
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective C–H chlorination: towards the sequential difunctionalization of phenol derivatives and late-stage chlorination of bioactive compounds†
Chao Gaob,
Hongchen Lib,
Miaochang Liub,
Jinchang Dingb,
Xiaobo Huang b,
Huayue Wu*b,
Wenxia Gaob and
Ge Wu*ac
b,
Huayue Wu*b,
Wenxia Gaob and
Ge Wu*ac
aSchool of Pharmaceutical Science, Wenzhou Medical University, Wenzhou 325035, People's Republic of China. E-mail: wuge@wmu.edu.cn
bCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, People's Republic of China. E-mail: huayuewu@wzu.edu.cn
cState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
First published on 3rd October 2017
Abstract
We have developed a protocol for the auxillary directed C–H chlorination of phenol derivatives using catalytic amounts of palladium acetate that is amenable to the late-stage chlorination of diflufenican and estrone. The 2-pyridine group allows for a highly efficient palladium-catalyzed chlorination and sequential ortho C–H functionalization reaction of phenol derivatives to produce a variety of symmetrical and unsymmetrical 2,4,6-trisubstituted phenols.
Introduction
Catalytic C–H activation/chlorination is one of the most efficient pathways to prepare aryl chlorides.1,2 The resulting aryl chlorides serve as synthetic handles that participate in transition metal catalyzed cross-coupling reactions,3 and they also function as precursors of organometallic reagents utilized for nucleophilic addition and substitution reactions.4 Currently, their use in the development of site-selective chlorination of phenol is highly attractive as these compounds are prevalent in numerous pharmaceuticals and agrochemicals, such as Nitrofungin,5a Lofexidine,5b Chloroxynil5c and Sportak5d (Scheme 1a). | ||
| Scheme 1 Synthesis and application of chloro-containing multifunctionalization phenols. | ||
Currently, electrophilic aromatic substitution represents the leading strategy to obtain chlorinated phenols (Scheme 1b), yet producing a mixture of ortho and para substituted products. In addition, many efforts have been devoted toward the development of new routes to ortho-chlorinated phenols, including dehalogenation,6 arene oxidation,7 or O-methoxymethyl directed lithiation.8 However, these methods suffer from limitations, such as requirement of harsh reaction condition and limited substrate scope. Recently, efficient methods have been developed. Snider demonstrated that bulky amine catalyzed ortho-chlorination of phenols by sulfuryl chloride.9 Gustafson reported the preparation of ortho-chlorinated phenols by employing Nagasawa's bis-thiourea catalyst.10 However, the former was only effective for electron-deficient phenols, whereas, the later often gave the undesired para-substituted phenols. Moreover, palladium-catalyzed C–H chlorination of phenyl carbamate was also reported.2j Up to now, the metal-catalyzed double C–H functionalization of phenol and their derivatives has not been explored and remains a great challenge. Therefore, an efficient and general methodology for the synthesis of densely functionalized phenol and derivatives is highly desired.
2-Aryloxypyridines are ubiquitous motif found in numerous biologically active molecules and pesticide.11 During the past few years, palladium-catalyzed ortho mono-arylation,12 -nitration,13 -alkenylation,14 -acylation,15 -fluorination,16 -acetoxylation17 -alkoxylation,18 and sulfonylation19 of 2-aryloxypyridines have been developed. However, to the best of our knowledge, selective chlorination, bromination, iodination and borylation of 2-aryloxypyridines have not been reported. Herein, we report a new protocol for double symmetrical and unsymmetrical C–H functionalization of phenols, directed by a removable 2-pyridine group, enabling the introduction of two Cl or different functional groups (Cl/F, Br, I, NO2 and Bpin) into ortho positions of phenols (Scheme 1c).
Results and discussion
At the onset of this project, we selected 2-phenoxypyridine (1a) and NCS as a model substrates to examine the feasibility of palladium-catalyzed C–H chlorination reaction. As shown in Table 1, we observed that the choice of additive had a considerable impact on the chlorination reactions. Further exploration confirmed that TsOH was the most efficient promoter. However, no significant results could be achieved when K2S2O8 or Na2S2O8 used as the additive.2h It is worth noting that when the reaction was conducted in a coordinating solvent, such as DMF and dioxane, the desired product was not observed, and the starting material was recoveried. Lowering the amount of NCS decrease the yield of 3a to 58%, and 10% of the monochlorinated product was isolated. Finally, the optimal yield of dichlorinated product 3a was obtained when 2-phenoxypyridine and NCS (molar ration 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.0) were stirred in EtOAc in the presence of Pd(OAc)2 (10 mol%) and TsOH (10 mol%) at 100 °C for 6 h. The practical utilization of current method was demonstrated by scaling up the reaction: when 1a was subjected to dichlorination on 10 mmol scale, 3a was obtained in 69% yield (entry 1). Interestingly, we found that when 2.0 equiv. of NCS in DMF, only para-chlorination product (3a′) was obtained (Scheme 2) in 90% yield without any trace of isomers.
:3.0) were stirred in EtOAc in the presence of Pd(OAc)2 (10 mol%) and TsOH (10 mol%) at 100 °C for 6 h. The practical utilization of current method was demonstrated by scaling up the reaction: when 1a was subjected to dichlorination on 10 mmol scale, 3a was obtained in 69% yield (entry 1). Interestingly, we found that when 2.0 equiv. of NCS in DMF, only para-chlorination product (3a′) was obtained (Scheme 2) in 90% yield without any trace of isomers.
|
||||
|---|---|---|---|---|
| Entry | Additive | Equiv. of NCS | Solvent | Yieldb% |
| a Conditions: 1 (0.2 mmol), NCS (3.0 equiv.), Pd(OAc)2 (10 mol%), additive (10 mol%), solvent (2.0 mL), 110 °C, under N2, 6 h.b Isolated yields.c 10 mmol gram-scalable reaction. | ||||
| 1 | TsOH | 3.0 | EtOAc | 83 (69)c |
| 2 | AcOH | 3.0 | EtOAc | 22 |
| 3 | K2S2O8 | 3.0 | EtOAc | 0 |
| 4 | Na2S2O8 | 3.0 | EtOAc | 0 |
| 5 | AgOAc | 3.0 | EtOAc | 0 |
| 6 | TsOH | 3.0 | DMF | Trace |
| 7 | TsOH | 3.0 | Dioxane | Trace |
| 8 | TsOH | 3.0 | Toluene | 62 |
| 9 | TsOH | 1.5 | EtOAc | 58 |
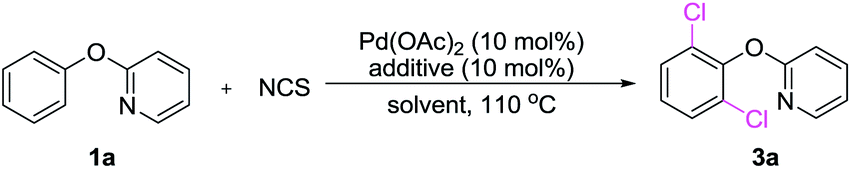
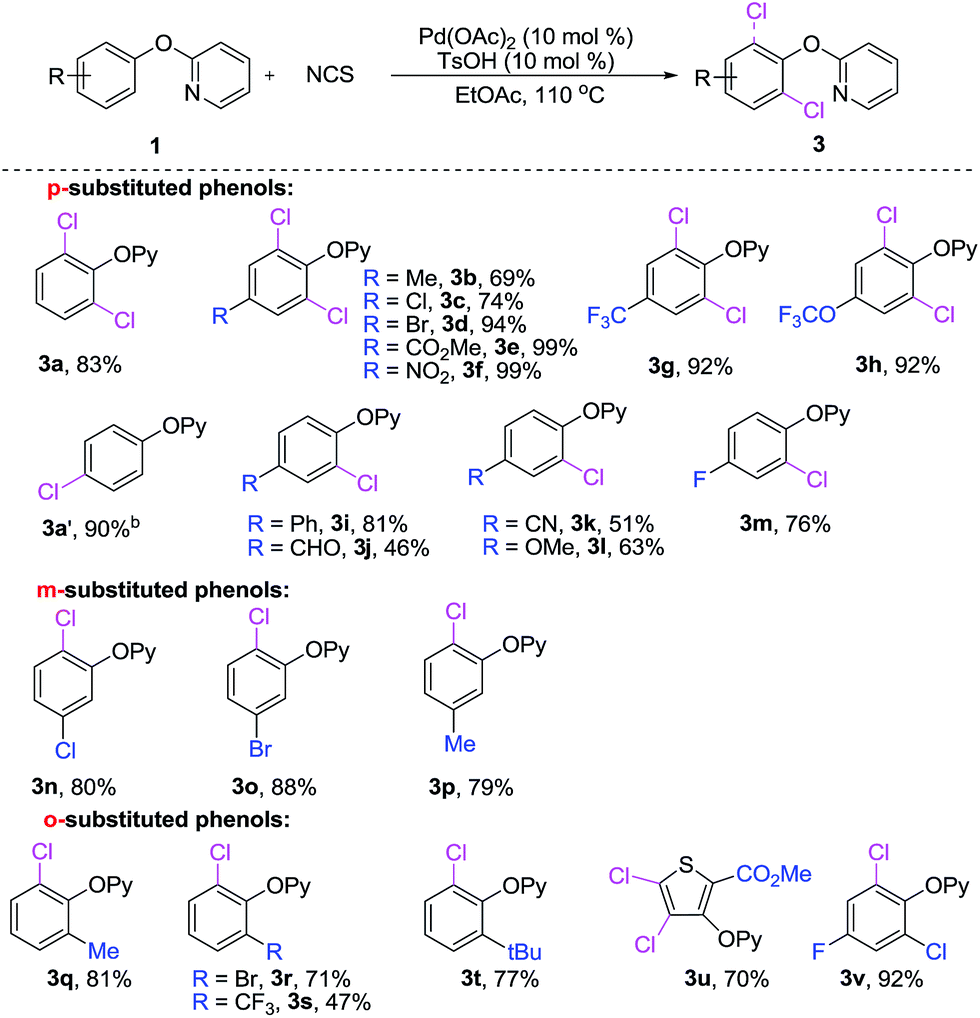 | ||
| Scheme 2 C–H chlorination of 2-aryloxylpyridine. aConditions: 1 (0.2 mmol), NCS (3.0 equiv.), Pd(OAc)2 (10 mol%), TsOH (10 mol%), EtOAc (2.0 mL), 110 °C, under N2, 6 h, isolated yields. bNCS (2.0 equiv.), DMF (2.0 mL), 100 °C, under open air, 2 h, isolated yields. | ||
With the optimal conditions in hand, we next examined the scope of 2-aryloxypyridines. As shown in Scheme 2, both electron-donating and -withdrawing groups at ortho-, meta-, or para-position of phenyl groups were well tolerated and afforded the corresponding chlorinated products in good to excellent yields. It is worth mentioning that the degree of chlorination is dependent on the substitutent on phenyl ring of 2-phenoxypyridine derivatives. When para-positions of 2-phenoxypyridine were substituted by a methyl (3b), chloride (3c), bromide (3d), ester (3e), nitro (3f), trifluoromethyl (3g) and trifluoromethoxy (3h), the corresponding dichlorination products were yields. In contrast, when 2-aryloxypyridine bearing a phenyl (3i), aldehyde (3j), nitrile (3k), methoxy (3l) and fluoro (3m) in para-position, affording the monochlorination products. Notably, the chlorination reaction was highly steric sensitive, in the cases of 1n–p, the less congested C–H bonds of the meta-position of 2-phenoxypyridines (3n–p) were regioselectively chlorinated. Furthermore, the ortho-substituted 2-aryloxypyridines (3q–v) are also viable substrates in the current reaction, giving the corresponding products in good yields.
The diversity of 2-phenoxypyridines for dichlorination was examined. As shown in Scheme 3, we found that the dichlorination of 2-phenoxypyridine derivatives bearing electron-rich substitutents on pyridine rings reacted smoothly providing the corresponding dichlorinated products in fair to excellent yields (5a–i). The reactions of electron-deficient substrates, gave lower yields of products (5a, 5g). Probably containing the electron-withdrawing substituents 2-phenoxypyridines weakens their coordinating abilities and lowers their activities of phenol's C–H bonds.
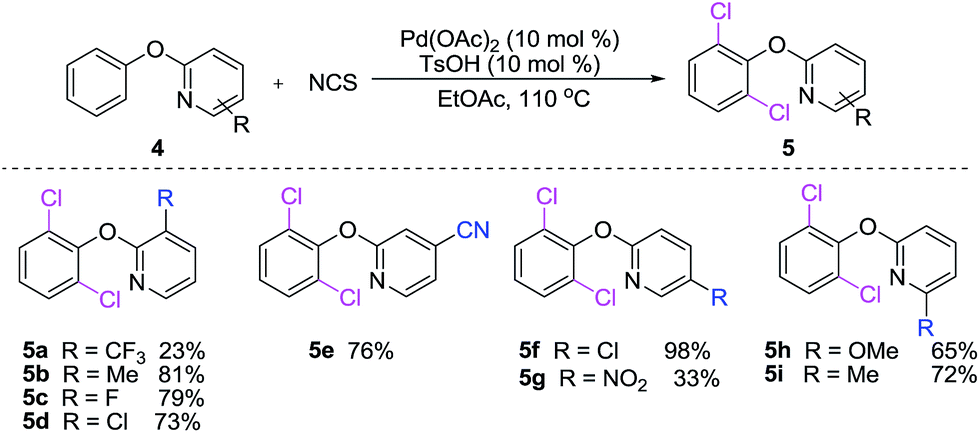 | ||
| Scheme 3 C–H chlorination of 2-phenoxypyridine derivatives. | ||
The advantage of 2-pyridyl directing group lies in the possibility of their removal to provide the structurally diversified 2,6-dichlorinated and 2-chlorinated phenols (Scheme 4).12a Significantly, the current reaction offer opportunities to synthesis ortho-chlorination phenols with electron-withdrawing groups (6b–f, 6j) and electron-donating groups (6a, 6g–i), which nicely complements the aforementioned approaches (Scheme 5).
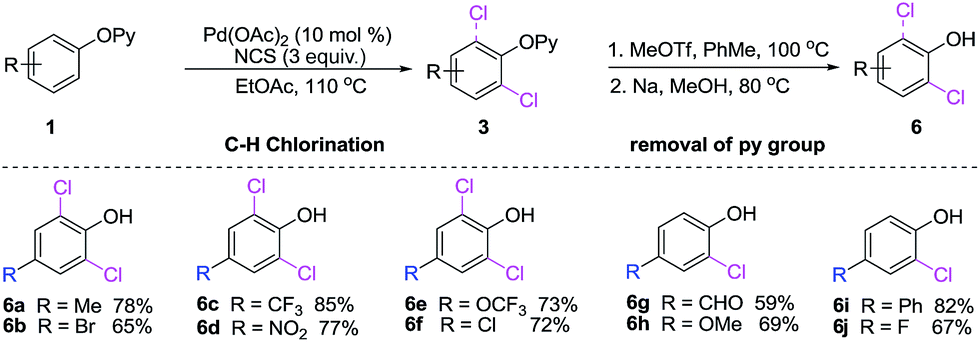 | ||
| Scheme 4 Removal of pyridyl group. | ||
 | ||
| Scheme 5 Sequential C–H functionalization of 2-phenoxypyridine. aReaction conditions: (a) 3l (0.2 mmol), NFSI (3.0 equiv.), Pd(OAc)2 (10 mol%), EtOAc (2.0 mL), 110 °C, under N2, 6 h, isolated yields. (b) 3l (0.2 mmol), AgNO2 (2.0 equiv.), Pd(OAc)2 (10 mol%), K2S2O8 (2.0 equiv.), DCE (2.0 mL), 110 °C, under N2, 48 h, isolated yields. (c) 3l (0.2 mmol), B2pin2 (2.0 equiv.), [RhCp*Cl2]2 (5 mol%), PCy3 (30 mol%), EtOAc (2.0 mL), 100 °C, under N2, 24 h, isolated yields. (d) 3l (0.2 mmol), NBS (3.0 equiv.), Pd(OAc)2 (10 mol%), TsOH (10 mol%), EtOAc (2.0 mL), 110 °C, under N2, 6 h, isolated yields. (e) 3l (0.2 mmol), NIS (3.0 equiv.), Pd(OAc)2 (10 mol%), TsOH (10 mol%), EtOAc (2.0 mL), 110 °C, under N2, 6 h, isolated yields. | ||
With the palladium-catalyzed C–H chlorination protocol in hand, we tried to achieve sequential C–H functionalization access to a variety of polysubstituted phenols. We began using our current monoselective C–H chlorination, which is compatible with various substitutents (3i–m). Three grams of monochlorinated 3l could be prepared in one pot via coupling of 1l with NCS, further functionalizations of 3l were explored. Subsequential C–H fluorination (7a),17 nitration (7b),14 bromination (7d) and iodination (7e) were quite successful, and the highly polysubstituted phenols 7a–e were obtained in good yields. We also developed the Cp*Rh(III)-catalyzed C–H bond borylation of 3l in the presence of PCy3 at 100 °C in EtOAc within 12 h, and 7c was afforded in 57% yield.
Next, we evaluated the utility of this work in the context of late-stage functionalization of known bioactive molecular (Scheme 6). Selective C–H functionalization of a phenyl ring is always a ticklish problem. Diflufenican acts as residual and foliar herbicide, contains two potential directing groups, a phenoxy pyridine and amide functionality. To our delight, its chlorination under the optimized conditions selectively occurred at the para position of aryloxy group gave the monochlorinated product 8a in 94% yield. Meanwhile, to illustrate the chemoselectivity, the current palladium-catalyzed chlorination reaction and direct chlorination in DMF of estrone were comparatively studied (Scheme 6 eqn (2) and eqn (3)), in the presence of the palladium catalyst, the desired chlorinated product 8b was isolated in 55% yield, in contrast, utilizing the aforementioned DMF reaction condition, we didn't observe appreciable chlorination.
 | ||
| Scheme 6 Late-stage C–H chlorination of diflufenican and estrone. | ||
A few control experiments were conducted to shed light on the mechanism of dichlorination reaction. Kinetic isotope effect (KIE) studies, between 2-phenoxypyridine and five-deuterated 2-phenoxypyridine showed a KIE of 1.8 (Scheme 7, eqn (1)). It suggested that the C–H dichlorination of phenols might proceeds the concerted metalation and deprotonation mechanism.20 When complex A12a was used as the catalyst, 2-phenoxypyridine could be smoothly converted to 3a with NCS (Scheme 7, eqn (2)), which suggesting that complex A is probably the catalytically active species.
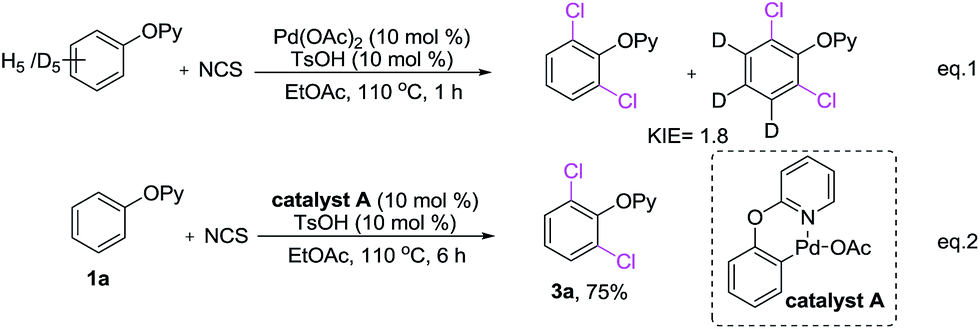 | ||
| Scheme 7 Mechanistic studies. | ||
On the basis of these results and previous literatures, a plausible reaction mechanism was proposed in Scheme 8. The reaction begins with the pyridine-assisted ortho C–H activation of 2-aryloxypyridine to form cyclopalladate complex A, subsequently oxidative addition with NCS generated Pd(IV) intermediate B. Finally, reductive elimination of B afforded the chlorinated product and regenerates the catalyst. PTSA2f is probably to play dual roles in the activation N–Cl bond by protonating a carbonyl group of the NCS, and increasing the electrophilicity of the Pd(II) center by replacement of AcO− with TsO−.
 | ||
| Scheme 8 Proposed reaction mechanism. | ||
Conclusions
In this work, we have described a convenient and straightforward strategy for C–H chlorination/sequential C–H functionalization of phenols, employing 2-pyridyl as the removable group. A variety of 2,4,6-trisubstituted phenols could be readily accessed through this step-by-step difunctionalization of both ortho C–H bonds of phenols. The present protocol could be applied to the late-stage of diflufenican and estrone, to facilitate drug development, especially for new herbicide agent.General information
1H NMR (500 MHz), 13C NMR (125 MHz) and 19F NMR (470 MHz) spectra were recorded in CDCl3 solutions using a 500 MHz spectrometer. Alternatively, 1H NMR (400 MHz), 13C NMR (100 MHz) and 19F NMR (377 MHz) spectra were recorded in CDCl3 solutions using a 400 MHz spectrometer. High-resolution mass spectra were recorded on an ESI-Q-TOF mass spectrometer. All reactions were conducted using standard Schlenk techniques. Column chromatography was performed using EM silica gel 60 (300–400 mesh). 1H NMR and 13C NMR spectra are provided as ESI.† 2-phenoxy pyridine derivatives21 were prepared according to the reported procedures. 1H and 13C spectra of known compounds were in accordance with those described in the literature.General procedure of palladium-catalyzed C–H chlorination of 2-aryloxylpyridine
A 25 mL Schlenk tube equipped with a stir bar was charged with 2-aryloxylpyridine (0.2 mmol), NCS (0.6 mmol), Pd(OAc)2 (10 mol%), TsOH (10 mol%). The tube was fitted with a rubber septum, and then it was evacuated and refilled with nitrogen three times. Under nitrogen, EtOAc (2 mL) were added in turn to the Schlenk tube through the rubber septum using syringes, and then the septum was replaced by a Teflon screw cap under nitrogen flow. The reaction mixture was stirred at 100 °C for 6 h. After cooling down, the reaction mixture was diluted with 10 mL of ethyl ether, filtered through a pad of silica gel, concentrated under reduced pressure. The residue was then purified by flash chromatography on silica gel to provide the corresponding product.General procedure of DMF promoted C–H chlorination of 2-aryloxylpyridine to afford the para-chlorination product
A 25 mL Schlenk tube equipped with a stir bar was charged with 2-aryloxylpyridine NCS (2.0 equiv.), DMF (2 mL) were added in the Schlenk under open air, then obturated with Teflon screwcap. The reaction mixture was stirred at 100 °C for 2 h. After it was cooled, the reaction mixture was diluted with 10 mL of ethyl ether, and filtered through a pad of silica gel, followed by washing the pad of silica gel with the same solvent (20 mL). The filtrate was washed with water (3 × 15 mL). The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was then purified by flash chromatography on silica gel to provide the corresponding product.General procedure of removal of pyridyl group
To a solution of 2-(2,6-dichloro-4-methylphenoxy)pyridine (3b) (101 mg, 0.4 mmol) in dry toluene (10 mL), MeOTf (144 mg, 0.88 mmol) was added. The solution was stirred at 100 °C under N2 atmosphere for 2 h. The reaction mixture was cooled to ambient temperature and the solvent was evaporated under vacuum. The crude product was dissolved in dry methanol (2.0 mL) and then added to a solution of Na (276 mg, 12 mmol) in dry methanol (10 mL) under N2 atmosphere. The reaction mixture was heated to reflux for 30 min, cooled to room temperature. After evaporating the solvent under vacuum, water (30 mL) was added, and the aqueous solution was extracted with EtOAc. The combined organic layer was dried over anhydrous Na2SO4. The solution was concentrated by vacuum and the residue was purified by column chromatography on silica gel (hexane/EtOAc: 10/1) to give the corresponding product 6a.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (40 mg, 83% yield). 1H NMR (500 MHz, CDCl3): δ 8.10 (d, J = 3.5 Hz, 1H), 7.33 (t, J = 7.0 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.14 (t, J = 8.5 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 7.00 (t, J = 6.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 162.0, 147.4, 146.4, 139.6, 129.8, 128.8, 126.4, 118.7, 110.6; HRMS (TIC): calcd for C11H8Cl2NO [M + H]+ 239.9978, found 239.9976.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (37 mg, 90% yield). 1H NMR (500 MHz, CDCl3): δ 8.16–8.15 (m, 1H), 7.66 (t, J = 7.3 Hz, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.06 (d, J = 8.5 Hz, 2H), 6.97 (t, J = 5.6 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 163.4, 152.7, 147.6, 139.6, 129.8, 129.6, 122.6, 118.8, 111.7; HRMS (TIC): calcd for C11H8ClNO [M + H]+ 206.0367, found 206.0365.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (35.1 mg, 69% yield). 1H NMR (500 MHz, CDCl3): δ 8.03 (d, J = 4.5 Hz, 1H), 7.65 (t, J = 7.0 Hz, 1H), 7.13 (s, 2H), 6.97–6.91 (m, 2H), 2.26 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 161.1, 146.4, 142.9, 138.5, 135.7, 128.3, 128.1, 117.6, 109.6, 19.7; HRMS (TIC): calcd for C12H10Cl2NO [M + H]+ 254.0134, found 254.0131.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (35.1 mg, 69% yield). 1H NMR (500 MHz, CDCl3): δ 8.08 (d, J = 4.5 Hz, 1H), 7.74 (t, J = 6.5 Hz, 1H), 7.40 (s, 2H), 7.08–7.01 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 161.7, 147.3, 145.4, 139.8, 131.0, 130.4, 128.7, 128.1, 121.7, 118.9, 110.7; HRMS (TIC): calcd for C11H7Cl3NO [M + H]+ 273.9588 found 273.9591.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (59.6 mg, 94% yield). 1H NMR (500 MHz, CDCl3): δ 8.09 (d, J = 4.5 Hz, 1H), 7.75 (t, J = 7.5 Hz, 1H), 7.55 (s, 2H), 7.07 (d, J = 8.0 Hz, 1H), 7.03 (t, J = 5.5 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 162.0, 147.4, 146.4, 139.6, 129.8, 128.8, 128.1, 16.4, 121.7, 118.7, 110.6; HRMS (TIC): calcd for C11H7BrCl2NO [M + H]+ 317.9083, found 317.9085.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (59.1 mg, 99% yield). 1H NMR (500 MHz, CDCl3): δ 8.07 (s, 3H), 7.76 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 7.03 (t, J = 5.0 Hz, 1H), 3.95 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 164.6, 161.7, 150.2, 147.3, 139.9, 130.1, 130.0, 128.6, 119.1, 110.8, 52.7; HRMS (TIC): calcd for C13H10Cl2NO3 [M + H]+ 298.0032, found 298.0030.:1 petroleum ether–EtOAc as the eluant afforded a white liquid (56.2 mg, 99% yield). 1H NMR (500 MHz, CDCl3): δ 8.30 (s, 2H), 8.04 (d, J = 6.0 Hz, 1H), 7.80 (t, J = 8.5 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 7.07 (t, J = 7.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 161.4, 152.1, 147.2, 144.8, 140.1, 131.1, 124.2, 119.5, 110.8; HRMS (TIC): calcd for C11H7Cl2N2O3 [M + H]+ 284.9828, found 284.9825.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (56.2 mg, 92% yield). 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 5.0 Hz, 1H), 7.77 (t, J = 6.5 Hz, 1H), 7.67 (s, 2H), 7.11 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 7.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 161.5, 149.5, 147.3, 140.0, 130.8, 128.9 (q, JF = 33.8 Hz), 126.0 (q, JF = 3.8 Hz), 122.7 (q, JF = 271.2 Hz), 119.2, 110.8. 19F NMR (470 MHz, CDCl3): δ −62.6 (s, 1F); HRMS (TIC): calcd for C12H7Cl2F3NO [M + H]+ 307.9852, found 307.9850.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (59.3 mg, 92% yield). 1H NMR (500 MHz, CDCl3): δ 8.09 (d, J = 6.5 Hz, 1H), 7.75 (t, J = 7.0 Hz, 1H), 7.30 (s, 2H), 7.08 (d, J = 8.5 Hz, 1H), 7.03 (t, J = 7.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 161.7, 147.3, 145.7 (q, JF = 1.2 Hz), 145.5, 139.8, 130.6, 121.6, 120.3 (q, JF = 256.2 Hz), 119.1, 110.7; 19F NMR (470 MHz, CDCl3): δ −58.1 (s, 3F); HRMS (TIC): calcd for C12H7Cl2F3NO2 [M + H]+ 323.9801, found 323.9800.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (45.6 mg, 81% yield). 1H NMR (500 MHz, CDCl3): δ 8.13 (d, J = 6.0 Hz, 1H), 7.75 (t, J = 8.5 Hz, 1H), 7.60–7.54 (m, 4H), 7.46–7.37 (m, 4H), 7.10–7.01 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 172.2, 162.1, 147.4, 145.5, 140.0, 137.7, 138.3, 129.9, 129.0, 128.2, 127.4, 127.0, 121.5, 118.8, 110.7; HRMS (TIC): calcd for C17H13ClNO [M + H]+ 282.0680, found 282.0684.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (21.3 mg, 46% yield). 1H NMR (500 MHz, CDCl3): δ 9.96 (s, 1H), 8.16 (d, J = 3.0 Hz, 1H), 8.01 (s, 1H), 7.84–7.76 (m, 2H), 7.36 (d, J = 8.5 Hz, 1H), 7.09–7.06 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 189.8, 162.2, 154.9, 147.1, 140.4, 133.6, 131.4, 129.2, 128.3, 123.7, 119.5, 111.8; HRMS (TIC): calcd for C12H9ClNO2 [M + H]+ 234.0317, found 234.0315.:1 petroleum ether–EtOAc as the eluant afforded white liquid (23.3 mg, 51% yield). 1H NMR (500 MHz, CDCl3): δ 8.14 (d, J = 3.5 Hz, 1H), 7.79–7.76 (m, 2H), 7.61–7.59 (m, 1H), 7.32 (d, J = 8.5 Hz, 1H), 7.09–7.07 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 162.0, 154.0, 147.4, 140.0, 134.3, 131.7, 128.3, 124.2, 119.6, 117.4, 111.8, 109.6; HRMS (TIC): calcd for C12H8ClN2O [M + H]+ 231.0320, found 231.0321.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (29.4 mg, 63% yield). 1H NMR (500 MHz, CDCl3): δ 8.14 (d, J = 5.5 Hz, 1H), 7.71–7.66 (m, 1H), 7.31–7.20 (m, 1H), 7.03–6.91 (m, 4H), 3.74 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 162.4, 156.8, 147.4, 140.0, 139.5, 129.8, 118.6, 114.5, 110.7, 110.5, 102.0, 55.9; HRMS (TIC): calcd for C12H11ClNO2 [M + H]+ 236.0473, found 236.0470.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (51.4 mg, 76% yield). 1H NMR (500 MHz, CDCl3): δ 8.18–8.12 (m, 1H), 7.72–7.66 (m, 1H), 7.23–7.17 (m, 1H), 7.10–6.97 (m, 3H), 6.90 (d, J = 8.5 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 163.3 (d, JF = 98.8 Hz), 160.4 (d, JF = 22.5 Hz), 158.5 (d, JF = 26.2 Hz), 147.5 (d, JF = 26.2 Hz), 139.5 (d, JF = 15.0 Hz), 124.7 (d, JF = 10.0 Hz), 122.7 (d, JF = 8.8 Hz), 118.6 (d, JF = 20.0 Hz), 117.6 (d, JF = 26.2 Hz), 116.2 (d, JF = 22.5 Hz), 111.2 (d, JF = 46.2 Hz); 19F NMR (470 MHz, CDCl3): δ −118.5 (s, 1F); HRMS (TIC): calcd for C11H8ClFNO [M + H]+ 224.0273, found 224.0275.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (38.1 mg, 80% yield). 1H NMR (400 MHz, CDCl3): δ 8.26–8.21 (m, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.54–7.26 (m, 3H), 7.04–6.95 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 162.5, 150.4, 147.5, 139.8, 133.0, 131.1, 128.3, 126.2, 124.2, 119.0, 111.3; HRMS (TIC): calcd for C11H8Cl2NO [M + H]+ 239.9978, found 239.9976.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (49.7 mg, 88% yield). 1H NMR (400 MHz, CDCl3): δ 8.19 (s, 1H), 7.77 (t, J = 7.2 Hz, 1H), 7.42 (s, 1H), 7.39–7.34 (m, 2H), 7.06–7.04 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 162.5, 150.3, 147.5, 139.8, 131.5, 129.1, 127.0, 126.6, 120.4, 119.1, 111.5; HRMS (TIC): calcd for C11H8BrClNO [M + H]+ 283.9473, found 283.9474.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (34.8 mg, 79% yield). 1H NMR (400 MHz, CDCl3): δ 8.27–8.22 (m, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.41–7.32 (m, 1H), 7.09–7.02 (m, 3H), 6.95 (d, J = 8.0 Hz, 1H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 162.1, 148.4, 146.5, 138.4, 137.2, 129.1, 125.9, 123.3, 123.1, 117.4, 110.0, 20.0; HRMS (TIC): calcd for C12H11ClNO [M + H]+ 220.0524, found 220.0523.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (35.5 mg, 81% yield). 1H NMR (400 MHz, CDCl3): δ 8.17 (s, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.30 (s, 1H), 7.16–7.12 (m, 1H), 7.01–6.99 (m, 2H), 2.23 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 161.5, 147.0, 146.5, 138.5, 132.7, 128.4, 126.9, 126.8, 124.9, 117.2, 109.2, 15.8; HRMS (TIC): calcd for C12H11ClNO [M + H]+ 220.0524, found 220.0523.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (40.1 mg, 71% yield). 1H NMR (500 MHz, CDCl3): δ 8.10 (d, J = 4.5 Hz, 1H), 7.74 (t, J = 7.0 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.09–6.99 (m, 3H); 13C NMR (125 MHz, CDCl3): δ 161.9, 147.5, 147.3, 139.6, 131.8, 129.7, 129.5, 126.9, 118.9, 118.7, 110.7; HRMS (TIC): calcd for C11H8BrClNO [M + H]+ 283.9473, found 283.9474.:1 petroleum ether–EtOAc as the eluant afforded yellow liquid (25.7 mg, 47% yield). 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 1H), 7.79 (t, J = 7.2 Hz, 1H), 7.70 (t, J = 8.4 Hz, 2H), 7.38–7.32 (m, 2H), 7.12 (d, J = 8.0 Hz, 1H), 7.06 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 163.1, 151.8, 147.5, 139.6, 138.4, 132.9, 127.1 (q, JF = 5.0 Hz), 124.5, 123.6, 123.0 (q, JF = 151.2 Hz), 119.0; 19F NMR (470 MHz, CDCl3): δ −61.8 (s, 3F); HRMS (TIC): calcd for C12H8ClF3NO [M + H]+ 274.0241, found 274.0240.:1 petroleum ether–EtOAc as the eluant afforded a brown liquid (40.1 mg, 77% yield). 1H NMR (500 MHz, DMSO-d6): δ 8.17 (d, J = 4.5 Hz, 1H), 7.85 (t, J = 7.5 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H), 7.22 (d, J = 7.5 Hz, 1H), 7.15–7.10 (m, 2H), 7.01–6.91 (m, 1H), 1.31 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 162.0, 147.1, 146.6, 143.7, 138.3, 128.0, 127.4, 124.9, 124.6, 116.9, 109.7, 34.3, 29.5, 28.2, 17.4; HRMS (TIC): calcd for C15H17ClNO [M + H]+ 262.0993, found 262.0990.:1 petroleum ether–EtOAc as the eluant afforded a brown liquid (42.3.3 mg, 70% yield). 1H NMR (500 MHz, CDCl3): δ 8.01 (d, J = 5.0 Hz, 1H), 7.66 (t, J = 6.5 Hz, 1H), 7.33 (s, 1H), 7.00 (d, J = 8.5 Hz, 1H), 6.93 (t, J = 7.0 Hz, 1H), 3.65 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 161.5, 159.4, 148.7, 146.2, 138.6, 123.5, 121.4, 118.6, 117.8, 109.8, 51.1; HRMS (TIC): calcd for C11H8Cl2NO3S [M + H]+ 303.9597, found 303.9595.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (48.4 mg, 92% yield). 1H NMR (500 MHz, CDCl3): δ 8.00 (d, J = 4.0 Hz, 1H), 7.66 (t, J = 7.0 Hz, 1H), 7.08 (t, J = 8.0 Hz, 2H), 6.99–6.92 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 160.8, 158.7, 156.7, 146.2, 142.1, 138.7, 129.3 (d, JF = 12.5 Hz), 117.9, 115.2 (d, JF = 25.0 Hz), 109.6; 19F NMR (470 MHz, CDCl3): δ −113.9 (s, 1F); HRMS (TIC): calcd for C11H7Cl2FNO [M + H]+ 257.9883, found 257.9885.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (14.1 mg, 23% yield). 1H NMR (500 MHz, CDCl3): δ 8.25 (d, J = 4.5 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 7.0 Hz, 1H), 7.42–7.33 (m, 1H), 7.25–7.22 (m, 1H), 7.11–7.09 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 159.6, 150.7, 149.0, 137.1 (q, J F = 5.0 Hz), 130.6, 128.1 (q, J F = 108.8 Hz), 127.9, 126.7, 120.0 (q, J F = 271.2 Hz), 117.9; 19F NMR (470 MHz, CDCl3): δ −63.4 (s, 3F); HRMS (TIC): calcd for C12H7Cl2F3NO [M + H]+ 307.9852, found 307.9850.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (41.1 mg, 81% yield). 1H NMR (500 MHz, CDCl3): δ 7.81 (d, J = 5.0 Hz, 1H), 7.45 (d, J = 7.5 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.04 (t, J = 8.0 Hz, 1H), 6.84–6.82 (m, 1H), 2.35 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 159.3, 145.9, 143.4, 138.7, 128.7, 127.6, 125.1, 119.8, 117.8, 14.8; HRMS (TIC): calcd for C12H10Cl2NO [M + H]+ 254.0134, found 254.0131.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (40.8 mg, 79% yield). 1H NMR (500 MHz, CDCl3): δ 7.84 (s, 1H), 7.26–7.18 (m, 1H), 7.11 (t, J = 8.5 Hz, 3H), 7.00 (s, 1H); 13C NMR (125 MHz, CDCl3): δ 155.7 (d, JF = 6.2 Hz), 150.6 (d, JF = 200.0 Hz), 144.0 (d, JF = 75 Hz), 143.1, 129.9 (d, JF = 45 Hz), 126.8 (d, JF = 81.2 Hz), 124.4 (d, JF = 8.8 Hz), 123.4 (d, JF = 31.2 Hz), 118.8 (d, JF = 18.8 Hz); 19F NMR (470 MHz, CDCl3): δ −137.7 (s, 1F); HRMS (TIC): calcd for C11H7Cl2FNO [M + H]+ 257.9883, found 257.9885.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (40.1 mg, 73% yield). 1H NMR (500 MHz, CDCl3): δ 7.96 (d, J = 3.5 Hz, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.17 (t, J = 8.0 Hz, 1H), 7.01–6.98 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 157.3, 146.3, 144.9, 139.5, 129.6, 128.8, 126.7, 119.8, 118.3; HRMS (TIC): calcd for C11H7Cl3NO [M + H]+ 273.9588, found 273.9591.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (40.4 mg, 76% yield). 1H NMR (500 MHz, CDCl3): δ 8.40 (d, J = 2.0 Hz, 1H), 7.99 (d, J = 6.5 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.22–7.19 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 163.7, 151.9, 142.6, 129.4, 128.9, 127.2, 122.0, 120.9, 116.5, 111.5; HRMS (TIC): calcd for C12H7Cl2N2O [M + H]+ 264.9930, found 264.9927.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (53.3 mg, 98% yield). 1H NMR (500 MHz, CDCl3): δ 8.03 (d, J = 2.0 Hz, 1H), 7.71–7.68 (m, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.16 (t, J = 8.0 Hz, 1H), 7.03 (d, J = 9.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 160.3, 146.1, 145.7, 139.6, 129.7, 128.8, 126.7, 126.3, 111.7; HRMS (TIC): calcd for C11H7Cl3NO [M + H]+ 273.9588, found 273.9591.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (18.7 mg, 33% yield). 1H NMR (500 MHz, CDCl3): δ 8.98 (d, J = 2.5 Hz, 1H), 8.55 (d, J = 6.0 Hz, 1H), 7.43 (d, J = 8.5 Hz, 2H), 7.24–7.21 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 164.8, 145.6, 144.8, 141.0, 135.3, 129.3, 128.9, 127.3, 111.0; HRMS (TIC): calcd for C11H7Cl2N2O3 [M + H]+ 284.9828, found 284.9825.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (35.0 mg, 65% yield). 1H NMR (500 MHz, CDCl3): δ 8.06 (d, J = 3.5 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.05 (d, J = 9.0 Hz, 1H), 6.93 (d, J = 3.0 Hz, 2H), 6.90–6.76 (m, 2H), 3.72 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 156.0, 154.9, 153.6, 140.7, 129.2, 124.8, 121.1, 110.6, 109.2, 54.6; HRMS (TIC): calcd for C12H10Cl2NO2 [M + H]+ 270.0083, found 270.0084.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (36.6 mg, 68% yield). 1H NMR (400 MHz, CDCl3): δ 7.56 (t, J = 7.2 Hz, 1H), 7.41 (t, J = 6.4 Hz, 2H), 7.22–7.15 (m, 3H), 6.90 (d, J = 6.8 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 2.50 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 161.3, 154.4, 139.7, 129.7, 124.9, 124.6, 122.1, 120.2, 109.4, 22.4; HRMS (TIC): calcd for C12H10Cl2NO [M + H]+ 254.0134, found 254.0131.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (26.7 mg, 53% yield). 1H NMR (500 MHz, CDCl3): δ 8.12 (d, J = 4.5 Hz, 1H), 7.72 (t, J = 7.5 Hz, 1H), 7.05–7.00 (m, 2H), 6.95 (s, 1H), 6.82–6.68 (m, 1H), 3.80 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 163.2, 155.8, 146.3 (d, JF = 3.8 Hz), 138.6 (d, JF = 7.5 Hz), 134.5, 128.7 (d, JF = 23.8 Hz), 127.4, 117.7 (d, JF = 15.0 Hz), 113.5, 109.6 (d, JF = 27.5 Hz), 101.0 (d, JF = 22.5 Hz), 54.9; 19F NMR (470 MHz, CDCl3): δ −123.4 (s, 1F); HRMS (TIC): calcd for C12H10ClFNO2 [M + H]+ 254.0379, found 254.0382.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (35.3 mg, 68% yield). 1H NMR (500 MHz, CDCl3): δ 8.06 (d, J = 4.5 Hz, 1H), 7.60 (t, J = 8.0 Hz, 1H), 7.05 (d, J = 9.0 Hz, 1H), 6.93–6.87 (m, 2H), 6.78–6.76 (m, 1H), 3.72 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 161.2, 155.8, 146.4, 138.9, 138.5, 128.8, 124.4, 117.6, 113.5, 112.5, 109.5, 54.9; HRMS (TIC): calcd for C12H10ClN2O4 [M + H]+ 281.0324, found 281.0323.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (41.1 mg, 57% yield). 1H NMR (500 MHz, CDCl3): δ 8.05 (d, J = 4.5 Hz, 1H), 7.66 (t, J = 8.5 Hz, 1H), 6.96–6.92 (m, 2H), 6.88 (s, 2H), 3.74 (s, 3H), 1.51 (s, 6H), 1.18 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 162.3, 156.8, 147.5, 140.0, 139.5, 129.8, 126.1, 125.4, 118.6, 114.5, 113.6, 110.6, 83.5, 65.6, 56.7, 55.9, 25.0, 24.6; HRMS (TIC): calcd for C18H22BClNO4 [M + H]+ 362.1325, found 362.1323.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (32.3 mg, 52% yield). 1H NMR (500 MHz, CDCl3): δ 8.11 (s, 1H), 7.71 (t, J = 8.0 Hz, 1H), 7.04–6.98 (m, 2H), 6.94 (s, 2H), 3.80 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 162.3, 156.8, 147.4, 139.9, 139.5, 129.8, 125.4, 118.6, 114.5, 111.0, 110.6, 55.9; HRMS (TIC): calcd for C12H10BrClNO2 [M + H]+ 313.9578, found 313.9580.:1 petroleum ether–EtOAc as the eluant afforded a light yellow liquid (25.3 mg, 35% yield). 1H NMR (500 MHz, CDCl3): δ 8.05 (d, J = 5.0 Hz, 1H), 7.66 (t, J = 7.5 Hz, 1H), 6.97–6.93 (m, 2H), 6.88 (s, 2H), 3.74 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 163.4, 157.2, 147.5, 143.3, 139.4, 127.8, 124.4, 118.3, 115.6, 113.7, 110.8, 55.8; HRMS (TIC): calcd for C12H11ClINO2 [M + H]+ 361.9439, found 361.9437.:1 petroleum ether–EtOAc as the eluant afforded a light brown liquid (80.7 mg, 94% yield). 1H NMR (500 MHz, CDCl3): δ 9.82 (s, 1H), 8.71–8.69 (m, 1H), 8.52–8.47 (m, 1H), 8.24–8.22 (m, 1H), 7.68–7.55 (m, 3H), 7.29–7.26 (m, 1H), 6.95–6.87 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 161.0, 158.8 (dd, JF = 245.0, 11.2 Hz), 158.6, 152.9 (dd, JF = 246.2, 11.2 Hz), 150.4, 148.5, 142.9, 131.4, 131.2, 130.6 (q, JF = 33.8 Hz), 124.2, 123.9 (q, JF = 3.8 Hz), 123.1 (d, JF = 7.5 Hz), 122.8 (dd, JF = 10.0, 3.8 Hz), 122.0 (q, JF = 3.8 Hz), 120.4, 116.6, 111.3 (dd, JF = 21.2, 3.8 Hz), 103.6 (dd, JF = 26.2, 23.8 Hz); 19F NMR (470 MHz, CDCl3): δ −125.3 (s, 1F), −114.5 (s, 1F), −62.5 (s, 3F). HRMS (TIC): calcd for C19H11ClF5N2O2 [M + H]+ 429.0424, found 429.0423.:1 petroleum ether–EtOAc as the eluant afforded a yellow liquid (46.5 mg, 55% yield). 1H NMR (400 MHz, CDCl3): δ 8.22 (s, 1H), 7.77 (t, J = 6.8 Hz, 1H), 7.39 (s, 1H), 7.05–7.01 (m, 3H), 3.06–3.03 (m, 1H), 2.93 (s, 2H), 2.59–2.52 (m, 1H), 2.46–2.41 (m, 2H), 2.19–2.09 (m, 2H), 1.94–1.90 (m, 2H), 1.74–1.52 (m, 4H), 1.30 (s, 1H), 1.17 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 205.6, 162.9, 147.4, 147.2, 139.9, 137.4, 136.3, 127.4, 124.3, 123.8, 118.5, 111.2, 82.9, 47.3, 45.8, 45.7, 43.8, 36.5, 32.3, 28.7, 26.2, 25.2, 15.8; HRMS (TIC): calcd for C26H31ClNO2 [M + H]+ 424.2038, found 424.2039.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the National Natural Science Foundation of China (21602158, 21372177, and 21472140), State Key Laboratory of Structural Chemistry (No. 20170037), Zhejiang Provincial Natural Science Foundation (LY16B020011), Wenzhou Medical University start-up funding (QTJ15026) and Granted from the Opening Project of Zhejiang Provincial Top Key Discipline of Pharmaceutical Sciences (201723) are greatly appreciated.Notes and references
- Review: D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003 CrossRef CAS PubMed.
- For selected examples of palladium-catalyzed ortho C-H chlorniation, see: (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) X. Wan, Z. Ma, B. Li, K. Zhang, S. Cao, S. Zhang and Z. Shi, J. Am. Chem. Soc., 2006, 128, 7416 CrossRef CAS PubMed; (c) X. Zhao, E. Dimitrijević and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466 CrossRef CAS PubMed; (d) A. S. Dudnik, N. Chernyak, C. Huang and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 8729 CrossRef CAS PubMed; (e) R. B. Bedford, C. J. Mitchell and R. L. Webster, Chem. Commun., 2010, 46, 3095 RSC; (f) R. B. Bedford, M. F. Haddow, C. J. Mitchell and R. L. Webster, Angew. Chem., Int. Ed., 2011, 50, 5524 CrossRef CAS PubMed; (g) P. P. Singh, T. Thatikonda, K. A. A. Kumar, S. D. Sawant, B. Singh, A. K. Sharma, P. R. Sharma, D. Singh and R. A. Vishwakarma, J. Org. Chem., 2012, 77, 5823 CrossRef CAS PubMed; (h) X. Sun, G. Shan, Y. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440 CrossRef CAS PubMed; (i) P. Sadhu, S. K. Alla and T. Punniyamurthy, J. Org. Chem., 2013, 78, 6104 CrossRef CAS PubMed; (j) B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786 CrossRef CAS PubMed; (k) X. Sun, Y. Sun, C. Zhang and Y. Rao, Chem. Commun., 2014, 50, 1262 RSC; (l) F. M. Moghaddam, G. Tavakoli, B. Saeednia, P. Langer and B. Jafari, J. Org. Chem., 2016, 81, 3868 CrossRef CAS PubMed; (m) R. Das and M. Kapur, J. Org. Chem., 2017, 82, 1114 CrossRef CAS PubMed; (n) S. P. Zucker, F. Wossidlo, M. Weber, D. Lentz and C. C. Tzschucke, J. Org. Chem., 2017, 82, 5616 CrossRef CAS PubMed; (o) M. Dabiri, N. F. Lehi, S. K. Movahed and H. R. Khavasi, Org. Biomol. Chem., 2017, 15, 6264 RSC; Copper-catalyzed C-H chlorination, see: (p) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed; Nickel-catalyzed C-H chlorination, (q) B.-B. Zhan, Y.-H. Liu, F. Hu and B.-F. Shi, Chem. Commun., 2016, 52, 4934 RSC; Rhodium-catalyzed C-H chlorination, see: (r) G. Qian, X. Hong, B. Liu, H. Mao and B. Xu, Org. Lett., 2014, 16, 5294 CrossRef CAS PubMed; (s) F. Lied, A. Lerchen, T. Mück-Lichtenfeld, C. Knecht and F. Glorius, ACS Catal., 2016, 6, 7839 CrossRef CAS. Iron-catalyzed, see: (t) M. A. B. Mostafa, R. M. Bowley, D. T. Racys, M. C. Henry and A. Sutherland, J. Org. Chem., 2017, 82, 7529 CrossRef CAS PubMed.
- A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2004 Search PubMed.
- Handbook of Grignard Reagents, ed. G. S. Silverman and P. E. Rakita, Marcel Dekker, New York, 1996 Search PubMed.
- (a) M. Lang, P. Spiteller, V. Hellwig and W. Steglich, Angew. Chem., Int. Ed., 2001, 40, 1704 CrossRef CAS; (b) A. P. Vartak and P. A. Crooks, Org. Process Res. Dev., 2009, 13, 415 CrossRef CAS; (c) E. J. Peters and S. A. Lowance, Agron. J., 1970, 62, 400 CrossRef CAS; (d) M. Alam, M. Gleason, X. Zhang and A. Rehman, Phytopathology, 2016, 106, 184 Search PubMed.
- M. A. Keane, S. Gomez-Quero, F. Cardenas-Lizana and W. Shen, ChemCatChem, 2009, 1, 270 CrossRef CAS.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Chem.–Eur. J., 2015, 21, 2855 CrossRef CAS PubMed.
- E. Marzi, F. Mongin, A. Spitaleri and M. Schlosser, Eur. J. Org. Chem., 2001, 2911 CrossRef CAS.
- N. I. Saper and B. B. Snider, J. Org. Chem., 2014, 79, 809 CrossRef CAS PubMed.
- S. M. Maddox, A. N. Dinh, F. Armenta, J. Um and J. L. Gustafson, Org. Lett., 2016, 18, 5476 CrossRef CAS PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed.
- (a) J.-H. Chu, P.-S. Lin and M.-J. Wu, Organometallics, 2010, 29, 4058 CrossRef CAS; (b) L. Ackermann, E. Diers and A. Manvar, Org. Lett., 2012, 14, 1154 CrossRef CAS PubMed.
- W. Zhang, J. Zhang, S. Ren and Y. Liu, J. Org. Chem., 2014, 79, 11508 CrossRef CAS PubMed.
- B. Liu, H.-Z. Jiang and B.-F. Shi, J. Org. Chem., 2014, 79, 1521 CrossRef CAS PubMed.
- (a) J. Yao, R. Feng, Z. Wu, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2013, 355, 1517 CrossRef CAS; (b) D. C. McAteer, E. Javed, L. Huo and S. Huo, Org. Lett., 2017, 19, 1606 CrossRef CAS PubMed; (c) J.-H. Chu, S.-T. Chen, M.-F. Chiang and M.-J. Wu, Organometallics, 2015, 34, 953 CrossRef CAS.
- S.-J. Lou, Q. Chen, Y.-F. Wang, D.-Q. Xu, X.-H. Du, J.-Q. He, Y.-J. Mao and Z.-Y. Xu, ACS Catal., 2015, 5, 2846–2849 CrossRef CAS.
- L. Wang, L. Pan, Y. Huang, Q. Chen and M. He, Eur. J. Org. Chem., 2016, 18, 3113 CrossRef.
- C. Zhang and P. Sun, J. Org. Chem., 2014, 79, 8457 CrossRef CAS PubMed.
- Y. Xu, P. Liu, S.-L. Li and P. Sun, J. Org. Chem., 2015, 80, 1269 CrossRef CAS PubMed.
- (a) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754 CrossRef CAS PubMed; (b) D. Garcia-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef CAS PubMed.
- D. Maiti and S. L. Buchwald, J. Org. Chem., 2010, 75, 1791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectral data. See DOI: 10.1039/c7ra09939h |
| This journal is © The Royal Society of Chemistry 2017 |