 22.4, 19.3 and 11.3 mol%, respectively.
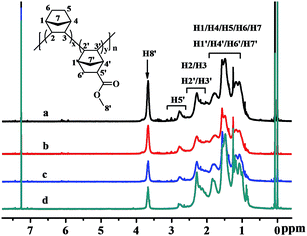 |
| | Fig. 5 1H NMR spectra of poly(NB-co-NB-COOCH3)s with (a) 24.9, (b) 22.4, (c) 19.3, and (d) 11.3 mol% NB-COOCH3 incorporation ratios. (a), (b), (c) and (d) obtained with Ni1–Ni4/B(C6F5)3 systems, respectively. | |
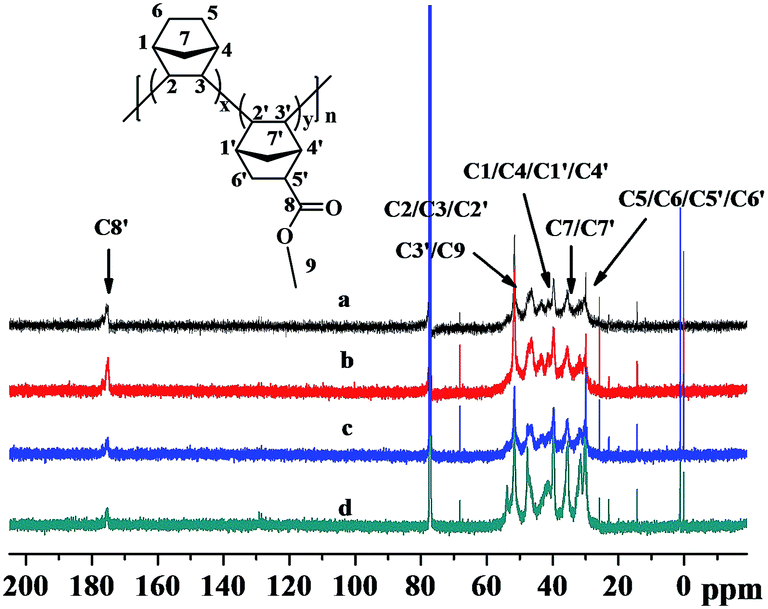
The 13C NMR spectra of the poly(NB-co-NB-COOCH3)s are shown in Fig. 6. Four groups of resonance peaks appeared between 28 and 56 ppm: 28.3–32.5, 33.9–37.8, 38.6–44.7 and 45.1–55.6 ppm, which could be attributed to the methyl, methene or methine carbons corresponding to (C5/C6/C5′/C6′), (C7/C7′), (C1/C4/C1′/C4′) and (C2/C3/C2′/C3′/C9), respectively. Further, the peaks at 174.2–177.8 ppm were assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbon corresponding to C8. The absence of CC double bond characteristic peaks at 120 ppm owing to the ROMP-type polymer structures further implied that the copolymerization of NB and NB-COOCH3 catalyzed by Ni1–Ni4/B(C6F5)3 was carried out via vinyl addition mechanism.
O carbon corresponding to C8. The absence of CC double bond characteristic peaks at 120 ppm owing to the ROMP-type polymer structures further implied that the copolymerization of NB and NB-COOCH3 catalyzed by Ni1–Ni4/B(C6F5)3 was carried out via vinyl addition mechanism.
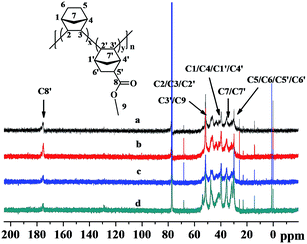 |
| | Fig. 6 13C NMR spectra of poly(NB-co-NB-COOCH3)s with (a) 24.9, (b) 22.4, (c) 19.3, and (d) 11.3 mol% NB-COOCH3 incorporation ratios. (a), (b), (c) and (d) obtained with Ni1–Ni4/B(C6F5)3 systems, respectively. | |
3.5 FTIR spectra of copolymers
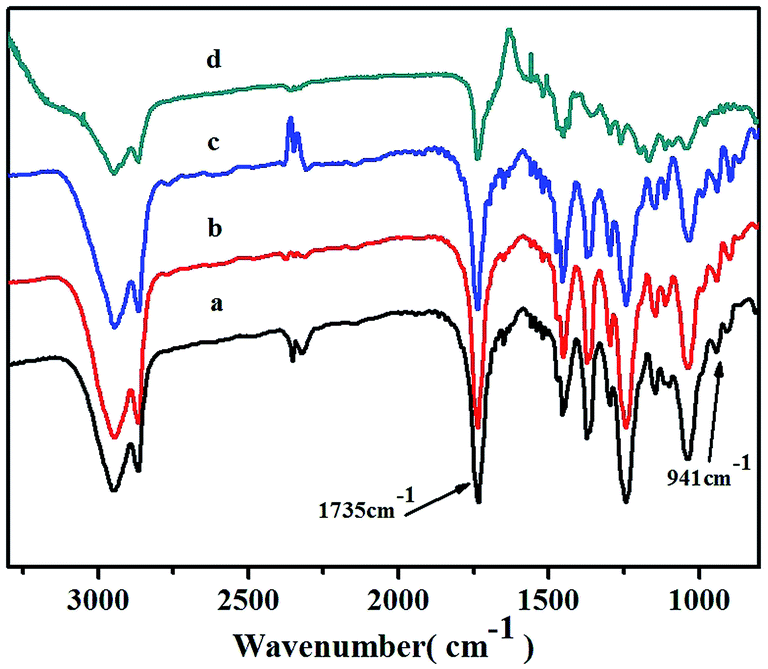
The obtained copolymers were also investigated by FTIR spectroscopy (Fig. 7). The characteristic absorption peak signals at about 941 cm−1 could be assigned to the ring of vinyl-type polynorbornene (PNB), as Kennedy and Makowski noted.42 There were no absorptions at about 1620–1680 cm−1 and 960 cm−1, which could be attributed to the characteristic trans form of stretching of the CC bond of the structure of ROMP-type PNB.43 These results indicated that the polymerization was carried out via vinyl addition mechanism rather than ROMP mechanism. A clear absorption signal of the carbonyl group (CO) at 1735 cm−1 in the FTIR spectrum further confirmed the incorporation of NB-COOCH3 and showed an increase in intensity as the NB-COOCH3 content increased in the co-monomer feed ratios.
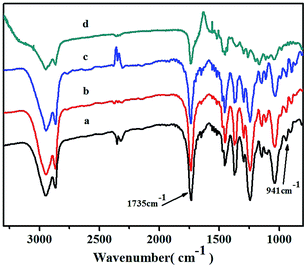 |
| | Fig. 7 FTIR spectra of poly(NB-co-NB-COOCH3)s with (a) 24.9, (b) 22.4, (c) 19.3, and (d) 11.3 mol% NB-COOCH3 incorporation ratios. (a), (b), (c) and (d) obtained with Ni1–Ni4/B(C6F5)3 systems, respectively. | |
3.6 TGA and DSC analyses of copolymers
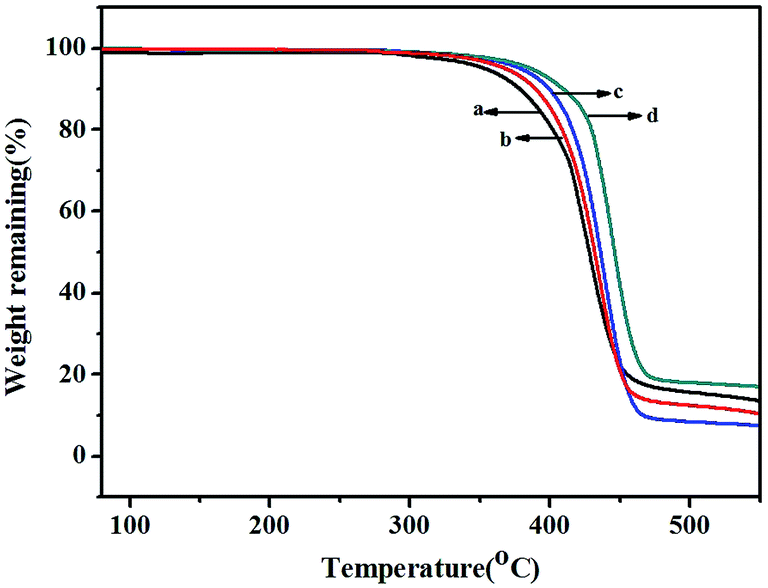
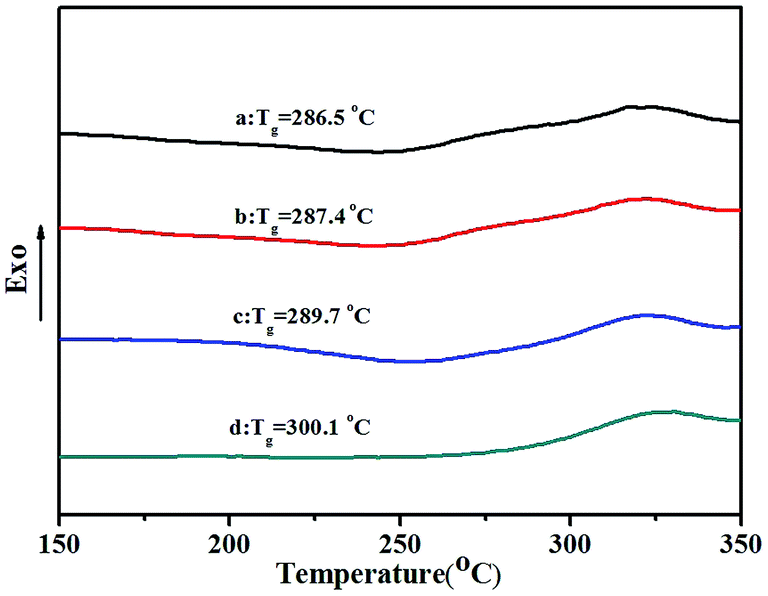
TGA thermograms of poly(NB-co-NB-COOCH3)s obtained by the Ni1–Ni4/B(C6F5)3 systems are shown in Fig. 8. All copolymers showed similar thermal stability with a decomposition temperature (Td) with 5% weight loss of about 370 °C that decreased with increasing NB-COOCH3 content in the copolymers. Glass transition temperatures (Tg) of poly(NB-co-NB-COOCH3)s achieved by different catalytic systems were analyzed by DSC and are shown in Fig. 9. The Tg values of the copolymers were about 290 °C and the trend was similar to that observed for Td. TGA and DSC analyses indicated that copolymers with different NB-COOCH3 contents obtained using the Ni1–Ni4/B(C6F5)3 systems exhibited good thermal stability under nitrogen.
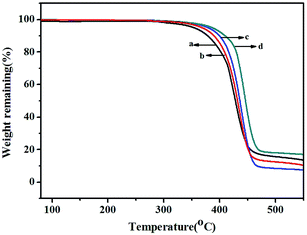 |
| | Fig. 8 The TGA thermograms of poly(NB-co-NB-COOCH3)s with (a) 24.9, (b) 22.4, (c) 19.3, and (d) 11.3 mol% NB-COOCH3 incorporation ratios. (a), (b), (c) and (d) obtained with Ni1–Ni4/B(C6F5)3 systems, respectively. | |
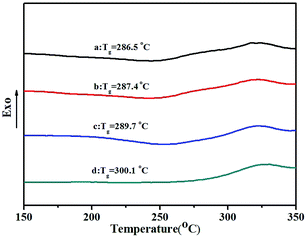 |
| | Fig. 9 The DSC curves of poly(NB-co-NB-COOCH3)s with (a) 24.9, (b) 22.4, (c) 19.3, and (d) 11.3 mol% NB-COOCH3 incorporation ratios. (a), (b), (c) and (d) obtained with Ni1–Ni4/B(C6F5)3 system, respectively. | |
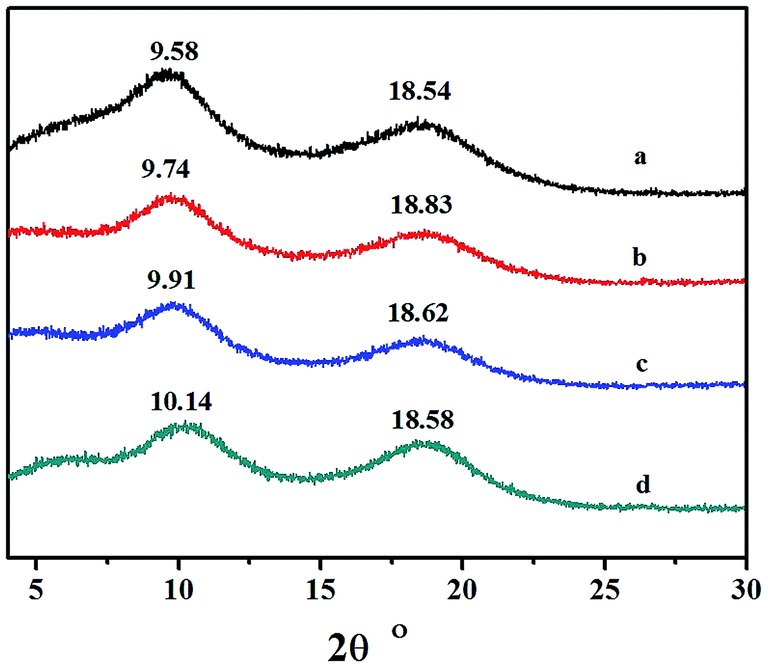
3.7 WXRD analyses of the copolymers
Wide-angle X-ray diffraction analysis of the copolymers catalyzed by the different catalytic systems are shown in Fig. 10. No traces of Bragg reflection in the characteristic crystalline regions were revealed, and therefore the copolymers were non-crystalline. The two halos at 2θ values of 9.58–10.14 and 18.54–18.83 were characteristic peaks for polynorbornene, and the corresponding distances were obtained according to the equation:
dinterchain = 1.22dBragg = 1.22λ/(2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ) sinθ) |
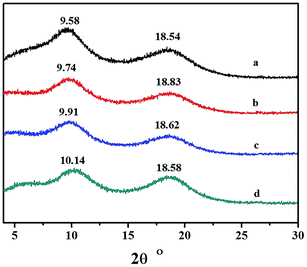 |
| | Fig. 10 XRD curves of the obtained NB-co-NB-COOCH3 copolymers with: (a) 24.9, (b) 22.4, (c) 19.3, and (d) 11.3 mol% NB-COOCH3 incorporation ratios. (a), (b), (c) and (d) obtained with Ni1–Ni4/B(C6F5)3 systems, respectively. | |
They were a: 11.25; b: 11.07; c: 10.88; and d: 10.63 Å, respectively. The results indicated that the packing density of copolymer obtained by different catalytic systems was fairly consistent.
4. Conclusions
Four N,O-chelating type bidentate ligand nickel complexes (Ni1–Ni4) bearing different electronic effects (–CH3, –OCH3, –F and –CF3) were synthesized. Ni2–Ni4 were characterized by single crystal X-ray diffraction, and the copolymerization of norbornene and 5-norbornene-2-carboxylic acid methyl ester in the presence of B(C6F5)3 was examined. All complexes showed high catalytic activity toward the copolymerization (up to 2.80 × 105 gpolymer molNi−1 h−1) and the activity order was Ni4 > Ni3 > Ni2 > Ni1, which illustrated that the electron-withdrawing effect was beneficial to the improvement of the catalytic performance and the activity was improved with increasing electron-attractive ability of the substituent. The analyses of the copolymer structures and properties indicated that the polymerization was carried out via vinyl addition mechanism rather than ROMP mechanism. The poly(NB-co-NB-COOCH3)s exhibited better solubility than that of the PNB and showed high molecular weights (up to a grade of 105) and thermal stability (Td > 370 °C). The poly(NB-co-NB-COOCH3)s were confirmed to be non-crystalline. Meanwhile, variation of the substituted electron group could slightly affect the properties of the above-mentioned copolymers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21164006 and 21674045).
References
- A. Ravasio, C. Zampa, L. Boggioni, I. Tritto, J. Hitzbleck and J. Okuda, Macromolecules, 2008, 41, 9565–9569 CrossRef CAS.
- X. H. He, Y. W. Chen, Y. M. Liu, S. X. Yu, S. G. Hong and Q. Wu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4733–4743 CrossRef CAS.
- I. Tritto, L. Boggion and D. R. Ferro, Coord. Chem. Rev., 2006, 250, 212–241 CrossRef CAS.
- Y. I. Denisova, M. L. Gringolts, A. V. Roenko, G. A. Shandryuk, E. S. Finkelshtein and Y. V. Kudryavtsev, Mendeleev Commun., 2017, 27, 416–418 CrossRef CAS.
- H. J. Yang, W. H. Sun, F. Chang and Y. Li, Appl. Catal., A, 2003, 252, 261–267 CrossRef CAS.
- C. Janiak and P. G. Lassahn, J. Mol. Catal. A: Chem., 2001, 16, 193–209 CrossRef.
- W. H. Sun, H. J. Yang, Z. L. Li and Y. Li, Organometallics, 2003, 22, 3678–3683 CrossRef CAS.
- X. H. He, Y. J. Deng, X. Jiang, Z. J. Wang, Y. P. Yang, Z. L. Han and D. F. Chen, Polym. Chem., 2017, 8, 2390–23396 RSC.
- M. Hollauf, M. Cajlakovic, M. Tscherner, S. Köstler, A.-C. Knall and G. Trimmel, Monatsh. Chem., 2017, 148, 121–129 CrossRef CAS PubMed.
- M. Chen and C. L. Chen, ACS Catal., 2017, 7, 1308–1312 CrossRef CAS.
- Z. X. Wu, M. Chen and C. L. Chen, Organometallics, 2016, 35, 1472–1479 CrossRef CAS.
- X. H. He, Y. J. Deng, Z. L. Han, Y. P. Yang and D. F. Chen, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3495–3505 CrossRef CAS.
- M. Schneider, J. Claverie, C. Graillat and T. F. Meckenna, J. Appl. Polym. Sci., 2002, 84, 1878–1896 CrossRef CAS.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 541–547 CrossRef CAS.
- L. P. He, J. Y. Liu, Y. G. Li, S. R. Liu and Y. S. Li, Macromolecules, 2009, 42, 8566–8570 CrossRef CAS.
- R. M. Golding, R. O. Pascual, C. Suvanprakorn and I. G. Dance, Bull. Korean Chem. Soc., 2006, 27, 1752–1756 CrossRef CAS.
- W. Kaminsky, S. Derlin and M. Hoff, Polymer, 2007, 48, 7271–7278 CrossRef CAS.
- H. J. Yang, Z. L. Li and W. H. Sun, J. Mol. Catal. A: Chem., 2003, 206, 23–28 CrossRef CAS.
- F. Bao, X. Lü and Y. Chen, Polym. Bull., 2007, 58, 495–502 CrossRef CAS.
- Y. M. Xu, K. Li, Y. H. Wang, W. Deng and Z. J. Yao, Polymers, 2017, 9, 105–115 CrossRef.
- A. A. Antonov, N. V. Semikolenova, V. A. Zakharov, W. J. Zhang, Y. H. Wang, W. H. Sun, E. P. Talsi and K. P. Bryliakov, Organometallics, 2012, 31, 1143–1149 CrossRef CAS.
- H. Y. Gao, L. X. Pei, Y. F. Li, J. K. Zhang and Q. Wu, J. Mol. Catal. A: Chem., 2008, 280, 81–86 CrossRef CAS.
- J. J. Lai, X. H. Hou, Y. W. Liu, C. Redshaw and W. H. Sun, J. Organomet. Chem., 2012, 702, 52–58 CrossRef CAS.
- X. F. Li and Y. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2680–2685 CrossRef CAS.
- D. P. Song, X. C. Shi, Y. X. Wang, J. X. Yang and Y. S. Li, Organometallics, 2012, 31, 966–975 CrossRef CAS.
- F. Bao, X. Q. Lue, Y. Q. Qiao, G. Q. Gui, H. Y. Gao and Q. Wu, Appl. Organomet. Chem., 2005, 19, 957–963 CrossRef CAS.
- H. Y. Gao, J. K. Zhang, Y. Chen, F. M. Zhu and Q. Wu, J. Mol. Catal. A: Chem., 2005, 240, 178–185 CAS.
- C. Carlini, M. Martinelli, G. Raspolli, M. Anna and G. Sbrana, J. Polym. Sci. Part A: Polym. Chem., 2006, 44, 1514–1521 CrossRef CAS.
- M. R. Radlauer, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 1478–1481 CrossRef CAS PubMed.
- J. W. Tian, H. Y. Zhu, J. Y. Liu, D. F. Chen and X. H. He, Appl. Organomet. Chem., 2014, 28, 702–711 CrossRef CAS.
- D. P. Song, W. P. Ye, Y. X. Wang, J. Y. Liu and Y. S. Li, Organometallics, 2009, 28, 5697–5704 CrossRef CAS.
- D. P. Song, J. Q. Wu, W. P. Ye, H. L. Mu and Y. S. Li, Organometallics, 2010, 29, 2306–2314 CrossRef CAS.
- Y. Z. Zhu, J. Y. Liu, Y. S. Li and Y. J. Tong, J. Organomet. Chem., 2004, 689, 1295–1303 CrossRef CAS.
- X. H. He, Y. Z. Yao, X. Luo, J. K. Zhang, Y. H. Liu, L. Zhang and Q. Wu, Organometallics, 2003, 22, 4952–4957 CrossRef CAS.
- Y. P. Xing, Y. W. Chen and X. H. He, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4425–4432 CrossRef CAS.
- Y. P. Xing, X. H. He, Y. W. Chen, H. R. Nie and Q. Wu, Polym. Bull., 2011, 66, 1149–1161 CrossRef CAS.
- X. H. He, Y. M. Liu, C. Lu, Y. W. Chen and D. F. Chen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4695–4704 CrossRef CAS.
- L. Chen, Z. C. Zhong, C. Chen, X. H. He and Y. W. Chen, J. Organomet. Chem., 2014, 752, 100–108 CrossRef CAS.
- J. W. Tian, X. H. He, J. Y. Liu, X. D. Deng and D. F. Chen, RSC Adv., 2015, 5, 61851–61860 RSC.
- X. H. He, Y. Z. Yao, X. Luo, J. K. Zhang, Y. H. Liu, L. Zhang and Q. Wu, Organometallics, 2003, 22, 4952–4957 CrossRef CAS.
- M. C. Sacchi, M. Sonzogni, S. Losio, F. Forlin, P. Locatelli, I. Tritto and M. Licchelli, Macromol. Chem. Phys., 2001, 202, 2052–2058 CrossRef CAS.
- J. P. Kennedy and H. S. Makowski, J. Macromol. Sci., Chem., 1967, 1, 345–370 CrossRef CAS.
- T. Tsujino, T. Saeguss and J. Furukawa, Macromol. Chem., 1965, 85, 71–79 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yingping Yanga,
Suli Wanga,
Zhilong Hana,
Guangshui Tua,
Feng Zhanga,
Shengmei Huangac,
Zijie Wanga and
Defu Chenb
*a,
Yingping Yanga,
Suli Wanga,
Zhilong Hana,
Guangshui Tua,
Feng Zhanga,
Shengmei Huangac,
Zijie Wanga and
Defu Chenb
 .
.